

Abstract
The title compound, C12H6Cl4S2, features an S—S bond [2.0252 (8) Å] that bridges two 2,3-dichlorophenyl rings with a C—S—S—C torsion angle of 88.35 (11)°. The benzene rings are normal one to the other with a dihedral angle of 89.83 (11)°. The crystal structure features intermolecular Cl⋯Cl [3.4763 (11) Å] and π–π stacking interactions [centroid–centroid distances = 3.696 (1) and 3.641 (2) Å]. Intramolecular C—H⋯S interactions are also observed.
Related literature
For applications of disulfide compounds, see: Crowley (1964 ▶); Hashash et al. (2002 ▶); Gomez-Benitez et al. (2006 ▶); Yu et al. (2010 ▶). For various methods of synthesizing disulfides, see: Xiao et al. (2009 ▶); Shaabani et al. (2008 ▶); Ogilby (2010 ▶). For similar compounds and their crystal structures, see: Deng et al. (2003 ▶); Korp & Bernal (1984 ▶); Tang et al. (2011 ▶). For disulfide bonds in proteins, see: Sevier & Kaiser (2006 ▶). For van der Waals radii, see: Bondi (1964 ▶).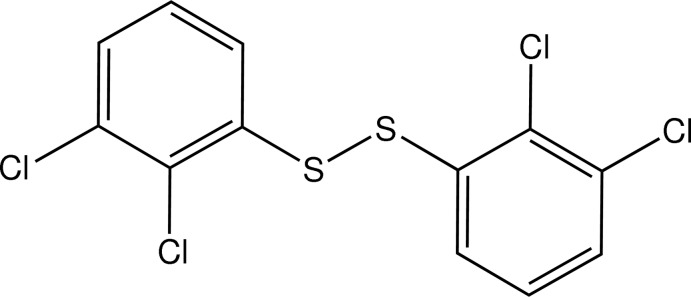
Experimental
Crystal data
C12H6Cl4S2
M r = 356.09
Triclinic,

a = 7.7149 (10) Å
b = 7.7326 (11) Å
c = 12.748 (2) Å
α = 91.472 (2)°
β = 91.233 (3)°
γ = 114.859 (2)°
V = 689.37 (18) Å3
Z = 2
Mo Kα radiation
μ = 1.14 mm−1
T = 298 K
0.37 × 0.24 × 0.14 mm
Data collection
Bruker SMART APEX CCD diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 2008 ▶) T min = 0.678, T max = 0.862
7044 measured reflections
3130 independent reflections
2594 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.035
wR(F 2) = 0.088
S = 1.03
3130 reflections
163 parameters
H-atom parameters constrained
Δρmax = 0.41 e Å−3
Δρmin = −0.30 e Å−3
Data collection: APEX2 (Bruker, 2012 ▶); cell refinement: SAINT (Bruker, 2012 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 2012 ▶) and DIAMOND (Brandenburg, 2006 ▶); software used to prepare material for publication: SHELXTL (Sheldrick, 2008 ▶) and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814007326/bx2456sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814007326/bx2456Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814007326/bx2456Isup3.cml
CCDC reference: 994982
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6⋯S2 | 0.93 | 2.70 | 3.202 (2) | 115 |
| C12—H12⋯S1 | 0.93 | 2.70 | 3.199 (2) | 115 |
Acknowledgments
This work was supported by CONACYT (grant No. CB2010–154732) and PAPIIT (grants IN201711–3 and IN213214–3). ESJ thanks PROMEP "Apoyo a perfil deseable". RRM and DMM thank Dr Ruben A. Toscano for technical assistance.
supplementary crystallographic information
1. Introduction
The disulfide bonds are found in proteins (Sevier and Kaiser, 2006), natural products and pharmacologically active compounds. Disulfide compounds have shown to exhibit activity as fungicide, mildew-proofing (Crowley, 1964) and antitumor agents (Hashash et al., 2002). In organic synthesis disulfides are used in cross-coupling reactions catalyzed by transition metal compounds such as palladium, nickel and copper (Gomez-Benitez et al., 2006; Yu et al., 2010).
Several methods for the synthesis of disulfides have been reported. These processes involve the oxidative coupling of mercaptans by various oxidants such as molecular oxygen, nitric oxide, solvent-free permanganate, metal ions and promoted by sulfonyl chloride in aqueous media (Xiao et al., 2009; Shaabani et al., 2008; Ogilby, 2010).
