

Summary
This protocol describes a cell free system to study vertebrate centromere and kinetochore formation. We reconstitute tandem arrays of centromere protein A (CENP-A) nucleosomes as a substrate for centromere and kinetochore assembly. These chromatin substrates are immobilized on magnetic beads and then incubated in Xenopus egg extracts that provide a source for centromere and kinetochore proteins and that can be cycled between mitotic and interphase cell cycle states. This cell free system lends itself to protein immunodepletion, complementation and drug inhibition as tools to perturb centromere and kinetochore assembly, cytoskeletal dynamics, DNA modification, and protein post-translational modification. This system provides a distinct advantage over cell-based investigations where perturbing centromere and kinetochore function often results in lethality. Reconstituted CENP-A chromatin specifically assembles centromere and kinetochore proteins after incubation in egg extract that locally stabilize microtubules and, upon microtubule depolymerization with nocodazole, activate the mitotic checkpoint. A typical experiment occupies 3 days.
Introduction
Development of the protocol
Chromosome segregation requires the interaction of each sister chromatid pair with the microtubules of the mitotic spindle. The kinetochore is the primary microtubule attachment site on each chromosome that enables chromosome segregation and ensures the proper attachment and alignment of all chromosomes on the spindle through the activity of the mitotic checkpoint. Kinetochores assemble during mitosis on a specialized chromatin region called the centromere, which is specified by the replacement of histone H3 in centromeric nucleosomes with a centromere-specific histone H3 variant CENP-A (reviewed in 1). A detailed understanding of how CENP-A chromatin promotes centromere and kinetochore formation in vertebrates has been hampered by the fact that vertebrate centromeres are complex in their organization (spanning hundreds of kilobases of repetitive DNA with domains of interspersed histone H3 and CENP-A nucleosomes), have been impossible to isolate as intact functional complexes from cells and when perturbed often result in cell death due to chromosome missegregation.
To overcome these limitations, we developed a method for centromere assembly in vitro by reconstituting CENP-A chromatin from purified components, then adding this chromatin to Xenopus egg extracts to assemble centromere and kinetochore proteins from the extract 2. Briefly, we generate CENP-A chromatin (and H3 chromatin as a control) on biotinylated DNA containing tandem repeats of high affinity nucleosome positioning sequences using the salt dialysis method 3,4. We then immobilize the chromatin on magnetic beads, incubate the chromatin-coated beads in Xenopus egg extract and, after retrieving the beads using a magnet, we analyze centromere and kinetochore assembly via quantitative immunofluoresence. We assay kinetochore function by analyzing the ability of the immobilized chromatin arrays to promote microtubule polymerization and stabilization as well the activation of the mitotic checkpoint in response to microtubule depolymerization 2.
Xenopus egg extracts are a powerful in vitro system for investigating the molecular mechanisms of cell cycle control, chromosome segregation, spindle assembly, and kinetochore assembly 5–8. These extracts are naturally arrested in metaphase of meiosis II by the activity of cytostatic factor (CSF) and can be cycled in vitro from metaphase into interphase by adding calcium and then back into metaphase with fresh meiotic extract. Egg extracts are also biochemically accessible and allow protein depletion by specific antibodies, inhibition with dominant-negative proteins or antibodies, and perturbation by the addition of small molecules 9,10.
The addition of demembranated Xenopus sperm to Xenopus egg extracts supports kinetochore formation and spindle assembly. If sperm chromatin is added to CSF arrested metaphase extracts meiotic spindles form around unreplicated, haploid sperm DNA with functional kinetochores but lacking paired sister chromatids. If these extracts are driven out of metaphase with calcium, through interphase DNA replication and back into metaphase, replicated chromosomes assemble bipolar spindles with attached sister chromatids that support anaphase chromosome segregation. Spindle formation in egg extracts does not require kinetochores as lambda DNA coupled to beads is assembled into chromatin and supports spindle assembly 6,10. Although sperm chromatin contains centromeres that give rise to functional kinetochores, manipulating the composition of these centromeres presents the same challenges as manipulating any other complex centromere.
To generate experimentally manipulatable centromeric chromatin we assembled chromatin with bacterially expressed histones and biotinylated arrays of nucleosome positioning sequence DNA (19X601) using established chromatin reconstitution methods 3,4,11–14. We bound biotinylated chromatin arrays to streptavidin-coated magnetic beads so that they could be easily added to and recovered from frog egg extract. When added to frog egg extract, reconstituted CENP-A chromatin supported centromere assembly, kinetochore assembly, microtubule binding and mitotic checkpoint function (Figure 1). In this system, we assayed centromere and kinetochore assembly by quantifying proteins that specifically bound to CENP-A chromatin using immunofluorescence (or western blotting) (Figure 2a, 2b). We could control the cell cycle requirement for kinetochore assembly by cycling the extract between metaphase and interphase. We used two different assays to monitor microtubule binding and stabilization to chromatin either by quantifying bead-associated microtubule polymer using immunofluorescence or by cold shocking and nocodozole treatment to selectively depolymerize non-kinetochore microtubules (Figure 2c). Lastly, we measured the mitotic checkpoint response by quantifying checkpoint protein recruitment to chromatin after microtubule depolymerization to activate the checkpoint response 2,15 or by monitoring delays in the exit from mitosis by assessing the level of Wee-1 mitotic phosphorylation 16 (Figure 2b, 2d).
Figure 1. A cell free system for centromere and kinetochore reconstitution.

