

Abstract
Aim:
To compare the pharmacokinetic parameters of cefuroxime lysine, a new second-generation of cephalosporin antibiotics, after intravenous (IV), intraperitoneal (IP), or intramuscular (IM) administration.
Methods:
Twelve male and 12 virgin female Sprague-Dawley rats, weighing from 200 to 250 g, were divided into three groups (n=4 for each gender in each group). The rats were administered a single dose (67.5 mg/kg) of cefuroxime lysine via IV bolus or IP or IM injection. Blood samples were collected and analyzed with a validated UFLC-MS/MS method. The concentration-time data were then calculated by compartmental and non-compartmental pharmacokinetic methods using DAS software.
Results:
After IV, IP or IM administration, the plasma cefuroxime lysine disposition was best described by a tri-compartmental, bi-compartmental or mono-compartmental open model, respectively, with first-order elimination. The plasma concentration profiles were similar through the 3 administration routes. The distribution process was rapid after IV administration [t1/2(d), 0.10±0.11 h vs 1.36±0.65 and 1.25±1.01 h]. The AUMC0–∞ is markedly larger, and mean residence time (MRT) is greatly longer after IP administration than that in IV, or IM routes (AUMC0–∞: 55.33±20.34 vs 16.84±4.85 and 36.17±13.24 mg·h2/L; MRT: 0.93±0.10 h vs 0.37±0.07 h and 0.65±0.05 h). The Cmax after IM injection was significantly higher than that in IP injection (73.51±12.46 vs 49.09±7.06 mg/L). The AUC0–∞ in male rats were significantly higher than that in female rats after IM administration (66.38±16.5 vs 44.23±6.37 mg·h/L). There was no significantly sex-related difference in other pharmacokinetic parameters of cefuroxime lysine between male and female rats.
Conclusion:
Cefuroxime lysine shows quick absorption after IV injection, a long retension after IP injection, and a high Cmax after IM injection. After IM administration the AUC0–∞ in male rats was significantly larger than that in female rats.
Keywords: cefuroxime lysine, intravenous administration, intramuscular administration, intraperitoneal administration, pharmacokinetics
Introduction
Cefuroxime lysine, a new second-generation cephalosporin, has been invented by Shenzhen Qingdazhong Biotech Co Ltd (patent No 201010191440.1). A related cephalosporin, cefuroxime sodium, has been listed in the Catalogue of Basic Medicines of the State in China for the treatment of patients with infections of soft tissue, respiratory tract, urinary tract, bone and joint tissues1,2,3. With the merits of greater water solubility and reduced irritation to the veins, cefuroxime lysine may become a suitable substitute for cefuroxime sodium in these indications. The two cefuroxime formulations currently used in clinic are the injectable cefuroxime sodium and cefuroxime axetil, for oral administration; both formulations must be converted into cefuroxime, in vivo, to act against most Gram-negative aerobic bacteria. There are numerous pharmacokinetic studies on cefuroxime sodium and cefuroxime axetil performed both in human beings and animals (rats, dogs, goats, and calves, etc)4,5,6,7. The elimination half-life in the studied species ranges from 0.5 to 2.3 h.
As a lyophilized crystalline powder, cefuroxime lysine may be administered by intravenous (IV), intramuscular (IM), or intraperitoneal (IP) injection routes in the clinic. However, there was only one report about its pharmacokinetic parameters after intravenous infusion of 108 mg/kg cefuroxime lysine in beagle dogs8. There were no reports about the other dosing routes. Therefore, the objective of the present study was to characterize the pharmacokinetics of cefuroxime lysine after IV, IM, and IP administration to rats as well as to investigate any sex-related differences in this species to provide enough information for further study of cefuroxime lysine.
Materials and methods
Chemicals and reagents
The cefuroxime reference standard (91.6% purity) and cefotaxime (86% purity, as internal standard) were obtained from the National Institute for Control of Pharmaceutical and Biological Products (Beijing, China). Cefuroxime lysine for injection (98% purity) was supplied by Shandong Luoxin Pharmacy Stock Co, Ltd (Shandong Province, China). Methanol, acetonitrile and formic acid (HPLC grade) were purchased from Fisher Scientific (NJ, USA). All other chemicals were of analytical grade. Water was purified by redistillation and filtered through a 0.22-μm membrane filter before use.