Thus, in this report we present the crystal structure of the bis(2,3-dichlorophenyl)disulfide obtained by a nucleophilic substitution reaction. The structure is represented in figure 1.
2. Experimental
2.1. Synthesis and crystallization
The title compound was obtained as a by-product of the reaction between 2-(chloromethyl)benzimidazole (0.2 g) and the lead salt of 2,3-dichlorobenzethiol ([Pb(SC6H3-2,3-Cl2)2]) (0.337 g) in toluene. The resulting reaction mixture was allowed to proceed under reflux by 8 h after which time the formation of PbCl2 was observed indicating completion of the reaction. The reaction mixture was then filtered through a short Celite plug to afford a colorless solution, the solvent was evaporated under vacuum and the residue column chromatographed (silica gel 60, eluted with 3/2 ethyl acetate/hexane system). Slow Evaporation of the first fraction collected produced crystals of the title compound suitable for X-ray diffraction analysis.
2.2. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 1.
H atoms were included in calculated positions (C—H = 0.93 A for aromatic H) and refined using a riding model with Uiso(H) = 1.2 Ueq of the carrier atoms.
In the refinement six reflections, (2 0 0), (0 1 3), (0 0 1), (-2 1 2), (2 -4 3) and (-1 0 1), were considered as disagreeable and were omitted.
3. Results and discussion
The asymmetric unit of the title compound consists of one molecule on the disulfide. The rings of the bis(2,3-dichlorophenyl)disulfide show a dihedral angle of 89.83° between the two planes and a torsion angle C1—S1—S2—C7 of 88.35 (11)°. The value of the C—S—S—C torsion angle is similar to those found in similar compounds, such as bis(pentachlorophenyl)disulfide (Deng et al., 2003), diphenyldisulfide (Korp & Bernal, 1984) and bis(4-amino-2-chlorophenyl)disulfide (Tang et al., 2011). The S—S distance is 2.0252 (8) Å, whereas the C—S distances are 1.784 (2) and 1.7835 (19) Å. These values are similar and close in value to compounds such as bis(pentachlorophenyl)disulfide with a S—S distance of 2.063 (2) Å and bis(4-amino-2-chlorophenyl)disulfide of 2.0671 (16) Å. The crystal packing is stabilized by π-π and Cl···Cl interactions (Figure 2). The π-π interactions of the 2,3-dichlorophenyl rings presents distances between centroids of 3.696 (1) and 3.641 (2) Å. The Cl1···Cl2 contact distance is of 3.476 Å that is close to the sum of the van der Waals radii of the chloride atoms (Bondi, 1964). The sulphur atoms present C—H···S intramolecular interactions, these values are in the table 1.
Figures
Fig. 1.

The molecular structure of the title compound showing 40% probability of displacement ellipsoids for the non-hydrogen atoms.
Fig. 2.

Representation of the π-π and Cl···Cl interactions shown by dashed lines. Hydrogen atoms are omitted.