A schematic showing the general experimental procedure and the corresponding steps in the protocol. CENP-A and H3 chromatin arrays are reconstituted from purified proteins and biotinylated high affinity nucleosome positioning sequences using the salt dialysis method. Assembled chromatin arrays are coupled to Streptavidin-coated magnetic beads. The chromatin arrays are incubated in Xenopus egg extracts. After recovery, centromere and kinetochore assembly and function is analyzed. (Figure is adapted from 2).
Figure 2. The cell free system can be used to analyze centromere assembly, kinetochore assembly, microtubule polymerization and mitotic checkpoint activation.

A schematic showing the four different types of assays described here analyzing the ability of reconstituted chromatin arrays coupled to beads to (A) assemble centromeres in CSF extracts (Step 119A) (B) assemble kinetochores in cycled extracts (Step 119B), (C) promote microtubule polymerization and stabilization in CSF extracts (Step 119C) and (D) activate the mitotic checkpoint in cycled egg extracts (Step 119D).
Application of the method
One of the key advantages of the reconstituted chromatin system we have developed is that it provides a mechanism to modify the protein and DNA composition of the assembly substrate, currently an extremely difficult task with vertebrate centromeres. We have used this advantage to generate chromatin containing chimeras of CENP-A and H3 to specifically test which CENP-A protein domains direct centromere and kinetochore assembly 2,11,12. Numerous additional applications using specific point mutations and deletions within the histone proteins are now accessible with this system. Furthermore, one can vary the histone composition to include interspersed H3 and CENP-A nucleosomes or other histone variants to assess the effects on centromere and kinetochore formation. In the same spirit, we can employ different lengths and sequences of DNA in the assembly system to test the contribution of the nucleic acid substrate to centromere formation. Finally, because we can control the cell cycle stage of the extract and recover the chromatin, we can assay cell cycle dependent changes in centromere protein composition and post-translational modifications.
Limitations of the system
Several steps must be carefully controlled in this system to ensure reproducibility. First, the CENP-A and H3 chromatin arrays must be close to saturation of the nucleosome positioning sequences with histones (not over- or under-saturated) to enable accurate comparison of different chromatin substrates. Undersaturation of chromatin arrays can result in the assembly of H3 nucleosomes from the Xenopus egg extracts as these extracts assemble chromatin on naked DNA 2,10,17. Oversaturation, on the other hand, causes chromatin aggregation and can alter the coupling efficiency of chromatin to the beads as well as precipitation of the chromatin. Centromere and kinetochore assembly have a strong dependence on the saturation and aggregation state that may reflects a density of CENP-A chromatin that is optimal for efficient assembly. We recommend careful titration to determine the ideal protein:DNA ratios for every new histone preparation and analysis of each chromatin assembly by restriction digest to ensure that the saturation level is comparable between CENP-A and H3 arrays. In our hands, ~90% saturated arrays and groups of 10–30 beads have been optimal for the assays described. The second variable that must be carefully monitored is the quality of Xenopus egg extracts. Xenopus egg extracts are known to be intrinsically variable. For example, it has been estimated that only 50–60% of cycled extracts release efficiently from mitotic arrest 10, and we estimate a similar rate of success for our spindle assembly checkpoint activation experiments (Figure 2d). Detailed methods for egg extract preparation have been developed that minimize the variability but the nature of the system requires that the quality of the extracts, their ability to maintain mitotic arrest and their ability to exit mitosis in response to calcium be assayed in every experiment.
Comparison with other methods
Currently, the method we describe here is the only available method for reconstituting functions of the vertebrate centromere and kinetochore on manipulatable chromatin templates. However, different experimental approaches complement the method presented here for studying the properties of centromeric chromatin that promote centromere and kinetochore formation. Methods for generating human artificial chromosomes using alphoid DNA sequences have demonstrated the importance of alpha satellite DNA sequences and the CENP-B DNA binding protein in promoting centromere formation 18–21. By interspersing tetracycline operator (TetO) sites throughout the repeated alpha satellite sequences and expressing tetracycline repressor (TetR) fusions to different proteins it has been possible to conditionally direct transcriptional activators or repressors to the HAC centromere to test the influence of transcriptional activity and chromatin state on centromere function 22,23. An analogous method using arrays of lac operators (lacO) integrated into noncentromeric chromosomal regions and the expression of CENP-A or other centromere protein fusion to the lac repressor (lacI) has been used to generate new exogenous centromeres in cells as well as to test the functions of other centromere binding proteins in centromere formation 24–26. These approaches make it possible to manipulate the DNA sequence of the centromere and to test the capacity of specific proteins to promote centromere formation at noncentromeric sites in cells and thus nicely complement the method described here.
The simplicity of the budding yeast centromere, which is specified genetically by a defined 125 bp sequence, has led to the development of elegant methods to analyze centromere and kinetochore function by reconstitution and purification. Yeast centromeric DNA, after incubation in yeast cell extracts, acquires the ability to bind to microtubules in a sequence dependent manner and this property has been used to identify key components of the yeast kinetochore 27–30. Biggins and colleagues have taken advantage of the simplicity of the yeast centromere sequence to purify plasmid based kinetochores from yeast cells by immunoprecipitation 31. These experiments have provided a route to the proteomic characterization of kinetochores and to biophysical measurements of kinetochore activity 31,32. It is clear that analogous reconstitution and purification approaches are required to dissect similarities and differences between the genetically defined centromeres of budding yeast and metazoan centromeres that are specified by the presence of CENP-A nucleosomes. Biochemical reconstitution of CENP-A nucleosomes and identification of CENP-A specific binding proteins has provided insight into how CENP-A is recognized by different centromere proteins and has formed the basis for developing the more complex centromere in vitro reconstitution system, described here 2,11,12.