Animals and treatments
Twenty-four male and virgin female Sprague–Dawley rats, weighing from 200 to 250 g, were obtained from the Experimental Animal Research and Development Center of Guangzhou Institute of Pharmaceutical Industry (Guangzhou, China). The animals were housed four per cage in a 12 h light-dark cycle, with a constant temperature environment, and they were fed an antibiotic-free diet and water prior to study. Food was withheld 12 h before initiation of the experiment, but the animals had free access to water during this period. The study was carried out in accordance with the Guidelines for Animal Experimentation of Shenyang Pharmaceutical University (Shenyang, China) and the protocol was approved by the Animal Ethics Committee of the institute.
The rats were divided into three groups containing eight animals (half of each gender) in each group. Each animal received a single IV bolus or an IP or IM injection of cefuroxime lysine at a dose of 67.5 mg/kg.
A cefuroxime lysine solution (10 mL/kg) was injected into either the tail vein or intraperitoneal cavity of Sprague Dawley rats to assess the pharmacokinetics of IV or IP routes of administration. To assess the pharmacokinetics of the IM route, a 2 mL/kg solution of cefuroxime lysine was injected into the semimembranous muscle based on the body weight. Blood samples (0.3 mL) were drawn from the ophthalmic venous plexus at 0, 0.083, 0.167, 0.333, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, and 12 h after administration, and the samples were centrifuged at 2500×g for 10 min; the supernatant plasma was subsequently collected and frozen at −80 °C until analysis.
Analytical method
A UFLC-MS/MS method, modified from a similar protocol found in the literature, was applied to determine the plasma concentrations of cefuroxime lysine8,9,10. This protocol employed a protein precipitation method using a 2% formic acid in acetonitrile solution as the precipitator. A Shimadzu LC-20AD series UFLC system (Shimadzu, Japan), coupled to an Applied Biosystems Sciex Q-TRAP™ 4000 mass spectrometer (Foster City, CA, USA) via an electrospray ionization (ESI) source, was used for analysis. The mass spectrometer was operated in multiple reaction monitoring (MRM) mode with a negative ESI interface. Cefuroxime and IS cefotaxime were separated on a Shim-pack XR-ODS column (75 mm×3.0 mm, 2.2 μm) with Pre-filter (2 μm) (Shimadzu, Japan) using an isocratic elution of acetonitrile-0.1% formic acid water (40:60, v/v) at a flow rate of 400 μL/min. The linearities ranged from 0.01 to 1 μg/mL and 1 to 400 μg/mL (r>0.99) with a lower limit of quantification of 0.01 μg/mL. The relative standard deviations (RSDs) of intra- and inter-day precisions were both less than 11.5%, and the accuracy was between −7.1% and 2.2%. The mean extraction recovery was more than 83.5%, and mean matrix effect was between 98.4% and 109.5%. The analyte was stable under various plasma sample conditions including storage at −80 °C for a month, three freeze-thaw cycles at −80 °C, 6 h storage at room temperature and pretreating samples in autosampler for 4 h. RSD values of less than 10% were observed for all of these plasma conditions. The precision, accuracy, recovery, matrix effect and stability of this method met the requirements for analysis.
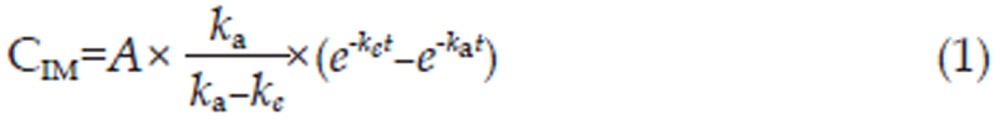 |
 |
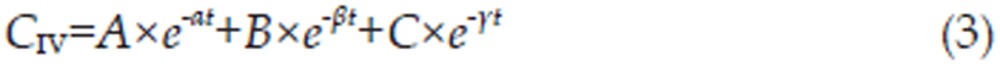 |
Pharmacokinetic analysis
A pharmacokinetic program named DRUG AND STATISTICS software (DAS, version 2.1.1, Mathematical Pharmacology Professional Committee of China) was used to analyze the plasma concentration-time curves of cefuroxime lysine by both non-compartmental and compartmental modeling approaches. The pharmacokinetic model with the best fit was determined by the application of Alkaike's Information Criterion (AIC)11. The mono-, bi-, and tri-compartment open models were depicted as follows12,13 where CIM, CIP, and CIV are the concentration of drug in the plasma; A, B, and C are the y intercepts of extrapolated lines for the central, tissue, and deep tissue compartments, respectively; and α, β, and γ are the first-order rate constants for the central, tissue, and deep tissue compartments, respectively. The variables ka and ke are the absorption rate constant and elimination rate constant, respectively.