Crystal data
| C12H6Cl4S2 | Z = 2 |
| Mr = 356.09 | F(000) = 356 |
| Triclinic, P1 | Dx = 1.715 Mg m−3 |
| a = 7.7149 (10) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.7326 (11) Å | Cell parameters from 4288 reflections |
| c = 12.748 (2) Å | θ = 2.8–27.5° |
| α = 91.472 (2)° | µ = 1.14 mm−1 |
| β = 91.233 (3)° | T = 298 K |
| γ = 114.859 (2)° | Prism, colourless |
| V = 689.37 (18) Å3 | 0.37 × 0.24 × 0.14 mm |
Data collection
| Bruker SMART APEX CCD diffractometer | 3130 independent reflections |
| Radiation source: fine-focus sealed tube | 2594 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.023 |
| Detector resolution: 8.333 pixels mm-1 | θmax = 27.5°, θmin = 2.9° |
| ω scans | h = −9→9 |
| Absorption correction: multi-scan (SADABS; Sheldrick, 2008) | k = −10→10 |
| Tmin = 0.678, Tmax = 0.862 | l = −16→16 |
| 7044 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.035 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.088 | H-atom parameters constrained |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0397P)2 + 0.1749P] where P = (Fo2 + 2Fc2)/3 |
| 3130 reflections | (Δ/σ)max < 0.001 |
| 163 parameters | Δρmax = 0.41 e Å−3 |
| 0 restraints | Δρmin = −0.30 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Cl1 | 0.81619 (10) | 0.72799 (9) | 0.12199 (5) | 0.06495 (19) | |
| Cl2 | 1.23381 (10) | 0.95252 (8) | 0.04969 (5) | 0.0714 (2) | |
| Cl3 | 0.64782 (10) | −0.26178 (7) | 0.37441 (5) | 0.06220 (18) | |
| Cl4 | 0.75120 (11) | −0.22491 (11) | 0.61516 (5) | 0.0772 (2) | |
| S1 | 0.67633 (8) | 0.32903 (8) | 0.20430 (5) | 0.05302 (16) | |
| S2 | 0.64526 (8) | 0.07120 (7) | 0.25388 (4) | 0.05061 (16) | |
| C1 | 0.9189 (3) | 0.4447 (3) | 0.16702 (13) | 0.0369 (4) | |
| C2 | 0.9766 (3) | 0.6267 (3) | 0.12843 (13) | 0.0372 (4) | |
| C3 | 1.1620 (3) | 0.7266 (3) | 0.09688 (15) | 0.0433 (4) | |
| C4 | 1.2912 (3) | 0.6466 (3) | 0.10343 (18) | 0.0539 (5) | |
| H4 | 1.4159 | 0.7138 | 0.0825 | 0.065* | |
| C5 | 1.2338 (3) | 0.4663 (3) | 0.14126 (18) | 0.0548 (5) | |
| H5 | 1.3207 | 0.4119 | 0.1455 | 0.066* | |
| C6 | 1.0495 (3) | 0.3650 (3) | 0.17301 (16) | 0.0462 (5) | |
| H6 | 1.0129 | 0.2434 | 0.1984 | 0.055* | |
| C7 | 0.7117 (3) | 0.1092 (3) | 0.39027 (14) | 0.0364 (4) | |
| C8 | 0.7107 (3) | −0.0468 (3) | 0.44284 (14) | 0.0378 (4) | |
| C9 | 0.7575 (3) | −0.0306 (3) | 0.54892 (16) | 0.0447 (5) | |
| C10 | 0.8069 (3) | 0.1407 (4) | 0.60354 (17) | 0.0564 (6) | |
| H10 | 0.8395 | 0.1517 | 0.6748 | 0.068* | |
| C11 | 0.8078 (3) | 0.2947 (3) | 0.55208 (18) | 0.0561 (6) | |
| H11 | 0.8405 | 0.4099 | 0.5891 | 0.067* | |
| C12 | 0.7608 (3) | 0.2809 (3) | 0.44603 (17) | 0.0460 (5) | |
| H12 | 0.7620 | 0.3864 | 0.4121 | 0.055* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Cl1 | 0.0859 (4) | 0.0712 (4) | 0.0671 (4) | 0.0592 (3) | 0.0296 (3) | 0.0276 (3) |
| Cl2 | 0.0806 (4) | 0.0444 (3) | 0.0751 (4) | 0.0115 (3) | 0.0078 (3) | 0.0195 (3) |