MATERIALS
REAGENTS
LB Agar Miller (EMD Millipore, catalogue # 1.10283)
Competent BL21(DE3) Codon Plus RIL (Agilent Technologies, catalogue # 230245)
Tryptone (BD Biosciences, catalogue # 211705)
Yeast extract (EMD Millipore, catalogue # 1.03753)
Chloramphenicol (Sigma-Aldrich, catalogue # C0378)
Carbenicillin (Sigma-Aldrich, catalogue # C1389)
IPTG (VWR, catalogue # 80056-920)
Triton X-100 (Sigma-Aldrich, catalogue # T8787)
Lysozyme (Sigma-Aldrich, catalogue # L6876)
Dimethyl sulfoxide (DMSO) (VWR, catalogue # JT9224-1) CAUTION: can enhance cell/skin permeability of other compounds. Avoid contact and use skin/eye protection.
Guanidine-HCl (Invitrogen, catalogue # 15502-016)
Dithiothreitol (DTT) (Sigma, catalogue # 0.154 g in 10ml water)
Urea (Sigma-Aldrich, catalogue # U5378)
NaCl (EMD Millipore, catalogue # SX0420-5)
Tris(hydroxymethyl) aminomethane (EMD Millipore, catalogue # 9210-OP)
EDTA (Sigma-Aldrich, catalogue # E9884)
EGTA (Sigma-Aldrich, catalogue # E3889)
Phenylmethanesulfonyl fluoride (PMSF) (Sigma, catalogue # P7626) CAUTION: PMSF powder is hazardous. Use skin/eye protection when preparing PMSF solutions.
AG-501-X8 anion/cation exchange resin (Bio-rad, catalogue # 142-6424)
Bradford reagent concentrate (Bio-rad, catalogue # 500-0006)
2-Mercaptoethanol (Sigma-Aldrich, catalogue # M6250) CAUTION: toxic.
Benzamidine (Sigma-Aldrich, catalogue # B6506)
Acrylamadie (J.T. Baker, catalogue # 4081) CAUTION: Toxic. Use skin and eye protection when preparing acrylamide solution and avoid inhalation.
Bis-acrylamide (BioExpress, catalogue # M104-250G) CAUTION: Toxic. Use skin and eye protection when preparing acrylamide solution and avoid inhalation.
AG 501-X8 Resin, ion exchange resin (BioRad, catalogue # 142-6424)
K2HPO4 (VWR, catalogue # EM-PX1570-1)
KH2PO4 (VWR, catalogue # EM-5108-2)
Potassium acetate (J.T. Baker, catalogue # 2914)
Tris-acetate (Sigma-Aldrich, catalogue # T1258)
Boric acid (Sigma-Aldrich, catalogue # B56307)
ATP (Sigma-Aldrich, catalogue # A2383)
BSA (Sigma-Aldrich, catalogue # A7906)
KOH (Sigma-Aldrich, catalogue # 221473) CAUTION: corrosive, causes burns, use eye and skin protection.
QIAquick PCR purification kit (Qiagen, catalogue # 28106)
Restriction enzymes EcoRI, DraI, XbaI, HaeII, AvaI (New England BioLabs, catalogue # R0101, R0129, R0145, R0107, R0152)
NEB 2-log DNA ladder (New England BioLabs, catalogue # N3200)
Agarose (GeneMate LE, catalogue # E-3120)
Biotin-14-ATP (Invitrogen, catalogue # 19524-016)
Alpha-thio-dTTP (Chemcyte, catalogue # CC-3004-1)
Alpha-thio-dGTP (Chemcyte, catalogue # CC-3003-1
dCTP (Invitrogen, catalogue # 10217016)
Klenow fragment (Invitrogen, catalogue # M0212)
Streptavidin-FITC at 0.2mg/ml (e.g. Invitrogen catalogue # 43-4311)
Polyethylene glycol (PEG) 4000 (TCI America, catalogue # P0885)
HCl (J.T. Baker, catalogue # 9530) CAUTION: corrosive, causes burns, use eye and skin protection and a chemical fume hood.
40% Acrylamide/Bis Solution, 29:1 (3.3%C) (Bio-Rad, catalogue # 161-0146) CAUTION: toxic.
Temed OmniPur (EMD Millipore, catalogue # 8930) CAUTION: corrosive, toxic, causes burns, use eye and skin protection.
Ethidium Bromide (EMD Millipore, catalogue # 4340) CAUTION: potentially mutagenic.
Dynabeads M-270 Streptavidin (Invitrogen, catalogue # 653-05)
Polyvinyl alcohol (PVA) MW 30.000–70.000 (Sigma, catalogue # P8136)
Cysteine, free base (MP Biomedical, catalogue # 101444)
Xenopus laevis female frogs (Nasco)
Pregnant mare serum gonadotropin (PMSG) (100U/ml in H2O)(Sigma, catalogue # G4877); stored at −20°C
Human chorionic gonadotropin (hCG), 1000U/ml in H2O (Sigma G4877), stored at −20°C
Creatine phosphate (Roche Molecular Biochemicals, catalogue # 0621714)
Cytochalasin D (Sigma-Aldrich, catalogue # C8273) CAUTION: highly toxic, use skin and eye protection and avoid inhalation.
Cytochalasin B (Sigma-Aldrich, catalogue # C6762) CAUTION: highly toxic, use skin and eye protection and avoid inhalation.
NaOH, Pellets (J.T. Baker, catalogue # 3722-01) corrosive, causes burns, use eye and skin protection.