For each rat, the maximum plasma concentration (Cmax), and its corresponding time (Tmax), for cefuroxime lysine was determined by visual inspection of the profiles. The apparent terminal elimination rate constant (λ) was calculated by linear regression of the natural logarithms of the terminal plasma concentrations. The terminal half-life (t1/2) was derived from 0.693/λ14. The area under the curve to the last measured point (AUC0–t) was calculated using the trapezoidal rule. The area under the plasma concentration versus time curve from 0 h to infinite time (AUC0–∞) was calculated as the sum of AUC0–t and Ct/λ, where Ct was the last quantifiable concentration15. Total body systemic clearance (Cl) was determined as the given dose divided by the AUC, and this value was normalized to the body weight. The total body weight-normalized apparent volume of distribution (V) was calculated as CL/(λ×W), where W was the body weight of the rat. The mean residence time (MRT) was calculated by MRT=AUMC0–∞/AUC0–∞. The mean absorption times were calculated as MAT= MRTIP,IM–MRTIV. The absolute bioavailability (F) of cefuroxime lysine was calculated using the following equation14,16:
where AUCIV, AUCIP, AUCIM, and t1/2e(IV), t1/2e(IP), t1/2e(IM) are the areas under the concentration-time curves and the elimination half-life for the IV, IP, and IM routes of administration, respectively.
Statistical analysis
The statistical analyses were performed by using SPSS software (version 16.0; SPSS Inc, Chicago, IL). A one-way analysis of variance (ANOVA) was used to determine the difference between the dosing routes and sexes. The values for Tmax were compared using the non-parametric Wilcoxon two-sample test. Comparisons of the pharmacokinetic parameters between the different sexes were evaluated by paired t-test. Descriptive statistics were expressed as the means±standard deviation (SD) values. A P-value of less than 0.05 was considered statistically significant17,18.
Results
Mono-, bi-, and tri-compartment models with first-order elimination were used to describe the cefuroxime lysine plasma concentration data to assess the disposition characteristics after three different routes of administration. The results indicated that the plasma cefuroxime lysine disposition was best fit by a tri-compartmental, bi-compartmental and mono-compartmental open model with first-order elimination after IV, IP, and IM administration, respectively. The observed cefuroxime lysine concentrations exhibited excellent agreement with the predictions obtained from the selected model. The correlation coefficient (r2) of the curves deriving from the predicted versus observed drug concentrations was greater than 0.99, demonstrating that the tri-, bi-, and mono-compartment open models could predict the plasma concentration of cefuroxime lysine after IV, IP, and IM administration well.
The final, corresponding pharmacokinetic model is mathematically described by the following set of equations
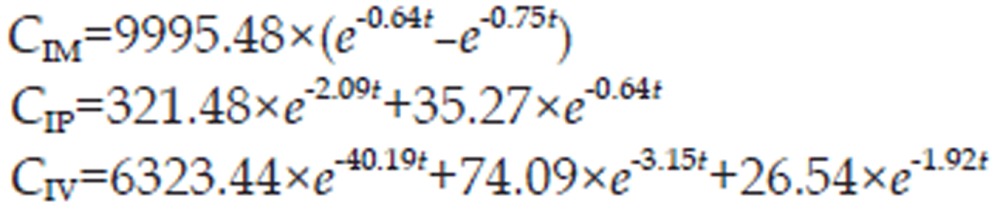 |
Cefuroxime lysine had the characteristics of extensive distribution, high clearance and short half-life after all three administration routes. The plasma concentration increased quickly during the first 0.333 h after IV administration when compared to IM or IP administration (Figure 1).
Figure 1.