| Cl3 | 0.0915 (5) | 0.0416 (3) | 0.0596 (4) | 0.0333 (3) | 0.0146 (3) | 0.0022 (2) |
| Cl4 | 0.0910 (5) | 0.0889 (5) | 0.0677 (4) | 0.0512 (4) | 0.0099 (3) | 0.0389 (3) |
| S1 | 0.0447 (3) | 0.0628 (3) | 0.0570 (3) | 0.0262 (3) | 0.0104 (2) | 0.0284 (3) |
| S2 | 0.0609 (3) | 0.0430 (3) | 0.0377 (3) | 0.0117 (2) | −0.0002 (2) | 0.0080 (2) |
| C1 | 0.0414 (10) | 0.0419 (9) | 0.0290 (9) | 0.0188 (8) | 0.0000 (7) | 0.0059 (7) |
| C2 | 0.0487 (11) | 0.0412 (9) | 0.0274 (9) | 0.0246 (9) | 0.0011 (7) | 0.0022 (7) |
| C3 | 0.0511 (12) | 0.0395 (10) | 0.0329 (9) | 0.0128 (9) | −0.0014 (8) | 0.0030 (7) |
| C4 | 0.0364 (11) | 0.0659 (14) | 0.0526 (13) | 0.0148 (10) | −0.0018 (9) | 0.0069 (11) |
| C5 | 0.0465 (12) | 0.0707 (14) | 0.0576 (13) | 0.0348 (11) | −0.0038 (10) | 0.0085 (11) |
| C6 | 0.0476 (11) | 0.0481 (11) | 0.0481 (11) | 0.0251 (9) | −0.0026 (9) | 0.0112 (9) |
| C7 | 0.0341 (9) | 0.0373 (9) | 0.0347 (9) | 0.0116 (7) | 0.0058 (7) | 0.0057 (7) |
| C8 | 0.0383 (10) | 0.0372 (9) | 0.0396 (10) | 0.0168 (8) | 0.0093 (8) | 0.0062 (7) |
| C9 | 0.0377 (10) | 0.0543 (12) | 0.0433 (11) | 0.0197 (9) | 0.0065 (8) | 0.0144 (9) |
| C10 | 0.0462 (12) | 0.0723 (15) | 0.0395 (11) | 0.0141 (11) | −0.0012 (9) | 0.0010 (10) |
| C11 | 0.0520 (13) | 0.0469 (12) | 0.0547 (13) | 0.0070 (10) | 0.0028 (10) | −0.0125 (10) |
| C12 | 0.0456 (11) | 0.0338 (9) | 0.0536 (12) | 0.0115 (8) | 0.0046 (9) | 0.0054 (8) |
Geometric parameters (Å, º)
| Cl1—C2 | 1.7224 (19) | C5—C6 | 1.380 (3) |
| Cl2—C3 | 1.725 (2) | C5—H5 | 0.9300 |
| Cl3—C8 | 1.7291 (19) | C6—H6 | 0.9300 |
| Cl4—C9 | 1.726 (2) | C7—C12 | 1.389 (3) |
| S1—C1 | 1.784 (2) | C7—C8 | 1.392 (3) |
| S1—S2 | 2.0252 (8) | C8—C9 | 1.381 (3) |
| S2—C7 | 1.7834 (19) | C9—C10 | 1.378 (3) |
| C1—C6 | 1.386 (3) | C10—C11 | 1.372 (3) |
| C1—C2 | 1.393 (2) | C10—H10 | 0.9300 |
| C2—C3 | 1.384 (3) | C11—C12 | 1.382 (3) |
| C3—C4 | 1.378 (3) | C11—H11 | 0.9300 |
| C4—C5 | 1.378 (3) | C12—H12 | 0.9300 |
| C4—H4 | 0.9300 | ||
| C1—S1—S2 | 105.09 (7) | C1—C6—H6 | 120.0 |
| C7—S2—S1 | 105.02 (7) | C12—C7—C8 | 119.02 (17) |
| C6—C1—C2 | 119.06 (18) | C12—C7—S2 | 124.42 (15) |
| C6—C1—S1 | 124.55 (15) | C8—C7—S2 | 116.55 (14) |
| C2—C1—S1 | 116.38 (14) | C9—C8—C7 | 120.32 (17) |
| C3—C2—C1 | 120.36 (18) | C9—C8—Cl3 | 120.28 (15) |
| C3—C2—Cl1 | 120.22 (14) | C7—C8—Cl3 | 119.39 (14) |
| C1—C2—Cl1 | 119.42 (15) | C10—C9—C8 | 120.27 (19) |
| C4—C3—C2 | 120.23 (18) | C10—C9—Cl4 | 119.19 (17) |
| C4—C3—Cl2 | 119.41 (17) | C8—C9—Cl4 | 120.53 (16) |
| C2—C3—Cl2 | 120.36 (16) | C11—C10—C9 | 119.6 (2) |
| C5—C4—C3 | 119.4 (2) | C11—C10—H10 | 120.2 |
| C5—C4—H4 | 120.3 | C9—C10—H10 | 120.2 |
| C3—C4—H4 | 120.3 | C10—C11—C12 | 120.9 (2) |
| C4—C5—C6 | 121.0 (2) | C10—C11—H11 | 119.5 |
| C4—C5—H5 | 119.5 | C12—C11—H11 | 119.5 |
| C6—C5—H5 | 119.5 | C11—C12—C7 | 119.85 (19) |