Sucrose (EMD Millipore, catalogue # 8510)
Gelatin (Difco, catalogue # 214340)
Hepes, free acid (J.T. Baker, catalogue # 4018)
MgCl2 × 6H2O (J.T. Baker, catalogue # 2448)
CaCl2 × 2H2O (J.T. Baker, catalogue # 1332)
KCl (J.T. Baker, catalogue # 3040)
Leupeptin (Sigma-Aldrich, catalogue # L2023)
Pepstatin A (Sigma-Aldrich, catalogue # P5318)
Chymostatin (Sigma-Aldrich, catalogue # C7268)
Creatine Phosphate (Roche Molecular Biochemicals, catalogue # 0621714)
ATP (Sigma, catalogue # A2383)
Nocodazole (EMD Millipore, catalogue # 487928)
Formaldehyde solution 37% (w/v) (J.T.Baker catalogue # 2106) CAUTION: toxic, carcinogenic, use skin and eye protection and a chemical fume hood.
Guanidine Hydrochloride (Sigma, catalogue # G3272)
Pipes, free acid (EMD Millipore, catalogue # 528131)
Glycerol (EMD Millipore, catalogue # 56815)
Anti-tubulin antibody (DM1α) (Sigma-Aldrich, catalogue # T9026)
CAPS (Sigma-Aldrich, catalogue # C2632)
Methanol (J.T. Baker, catalogue # 9070) CAUTION: flammable, toxic.
NEB pre-stained marker (Bio-rad, catalogue # 161-0305)
Ammonium Persulfate (APS) (J.T. Baker, catalogue # 4030)
40% Acrylamide/Bis Solution 29:1 (3.3%C) (BioRad, catalogue # 161-0146)
Sodium Dodecylsulfate (SDS) (J.T. Baker, catalogue # 4095)
Instant nonfat dry milk (many suppliers, e.g Safeway)
P-Wee1 antibody 16. The antibody was a gift from J. E. Ferrell.
Goat-anti-rabbit-Alexa 647 (Invitrogen, catalogue # A21245)
Goat-anti-mouse-Alexa 488 (Invitrogen, catalogue # A11029)
DNA dye Bisbenzimide/Hoechst 33258 (Sigma-Aldrich, catalogue # B2883) CAUTION: potentially mutagenic.
Propidium Iodide (VWR, catalogue # IC19545880)
Specific antibodies for centromere and kinetochore proteins of interest
Anti-Histone H4 (Abcam, catalogue # ab7311)
Rabbit IgG whole molecule as blocking agent (Jackson Immuno Research, catalogue # 011-000-003)
Poly-L-lysine hydrobromide (Sigma-Aldrich, catalogue # P1524)
Nail polish (many suppliers, e.g. Walgreens Pharmacy)
p-phenylenediamine (Sigma, # P6001)
SURE 2 Supercompetent Cells (Stratagene, # 200152)
Histone H2A expression plasmid 4. (Plasmid was a gift from Karolin Luger)
Histone H2B expression plasmid 4. (Plasmid was a gift from Karolin Luger)
Histone H3 expression plasmid 4. (Plasmid was a gift from Karolin Luger)
-
Histone H4 expression plasmid 4. (Plasmid was a gift from Karolin Luger)
CRITICAL: H3/H4 tetramers and H2A/H2B dimmers are assembled from denatured histone H3, H4, H2A and H2B expressed in E. coli and extracted from bacterial inclusion bodies as previously described 12,33,34 (see Steps 1–44).
-
Bicistronic CENPA/H4 expression plasmid 12. Codon optimized Xenopus H4 and human CENP-A were cloned into the XbaI/BamHI and EcoRI/HindIII restriction sites, respectively, of the bicistronic pST39 expression system 35,36. (The pST39 plasmid was a gift from Song Tan.)
CRITICAL: CENP-A/H4 tetramers are expressed as soluble tetramers using a bicistronic expression construct and purified by sequential hydroxyapatite and cation exchange chromatography 12,37 (see Steps 45–70).
Plasmid containing 19 repeats of the 601 nucleosome positioning sequence with a pUC18 backbone 3,13,38. (Plasmid was a gift from Daniela Rhodes.)
EQUIPMENT
Dialysis tubing 3500 MWCO (VWR, catalogue # 25219-164)
Amicon centrifugal filter concentrator, 10000 MWCO (EMD Millipore, catalogue # UFC901008)
Qiagen Tip-10000 (Qiagen, catalogue # 10091)
Dialysis membrane (Spectrum Spectra/Por 6000–8000 MWCO 32mm, Catalogue # 132 655)
PVDF immuoblot-membrane (Bio-rad, catalogue #162-0177)
Sonicator
Spectrophotometer
Large capacity centrifuge, rotor (JS-4.2)
Oakridge tubes, rotor (JA-20), high-speed centrifuge
Peristaltic pump
FPLC
Columns, 5ml HiTrap Q FF, 5ml HiTrap SP FF, Superdex 200 16/60, 60ml Hydroxyapatite (HA) column, 1ml HiTrap SP FF
Conductivity meter
pH meter
Vortex
Spectrophotometer (e.g. NanoDrop ND-1000)
Tabletop centrifuge
Dialysis buttons (Hampton Research; 50μl, catalogue # HR3-326; 200μl, catalogue # HR3-330)
100μl Hamilton syringe (Fisher, catalogue # 14-813-138)
Typhoon 9400 Variable Mode Imager (Amersham Biosciences)
Slide-a-lyzer mini dialysis units, 7000 MWCO (Thermo Scientific #69560)
Peristaltic pump (Gilson Minipuls 2)
Yellow-blue tubing for peristaltic pump (0.06 in ID)
1ml plastic pipettes
Typhoon 9400 Variable Mode Imager (Amersham Biosciences)
Magnetic stir plate
Magnetic particle concentrator for microcentrifuge tubes (Invitrogen, Dynal MPC-S, catalogue # 120.20D)
Aspirator
1ml disposable Syringes with 16 and 25-gauge needles (multiple suppliers)
Clinical centrifuge
Forceps
SW-55 swinging bucket rotor
SW-55 centrifuge tubes, e.g. (Beckmann, catalogue # 326819 or # 344057)
Pasteur pipette, cut-off and firepolished
13ml plastic tubes (BD Biosciences, catalogue # 352063)
16°C incubator
4l and 2l plastic frog containers with lids containing small holes for ventilation
Pyrex dish 15×75 (Pyrex, catalogue # 3140)
16 °C – 20 °C water bath. CRITICAL: A P1000 tip box works well for 0.6ml tubes for volumes between 20–100μl. For spindle assembly reactions, a floating rack and 1.5ml Eppendorf tubes have been used.