Plasma concentrations (mean±SD) of cefuroxime lysine at a single dose of 67.5 mg/kg body weight (n=8) by IV, IP, and IM administration routes plotted on semi-logarithmic plots.
The distribution process was rapid after IV administration [t1/2(d), 0.10±0.11]. The AUMC0–∞ is markedly higher and MRT is greatly longer after IP administration than that in IV, or IM routes indicating cefuroxime lysine has a long retention after IP injection. The Cmax after IM injection was significantly higher than that in IP injection (73.51±12.46 vs 49.09±7.06 mg/L, Table 1).
Table 1. Pharmacokinetic parameters (mean±SD) of cefuroxime lysine in rats after IV, IP, and IM administration at a single dose of 67.5 mg/kg body weight (n=8). bP<0.05 vs IV. eP<0.05 vs IP.
| Parameters | IV | IP | IM |
|---|---|---|---|
| Cl (L·h−1·kg−1) | 1.51±0.20 | – | – |
| V (L/kg) | 1.20±0.64 | – | – |
| AUC0–∞ (mg·h/L) | 45.29±5.42 | 58.12±15.47 | 55.31±16.57 |
| AUMC0–∞ (mg·h2/L) | 16.84±4.85 | 55.33±20.34b | 36.17±13.24be |
| MRT (h) | 0.37±0.07 | 0.93±0.10b | 0.65±0.05be |
| t1/2(a) (h) | – | 0.61±0.51 | 0.77±0.69 |
| t1/2(d) (h) | 0.10±0.11 | 1.36±0.65b | 1.25±1.01b |
| t1/2(e) (h) | 0.56±0.32 | 0.62±0.64 | 0.75±0.55 |
| Cmax (mg/L) | – | 49.09±7.06 | 73.51±12.46e |
| tmax (h) | – | 0.37±0.15 | 0.16±0.03e |
| MAT (h) | – | 0.565±0.103 | 0.283±0.062e |
| F (%) | – | 86.5±33.9 | 90.8±29.2 |
Cl, the total body clearance of drug from the plasma; V, the apparent volume of distribution; AUC0–∞, the area under the plasma concentration-time curve from zero to infinity; AUMC, area under the moment curve. MRT, mean residence time; t1/2(a), absorption half-life; t1/2(d), distribution half-life; t1/2(e), elimination half-life; Cmax, peak plasma concentration. tmax, the time to reach peak concentration; MAT, mean absorption time; F, the fraction of the administered dose systemically available (bioavailability).
The AUC0–∞ in male rats were significantly higher than that in female rats after IM administration (66.38±16.5 vs 44.23±6.37 mg·h/L). There was no significantly sex-related difference in other pharmacokinetic parameters of cefuroxime lysine between male and female rats (Table 2).
Table 2. Pharmacokinetic parameters observed in male and female rats after IV, IP, and IM administration of 67.5 mg/kg cefuroxime lysine (n=4, mean±SD). bP<0.05 vs male.
| Parameters | IV |
IP |
IM |
|||
|---|---|---|---|---|---|---|
| Males | Females | Males | Females | Males | Females | |
| tmax (h) | – | – | 0.42±0.10 | 0.31±0.21 | 0.15±0.04 | 0.17±0.00 |
| Cmax (mg/L) | – | – | 51.10±4.48 | 49.45±9.88 | 81.8±10.1 | 65.25±8.85 |
| t1/2(e) (h) | 0.46±0.08 | 0.65±0.42 | 0.57±0.14 | 0.73±0.11 | 0.64±0.24 | 0.86±0.78 |
| Cl (L·h−1·kg−1) | 1.38±0.09 | 1.61±0.20 | – | – | – | – |
| V (L/kg) | 0.91±0.18 | 1.42±0.80 | – | – | – | – |
| AUC0–∞ (mg·h/L) | 49.22±3.16 | 42.33±4.99 | 61.66±18.9 | 53.40±11.2 | 66.38±16.5 | 44.23±6.37b |
Cl, the total body clearance of drug from the plasma; V, the apparent volume of distribution; AUC0–∞, the area under the plasma concentration-time curve from zero to infinity; t1/2(e), elimination half-life; Cmax, peak plasma concentration; tmax, the time to reach peak concentration.