| C5—C6—C1 | 119.93 (19) | C11—C12—H12 | 120.1 |
| C5—C6—H6 | 120.0 | C7—C12—H12 | 120.1 |
| S2—S1—C1—C6 | 0.06 (18) | S1—S2—C7—C12 | 3.60 (18) |
| S2—S1—C1—C2 | 179.34 (12) | S1—S2—C7—C8 | −177.28 (13) |
| C6—C1—C2—C3 | −0.2 (3) | C12—C7—C8—C9 | 0.1 (3) |
| S1—C1—C2—C3 | −179.52 (14) | S2—C7—C8—C9 | −179.02 (14) |
| C6—C1—C2—Cl1 | −179.31 (14) | C12—C7—C8—Cl3 | 179.45 (14) |
| S1—C1—C2—Cl1 | 1.4 (2) | S2—C7—C8—Cl3 | 0.3 (2) |
| C1—C2—C3—C4 | 0.0 (3) | C7—C8—C9—C10 | −0.5 (3) |
| Cl1—C2—C3—C4 | 179.07 (16) | Cl3—C8—C9—C10 | −179.76 (16) |
| C1—C2—C3—Cl2 | −179.68 (14) | C7—C8—C9—Cl4 | 178.52 (14) |
| Cl1—C2—C3—Cl2 | −0.6 (2) | Cl3—C8—C9—Cl4 | −0.8 (2) |
| C2—C3—C4—C5 | 0.3 (3) | C8—C9—C10—C11 | 0.6 (3) |
| Cl2—C3—C4—C5 | 179.91 (17) | Cl4—C9—C10—C11 | −178.44 (17) |
| C3—C4—C5—C6 | −0.3 (3) | C9—C10—C11—C12 | −0.3 (3) |
| C4—C5—C6—C1 | 0.0 (3) | C10—C11—C12—C7 | 0.0 (3) |
| C2—C1—C6—C5 | 0.2 (3) | C8—C7—C12—C11 | 0.1 (3) |
| S1—C1—C6—C5 | 179.46 (16) | S2—C7—C12—C11 | 179.18 (16) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6···S2 | 0.93 | 2.70 | 3.202 (2) | 115 |
| C12—H12···S1 | 0.93 | 2.70 | 3.199 (2) | 115 |
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: BX2456).
References
- Bondi, A. (1964). J. Phys. Chem. 68, 441–451.
- Brandenburg, K. (2006). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2012). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Crowley, D. J. (1964). US Patent No. 3 150 186.
- Deng, S.-L., Long, L.-S., Xie, S.-Y., Huang, R.-B., Zheng, L.-S. & Ng, S. W. (2003). Acta Cryst. E59, o843–o844.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Gomez-Benitez, V., Baldovino-Pantaleon, O., Herrera-Alvarez, C., Toscano, R. A. & Morales-Morales, D. (2006). Tetrahedron Lett. 47, 5059–5062.
- Hashash, A., Kirkpatrick, D. L., Lazo, J. S. & Block, L. H. (2002). J. Pharm. Sci. 91, 1686–1692. [DOI] [PubMed]
- Korp, J. D. & Bernal, I. (1984). J. Mol. Struct. 118, 157–164.
- Ogilby, P. R. (2010). Chem. Soc. Rev. 39, 3181–3209. [DOI] [PubMed]
- Sevier, C. S. & Kaiser, C. A. (2006). Antioxid. Redox Signal. 8, 797–811. [DOI] [PubMed]
- Shaabani, A., Safari, N., Shoghpour, S. & Hossein, A. R. (2008). Monatsh. Chem. 139, 613–615.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Tang, J.-M., Feng, Z.-Q. & Cheng, W. (2011). Acta Cryst. E67, o1197. [DOI] [PMC free article] [PubMed]
- Xiao, H., Chen, J., Liu, M., Wu, H. & Ding, J. (2009). Phosphorus Sulfur Silicon Relat. Elem. 184, 2553–2559.
- Yu, C., Jin, B., Liu, Z. & Zhong, W. (2010). Can. J. Chem. 88, 485–491.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S1600536814007326/bx2456sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536814007326/bx2456Isup2.hkl
Supporting information file. DOI: 10.1107/S1600536814007326/bx2456Isup3.cml
CCDC reference: 994982
Additional supporting information: crystallographic information; 3D view; checkCIF report