Dynal MPC (Invitrogen, MPC-S Magentic partical concentrator)
Humidified chamber for coverslips, e.g., petri dish coated with parafilm. Cover with aluminium foil to protect from light and put wet paper towels inside to create a humid environment
Modified glass Corex tubes with spin-down adaptors and metal hook 9,10
Spin-down adaptors for Corex tubes (TAP Plastics, Mountain View)
JS 13.1 rotor
No. 1.5 12mm round glass coverslips (Fisher #12-545-81)
Freeze-drying lyophilizer (Labconco #7670021)
Fluorescence Microscope, Nikon eclipse 80i
60× 1.4NA Plan Apo oil immersion lens (Nikon)
Sedat quad-pass filter set (#89000, Chroma). CRITICAL: ensure that the wavelengths of the filter set match the fluorophores used for the staining and that there is no bleed-through between channels
CCD camera (Coolsnap HQ, Photometrics)
Motorized Z-drive (MFC-2000, Applied Scientific Instrumentation)
Microscope control software (Metamorph, MDS Analytical Technologies)
Matlab software with image processing toolbox (R2009b, MathWorks)
REAGENT SETUP
Chloramphenicol 1000x stock: 34mg/ml in ethanol and can be stored at −20°C for several months.
Carbenicillin 1000x stock: 100mg/ml in water and can be stored at −20°C for several months.
LB media: mix 19g Tryptone, 5g yeast extract, 10g NaCl in 1 L deionized water, pH to 7.0 with NaOH. Autoclave and store until use at room temperature (RT; ~21°C) for a few days. Add chloramphenicol (34μg/mL) and carbenicillin (100μg/mL) from 1000x stocks right before use.
LB Agar plates: Dissolve 37g LB Agar Miller in 1L deionized water, pH to 7.0 with NaOH, autoclave and add chloramphenicol (34μg/mL) and carbenicillin (100μg/mL) from 1000x stocks when the LB agar has cooled down but is still liquid. Pour plates and let LB agar solidify before use. Plates can be stored at 4°C for several months.
2xYT media: mix 16g tryptone, 10g yeast extract, and 5g NaCl in 1L deionized water, pH to 7.0. Autoclave and store until use at room temperature (RT; ~21°C) for a few days. Add chloramphenicol (34μg/mL) and carbenicillin (100μg/mL) from 1000x stocks right before use.
200mM IPTG: Dissolve 0.48g IPTG in 10ml in water. Aliquot and store −20°C for several months.
1M Tris-HCl: Dissolve 60.56 g Tris(hydroxymethyl) aminomethane in 500ml water. Adjust pH to 7.5 with HCl, filter sterilize or autoclave, store at RT indefinitely.
PMSF: 100mM in ethanol. Make this fresh each day: add PMSF to buffers just before use. CAUTION: PMSF powder is hazardous. Use skin/eye protection when preparing PMSF solutions.
5M NaCl: Dissolve 29.22g NaCl in 500ml water. Filter sterilize or autoclave, store at RT indefinitely
0.5M EDTA: Dissolve 14.6g EDTA in 100ml water, filter sterilize or autoclave, store at RT indefinitely.
W buffer: 50mM Tris-HCl pH 7.6, 100mM NaCl, 1mM EDTA, 5mM 2-Mercaptoethanol, 0.2mM PMSF. Make up freshly and add 2-Mercaptoethanol and PMSF right before use.
TW buffer: W buffer with 0.1% (vol/vol) triton X-100. Make up freshly and add 2-Mercaptoethanol and PMSF right before use.
8M Urea: For 2L, add 960.96 g urea to 1L water. Stir to dissolve, and add water to a final volume of 2L.
Deionized urea (8M): Add 20g/L of AG-501-X8 resin to 8M Urea and stir for 1 hour at RT to deionize. (Make 1L for a single histone prep.) Filter to remove resin. Store this at RT and use within a day to make the urea buffer.
100mM Dithiothreitol (DTT): Dissolve 0.154g DTT in 10ml water. Store in aliquots at −20°C for up to several years.
8M guanidine-HCl: Dissolve 7.6g in 10ml water. Store at RT indefinitely.
Unfolding buffer: 7M guanidine-HCl, 20mM Tris-HCl pH 7.5, 10mM DTT. Prepare fresh for each protein preparation.
1M K2HPO4: Dissolve 87.09g in 500ml water, filter-sterilize and store at RT indefinitely.