Discussion
This is the first report detailing the pharmacokinetic study of cefuroxime lysine after IV, IP, and IM administration routes in rats. The tri-compartment model obtained after IV bolus administration is in agreement with that obtained following IV infusion to beagle dogs and an IV bolus of cefuroxime sodium in humans8,9. However, these results differ from other pharmacokinetic studies for cefuroxime sodium in humans, rats and goats that observed that a bi-compartmental model provided the best fit19,20,21,22. The reasons for this disparity may be related to interspecies variation, health status, or assay method employed23.
After IV administration, the distribution process was rapid (t1/2(d), 0.10±0.11). The elimination process of cefuroxime lysine in rats after IV bolus seems to be faster than in dogs following IV infusion, reflected by the elimination half-life (t1/2(e), 0.56±0.32 h vs 0.91±0.10 h) and total body clearance (Cl, 1.51±0.20 L·h−1·kg−1 vs 0.319±0.031 L·h−1·kg−1)8. The possible reasons for this discrepancy include differing metabolic rates and wider distribution into the peripheral compartment. The rapid decline in plasma concentrations is likely due to the wide tissue distribution (1.20±0.64) L/kg and metabolic clearance (1.51±0.20) L·h−1·kg−1.
Following single IP administration of cefuroxime lysine at a dosage of 67.5 mg/kg, the plasma concentration increased quickly to reach its maximum concentration of 49.09±7.06 mg/L at 0.37±0.15 h post-injection and then declined gradually. The higher mean residence time (0.93±0.10 h) showed that cefuroxime lysine has the capability of long retention; the MRT is significantly longer than that in IV, or IM routes (0.37±0.07 h, 0.65±0.05 h). The area under the curve for the IP route was higher than the corresponding values after IM route; this finding indicates a better absorption for the IP route compared to the IM route. The significantly longer distribution half-life after IP administration, compared to that after IV injection, was the possible reasons of carry-over in the absorption phase. The inter-animal variation occurred in the absorption phase after IP administration, which is indicated by large variations (SD) in tmax (0.37±0.15 h), t1/2(a) (0.61±0.51 h), and MAT (0.565±0.103 h).
The mean observed peak concentration (Cmax) after IM injection was 73.51 mg/L, which was significantly higher than that in IP injection. There was a significantly longer distribution half-life (t1/2(d)) after IM injection compared to IV injection, possibly due to variation introduced by the difference in injection site. Similar to the pharmacokinetic phenomenon observed following IP administration, inter-animal variation was also found after IM absorption. This variation is most likely due to changes in local perfusion at the injection site24. For each administration route, there was no significantly difference between males and females except for the pharmacokinetics of AUC0–∞ in IM route.
The minimum inhibitory concentration (MIC) of cefuroxime against most (≥90%) strains of the microorganisms was 4 μg/mL or lower3,25. The results indicated that the antibacterial effects of cefuroxime lysine could last for no more than 2 h after IV bolus injection and no more than 3 h after IP and IM administrations in the present study. However, antibacterial effects lasted at least 4.5 h after intravenous infusion in beagle dogs8, which indicated that IP and IM administration may be an effective substitution for IV injection. The superior administration route was continuous infusion, which had the merits of a more reliable plasma drug concentration that was higher than the MIC of susceptible bacteria. The duration should be higher than the MIC with 40%–60% of the inter-dose interval to ensure therapeutic success26,27. Therefore, the recommended dosage regimen of cefuroxime lysine was 5–7.5 h, which is similar to that of cefuroxime sodium in the clinic.
A high bioavailability of 86.5% and 90.8% as well as a more extended mean residence time of 0.93 h and 0.65 h after IP and IM injections were observed in this s tudy; these findings indicate that cefuroxime lysine has almost complete absorption for improved therapeutic potential compared to cefuroxime sodium. However, these data should be considered cautiously as this study was not a randomized, cross-over trial28.
Conclusion
To our knowledge, this is the first report to thoroughly study the comparative pharmacokinetics of cefuroxime lysine in 24 healthy Sprague-Dawley rats after IV, IP, and IM injection. After all three administration routes, cefuroxime lysine had the characteristics of extensive distribution, high clearance and short half-life. The plasma concentration profiles were similar to each other, except for higher plasma concentration during the first 0.333 h after IV administration when compared to IM or IP administration. There were significant differences in MRT, t1/2(d) and AUMC0–∞ among the three routes of injection; there were also differences in the AUC0–∞ between male and female rats after IM administration. The present study suggests that this new cephalosporin, cefuroxime lysine, could be administered by either the intravenous, intraperitoneal, or intramuscular route.