1M KH2PO4: Dissolve 68.05g in 500ml water, filter-sterilize and store at RT indefinitely.
Lysis buffer for CENP-A/H4 tetramer purification (10ml/cell pellet from 1l bacterial cell culture): 20mM phosphate buffer pH 6.8 (for 1 L buffer use 9.94ml of 1M K2HPO4 and 10.06ml of 1M KH2PO4), 1M NaCl, 5mM 2-Mercaptoethanol, 1mM PMSF, 1mM Benzamidine, 0.05% NP-40, 0.2mg/ml lysozyme. Prepare fresh and add 2-Mercaptoethanol, Benzamidine and PMSF right before use, keep on ice.
Dialysis buffer for CENP-A/H4 tetramer purification (2×4l): 10mM Tris-HCl pH 7.4, 0.75 M NaCl, 10mM 2-Mercaptoethanol, 0.5mM EDTA. Prepare fresh and add 2-Mercaptoethanol right before use.
FPLC Buffers
CRITICAL: All buffers used with the FPLC should be filtered using a 0.2μm filter mesh to remove particulates.
Urea buffer A: 6M deionized urea, 200mM NaCl, 10mM Tris-HCl pH 8, 1mM EDTA, 5mM 2-Mercaptoethanol, 0.1mM PMSF. Prepare fresh for each protein preparation and add 2-Mercaptoethanol and PMSF right before use.
Urea buffer B: 6M deionized urea, 1M NaCl, 10mM Tris-HCl pH 8, 1mM EDTA, 5mM 2-Mercaptoethanol, 0.1mM PMSF. Prepare fresh for each protein preparation and add 2-Mercaptoethanol and PMSF right before use.
Refolding buffer: 2M NaCl, 10mM Tris-HCl pH 7.6, 1mM EDTA, 5mM 2-Mercaptoethanol. Prepare fresh and add 2-Mercaptoethanol and PMSF right before use.
Hydroxyapatite (HA) buffer A: 20mM potassium phosphate (pH 6.8), 5mM 2-Mercaptoethanol. This buffer can be prepared in advance and kept at 4°C. Add 2-Mercaptoethanol right before use.
Hydroxyapatite (HA) buffer B: 20mM potassium phosphate (pH 6.8), 5mM 2-Mercaptoethanol, 3.5M NaCl. This buffer can be prepared in advance and kept at 4°C. Add 2-mercaptoethanol right before use.
Hydroxyapatite (HA) column storage buffer: 0.5M potassium phosphate (pH 6.8). Store at 4°C indefinitely.
10M KOH: Dissolve 140g of KOH in 250ml water, sterile filter and store at RT indefinitely.
Hydroxyapatite (HA) column cleaning buffer: 0.5M KOH. Store at 4°C indefinitely.
S column (HiTrap) buffer A: 10mM Tris-HCl pH 7.4, 10mM 2-Mercaptoethanol, 0.5mM EDTA. Prepare fresh and add 2-Mercaptoethanol right before use.
S column (HiTrap) buffer B: 10mM Tris-HCl pH 7.4, 10mM 2-Mercaptoethanol, 0.5mM EDTA, 2M NaCl. Prepare fresh and add 2-Mercaptoethanol right before use.
30% acrylamide (w/vol): dissolve 150g acrylamide in 500ml water, add 20–50g deionizing resin, stir using a stir plate for 1h and sterile-filter. Store at 4°C for several months.
1% Bis-acrylamide (w/vol): dissolve 2.5g Bis-acrylamide in 250ml water, add 20–50g deionizing resin, stir using a stir plate for 1h and sterile-filter. Store at 4°C for several months.
Stack for SDS-PAGE gel: Mix 25ml 30% acrylamide, 20ml bis-acrylamide, 18.75 ml Tris-HCL pH 6.8 and add water to 150 ml final volume. Sterile filter and store at 4°C for several months.
20% (12.5%) SDS-PAGE: for 10ml volume, mix 6.7 ml (4.2 ml) 30% acrylamide, 0.66 ml (1.04 ml) 1% bis-acrylamide, 2.5ml Tris pH 8.8, 0.12 ml (2.24ml) water. Add 100μl 10% APS and 10μl Temed, pour gel into appropriate gel stand. Add ~500μl Isopropanol on top of the gel and let gel solidify. Remove Isopropanol, insert appropriate gel comb. Add 5μl Temed and 25μl 10% APS to 3ml Stack solution, mix and pipet on top of the solidified separating gel. Let the stacking gel solidify. Keep at RT and use the same day.
20% (w/vol) PEG (MW 4000), 0.5M NaCl: Dissolve 20g PEG and 2.9g NaCl in 100ml water, filter sterilize and store at RT indefinitely.
TE pH 8.0: 10mM Tris-HCl pH 8.0, 0.25mM EDTA, store at RT indefinitely.
TE + 2.5mM NaCl: 10mM Tris-HCl pH 8.0, 0.25mM EDTA, 2.5mM NaCl, store at RT indefinitely.
10x TE: 100mM Tris-HCl pH 7.5, 2.5mM EDTA, filter-sterilize or autoclave, store at RT indefinitely.
High salt (HS) assembly buffer: 10mM Tris-HCl, 0.25mM EDTA, 2M NaCl in cold H2O pH to 7.5 (while cold) with HCl. Prepare fresh for each assembly.
Low salt (LS) assembly buffer: 10mM Tris-HCl, 0.25mM EDTA, 2.5mM NaCl in cold H2O pH to 7.5 (while cold) with HCl. Prepare fresh for each assembly.
1M MgCl2: Dissolve 9.52g MgCl2 in water, sterile filter and store at RT indefinitely.
1M Potassium Acetate: Dissolve 9.8 g Potassium acetate in 50 mL water, bring to a final volume of 100 mL.
1M Tris-acetate: Dissolve 12.1 g tris in 75 mL water, pH to 7.9 with acetic acid. Bring to a final volume of 100 mL with water.
10x Array Digestion (AD) buffer: 500mM potassium, 200mM Tris-acetate, 10mM Dithiothreitol (DTT), 5 mM MgCl2, pH 7.9, store in aliquots at −20°C for up to several years.
5x TBE: 445mM Tris-Base, 445mM boric acid, 10 mM EDTA pH 8.0. Filter-sterilize, store at RT for up to several weeks (until significant precipitation of the buffer has occurred).
10% (w/vol) Ammonium persulfate (APS): Dissolve 1g APS in 9ml water, store at 4°C for up to several weeks.
5% native acrylamide gel: Mix 1.5 ml 40% Acrylamide, 4.5 ml water, 6 ml 1xTBE, 5–10μl Temed and 100μl 10% APS and pour into a preassembled polyacrylamide gel stand. Add the comb immediately (no stack gel is required) and let gel solidify before use.
0.7% agarose gel: Dissolve 0.7g agarose in 100ml water and microwave to dissolve the agarose. Let agarose cool down slightly, add 10μl EtBr at 10mg/ml and pour the gel into a gel tray. Let agrose gel solidify.
Ethidium Bromide: 10mg/ml in water for up to several years.
80% Glycerol (vol/vol): Mix 80ml Glycerole with 20ml water, filter-sterilize and store at RT indefinitely.
19×601 DNA (at ~2.5μg/μl or higher): For preparation see Steps 72–87, store at −20°C indefinitely.
4x Bead Buffer: 200mM Tris-HCl pH 8.0, 1mM EDTA, 300mM NaCl, 0.2% Triton, store at RT for several months.
1x Bead Buffer: 50mM Tris-HCl pH 8.0, 0.25mM EDTA, 75mM NaCl, 0.05% Triton, store at RT for several months.
Array buffer: 10mM Tris-HCl pH 7.5, 2.5mM NaCl, 0.25mM EDTA, store at RT indefinitely.
7.5 % (w/vol) Polyvinyl alcohol: Dissolve 7.5g PVA (MW 30.000–70.000) in 92.5ml water, filter sterilize, store at RT indefinitely.
1M CaCl2: Dissolve 73.5g CaCl2 in 500ml water, filter-sterilize and store at RT indefinitely.
12mM CaCl2 (to release Xenopus egg extracts from CSF arrest): Mix 120μl 1M CaCl2 with 9.88 ml water, filter-sterilize and store in small aliquots at −20°C indefinitely.
25x MMR: 150mM Na-Hepes pH 7.8, 2.5mM EDTA, 2.5M NaCl, 50mM KCl, 25mM MgCl2, 50mM CaCl2; For 4L, dissolve 142.99g Hepes, 584.4g NaCl, 14.91g KCl, 20.33g MgCl2, 29.4g CaCl2, add 20ml 0.5M EDTA and adjust pH to 7.8 with NaOH. Store at RT for up to several months.
10M NaOH: Dissolve 200g NaOH in 500ml water, store at RT indefinitely.
5% (w/vol) Gelatin: Mix 10 × 20g Gelatin with 80ml water in glass bottles, autoclave and store at RT indefinitely. Before use, liquefy the gelatin by microwaving brievely.
2M Sucrose: Dissolve 684.6g Sucrose in water and adjust final volume to 1L. Sterile filter and keep at RT and small aliquots at −20°C for up to several years.
1M K-Hepes pH 7.7: Dissolve 119.15g Hepes in water, adjust pH to 7.7 with KOH, adjust final volume to 500ml. Sterile filter and store at RT indefinitely.
0.5M K-EGTA: Dissolve 95.09g EGTA in water, adjust pH with KOH, adjust final volume to 500ml. Sterile-filter and store at RT indefinitely.
20x XB-salt: 2M KCl, 20mM MgCl2, 2mM CaCl2; sterile filter and keep at RT for several months.
1x CSF-XB buffer: 1xXB-salt (50ml of 20x stock), 50mM Sucrose, 10mM K-Hepes pH 7.7, 5mM K-EGTA, 1mM MgCl2, 110μl 10M NaOH. Prepare fresh.
1000x Protease inhibitors: 10mg/ml of leupetin, pepstatin A, and chymostatin in DMSO; aliquot and keep at −20°C indefinitely.
1x CSF-XB + protease inhibitors: Take 50ml of freshly prepared 1x CSF-XB buffer, add 50μl of 1000x stock protease inhibitors. Prepare right before use.
1x CSF-XB + 0.05% Triton-X-100. Prepare fresh.
20x energy mix: 150mM creatine phosphate, 20mM ATP, 20mM MgCl2, aliquot into 500μl aliquots and keep at −20°C for up to several months.
1000x Cytochalasin D: Prepare stock at 10mg/ml in DMSO, aliquot into 50μl aliquots and keep at −20°C for up to several months.
100x Cytochalasin B: Prepare stock at 10mg/ml in DMSO, aliquot into 50μl aliquots, keep at −20°C for up to several months.
Dejellying buffer: 2% (w/v) L-Cysteine in 1x MMR at pH 7.8 (adjusted with 10M NaOH). Prepare fresh and dissolve L-Cysteine right before use.