Author contribution
Kai-shun BI, Xiao-hui CHEN, Qing LI, and Ran YIN designed the research; Long-shan ZHAO, Bin-bin WEI, and Zhen-yuan JIANG performed the research; Long-shan ZHAO calculated and compared the pharmacokinetics as well as wrote this manuscript.
Acknowledgments
This study was supported by the National Key Scientific Project for New Drug Discovery and Development (No 2009 ZX09301-012). We wish to thank the excellent technical assistance of Dr Wei YANG and Bai-quan XIAO et al of Guangzhou Institute of Pharmaceutical Industry for their excellent work.
References
- Vazquez JC, Abalos E. Treatments for symptomatic urinary tract infections during pregnancy. Cochrane Database Syst Rev. 2011;1:CD002256. doi: 10.1002/14651858.CD002256.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Lee J, Lee J, Kim E, Lee S, Yu J, et al. Antimicrobial resistance in Haemophilus influenzae respiratory tract isolates in Korea: results of a nationwide acute respiratory infections surveillance. Antimicrob Agents Chemother. 2010;54:65–71. doi: 10.1128/AAC.00966-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M, Beck A, Frei P, Bischoff G. Pharmacokinetics of cefuroxime in traumatic wound secretion and antibacterial activity under vacuum therapy. J Chemother. 2010;22:92–7. doi: 10.1179/joc.2010.22.2.92. [DOI] [PubMed] [Google Scholar]
- Asiri YA, AI-Hadiya BM, Kadi AA, Al-Khamis KI, Mowafy HA, El-Sayed YM. Comparative bioavailability study of cefuroxime axetil (equivalent to 500 mg cefuroxime/table) tablets (Zednad® versus Zinnat®) in healthy male volunteers. Int J Clin Pharmacol Ther. 2011;49:571–6. doi: 10.5414/cp201376. [DOI] [PubMed] [Google Scholar]
- Ruiz-Carretero P, Merino-Sanjuán M, Nácher A, Casabó VG. Pharmacokinetic models for the saturable absorption of cefuroxime axetil and saturable elimination of cefuroxime. Eur J Pharm Sci. 2004;21:217–23. doi: 10.1016/j.ejps.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Abo El-sooud K, El-Banna HA, Hanafy MS, Goudah A. Pharmacokinetics and intramuscular bioavailability of cefuroxime sodium in goats. Res Vet Sci. 2000;69:219–24. doi: 10.1053/rvsc.2000.0412. [DOI] [PubMed] [Google Scholar]
- Chaudhary RK, Srivastava AK, Rampal S. Modification of the pharmacokinetics and dosage of cefuroxime by endotoxin-induced fever in buffalo calves. Vet Res Commun. 1999;23:361–8. doi: 10.1023/a:1006385624850. [DOI] [PubMed] [Google Scholar]
- Zhao LS, Zhao YL, Li Q, Chen XH, Xiao F, He BS, et al. A fast, sensitive, and high throughput method for the determination of cefuroxime lysine in dog plasma by UPLC-MS/MS. Talanta. 2012;89:84–90. doi: 10.1016/j.talanta.2011.11.063. [DOI] [PubMed] [Google Scholar]
- Viberg A, Sandström M, Jansson B. Determination of cefuroxime in human serum or plasma by liquid chromatography with electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:707–10. doi: 10.1002/rcm.1396. [DOI] [PubMed] [Google Scholar]
- Zhao LS, Li Q, Yang W, He BS, Wei BB, Liu R, et al. Determination of cefuroxime in liver-injured rat plasma by ultra-fast liquid chromatography-tandem mass spectrometry via acidified protein precipitation Chin J Chromatogr 2012. doi: 10.3724/SP.J.1123.2012.03001 [DOI] [PubMed]
- Yamaoka K, Nakagawa T, Uno T. Statistical moments in pharmacokientics. J Pharmacokinet Biopharm. 1978;6:547–58. doi: 10.1007/BF01062109. [DOI] [PubMed] [Google Scholar]
- Grasso S, Meinardi G, de Carneri I, Tamassia V. New in vitro model to study the effect of antibiotic concentration and rate of elimination on antibacterial activity. Antimicrob Agents Chemother. 1978;13:570–6. doi: 10.1128/aac.13.4.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Sakamoto H, Hirose T, Nishida M. New in vitro kinetic model for evaluting bactericidal efficacy of antibiotics. Antimicrob Agents Chemother. 1980;18:377–81. doi: 10.1128/aac.18.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibaldi M, Perrier D.Pharmacokinetics, second ed. New York: Marcel Dekker Inc; 1982
- Chen ML, Chiou WL. Tissue metabolism and distribution of methotrexate in rabbits. Drug Metab Dispos. 1982;10:706–7. [PubMed] [Google Scholar]
- Dimitrova D, Moutafchieva R, Kanelov I, Dinev T, Lashev L. Pharmacokinetics of tobramycin in ducks and sex-related differences. Vet J. 2009;179:462–4. doi: 10.1016/j.tvjl.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Powers J. Statistical analysis of pharmacokinetics data. J Vet Pharmacol Ther. 1990;13:113–20. doi: 10.1111/j.1365-2885.1990.tb00758.x. [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG.Statistical Methods, seventh ed. Ames (Iowa): Iowa State University Press; 1980. p39–63.
- Gower PE, Dash CH. The pharmacokinetics of cefuroxime after intravenous injection. Eur J Clin Pharmacol. 1977;12:221–7. doi: 10.1007/BF00609865. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Cheng FC, Chen KC, Chen YF, Chen CF. Simultaneous measurement of cefuroxime in rat blood and brain by microdialysis and microbore liquid chromatography. Application to pharmacokinetics. J Chromatogr B. 1999;735:25–31. doi: 10.1016/s0378-4347(99)00410-7. [DOI] [PubMed] [Google Scholar]
- Viberg A, Lannergård A, Larsson A, Cars O, Karlsson MO, Sandström M. A population pharmacokinetic model for cefuroxime using cystatin C as a marker of renal function. Br J Clin Pharmacol. 2006;62:297–303. doi: 10.1111/j.1365-2125.2006.02652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoderer CA, Saft SA, Walker SG, Rodefeld MD, Turrentine MW, Brown JW, et al. Cefuroxime pharmacokinetics in pediatric cardiovascular surgery patients undergoing cardiopulmonary bypass. J Cardiothorac Vasc Anesth. 2011;25:425–30. doi: 10.1053/j.jvca.2010.07.022. [DOI] [PubMed] [Google Scholar]
- Haddad NS, Pedersoli WM, Ravis WR, Fazeli MH, Carson RL. Combined pharmacokinetics of gentamicin in pony mares after a single intravenous and intramuscular administration. Am J Vet Res. 1985;46:2004–7. [PubMed] [Google Scholar]
- Albarellos GA, Ambros LA, Landoni MF. Pharmacokinetics of ceftazidime after intravenous and intramuscular administration to domestic cats. Vet J. 2008;178:238–43. doi: 10.1016/j.tvjl.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Sultana N, Arayne MS. In vitro activity of cefazoline and cefuroxime in presence of essential and trace elements. Pak J Pharm Sci. 2002;15:41–50. [PubMed] [Google Scholar]
- Toutain PL, Del Castillo JRE, Bousquet-Mélou A. The pharmacokinetic-pharmacodynamic approach to a rational dosage regimen for antibiotics. Res Vet Sci. 2002;73:105–14. doi: 10.1016/s0034-5288(02)00039-5. [DOI] [PubMed] [Google Scholar]
- McKellar QA, Sanchez Bruni SF, Jones DG. Pharmacokinetic/pharmacodynamic relationships of antimicrobial drugs used in veterinary medicine. J Vet Pharm Ther. 2004;27:503–14. doi: 10.1111/j.1365-2885.2004.00603.x. [DOI] [PubMed] [Google Scholar]
- Albarellos GA, Montoya L, Landoni MF. Pharmacokinetics of erythromycin after intravenous, intramuscular and oral administration to cats. Vet J. 2011;187:129–32. doi: 10.1016/j.tvjl.2009.09.019. [DOI] [PubMed] [Google Scholar]