10% (vol/vol) Triton-X-100:
Antibody Dilution Buffer (AbDil): 150mM NaCl, 20mM Tris-HCl pH 7.4, 0.1% (vol/vol) Triton X-100, 2% (w/vol) BSA, store at 4°C for up to a few weeks.
1mg/ml Nocodazole: Dissolve nocodazole at 1mg/ml in DMSO, store in small aliquots at −20°C indefinitely.
Xenopus sperm nuclei at 100 000μl−1: Prepared as described in 9 and store in small aliquots (2μl) at −80°C for up to several years.
1M Pipes ph 6.8: Prepare 1M Pipes stock solution by mixing 75.6g Pipes with water and start adding NaOH (otherwise Pipes will not dissolve). Adjust the final pH to 6.8 with NaOH and the final volume to 250ml. Sterile-filter and store at 4°C for several months.
5x BRB80: 400mM Pipes pH 6.8, 5mM MgCl2, 5mM K-EGTA, sterile filter and store at 4°C for up to several months.
Cushion: 1xBRB80, 40% Glycerol, sterile filter and store at RT for up to several months.
Dilution buffer: 1x BRB80, 0.05% Triton-X-100, 30% Glycerol, sterile filter and store at RT for up to several months.
CAPS 0.5M: pH to 11.3 with NaOH. Keep at RT for up to several months.
10% SDS: 10% SDS in water. Keep at RT for up to several years.
CAPS transfer buffer: 10mM CAPS pH 11.3, 1% SDS, optional: 20% MeOH. Prepare fresh. It is possible to store the used buffer at 4°C while stirring and re-use 2–3 times within one week.
5% milk in PBS-T: Keep at 4°C and use within one week.
Coverslips coated in poly-L-lysine
Wash coverslips by incubating them, covered, but not completely sealed in a solution of 1M HCl overnight at 50–60°C. Decant the acid solution into an appropriate waste container, and thoroughly rinse coverslips with several changes of distilled water until the pH returns to neutral. Prepare a solution of 1mg/mL poly-L-lysine in distilled water, and add the coverslips to this solution. Place the solution with coverslips on a rocker for at least 1h at RT. Decant the poly-L-lysine solution (this can be saved, stored frozen, and reused a few times), and rinse the coverslips thoroughly in 10 changes of distilled water. Rinse the coverslips in ethanol, and store submerged in 70% ethanol at RT indefinitely.
Mounting medium
20 mM Tris-HCl pH 8.8, 90% glycerol, 0.5% (w/v) p-phenylenediamine. Prepare the solution of Tris-HCl pH 8.8 and glycerol and bring to the final volume with water. Add the p-phenylenediamine solid (if obtained in medium size pieces or flakes, it can be helpful to gently break these into a powder before adding to the solution), and dissolve by gently bubbling nitrogen gas through the solution until no visible solid remains (generally a few hours). The solution may turn light brown during the dissolution of the p-phenylenediamine. Draw the mounting media into a syringe, attempting to minimize the amount of air trapped in the syringe. Store in the sealed syringe at −20 °C. Discard once the solution turns dark brown (typically a month or two).
PROCEDURE
H3/H4 tetramer or H2A/H2B dimer expression TIMING: ~5–6 days
-
1
For each histone (H2A, H2B, H3 and H4), transform the histone expression plasmid into BL21(DE3) pLysS bacteria using standard procedures: mix1μl of a mini-prep plasmid DNA with 40 μl competent bacteria on ice and incubate for 20 min. Heat shock for 45 sec at 42°C, transfer for 2min back on ice before adding 500 μl LB medium. Incubate with vigorous shaking (~200rpm) for 1h at 37°C. Plate ~ 100μl of the transformation and of the cells-only-control, respectively, each onto a LB/chloramphenicol/carbenicillin plate and incubate at 37°C overnight. Next day, pick a single colony and inoculate.
-
2
Plate on LB/chloramphenicol/carbenicillin plates and incubate overnight at 37°C.
-
3
Pick a single colony and inoculate into 5mL LB/chloramphenicol/carbenicillin overnight.
-
4
Dilute the overnight culture to OD600 = 0.005 (approximately 1:1000) in 2L 2xYT media.
-
5
Grow at 37 °C, shaking at 200rpm to an OD600 = 0.5 – 0.6. To measure the OD600 using a spectrophotometer, take 1ml of the culture and transfer into a plastic cuvette (LB media only is used as a blank). Measure absorbance at 600nm. Remove a 1 mL sample for an analytical gel in Step 34 as a control with no protein expression.
-
6
Add IPTG to a final concentration of 0.2mM from a 200mM stock in water to induce protein expression.
-
7
Grow for 3 hours at 37 °C with shaking.
-
8
Centrifuge at 2500 xg in 1L portions in a JS-4.2 rotor for 15 minutes.
-
9
Discard supernatant and resuspend each portion in 25mL W buffer.
-
10
Combine portions and freeze resuspended cells in liquid nitrogen.
PAUSE POINT: store frozen cells at −80 °C indefinitely.
-
11
Thaw cells in a 37°C water bath with constant shaking.
-
12
Sonicate six times using a probe sonicator with 30 seconds pulses separated by 1 minute intervals at 200W. Keep the cells in an ice bath the entire time.
-
13
Split the lysate into two tubes and centrifuge at 25400 xg for 20 minutes at 4 °C in a JA-20 rotor.
-
14
Decant the supernatant (save 50 μL for an analytical gel in Step 34), and resuspend the pellet in each tube in 25mL TW buffer.
CRITICAL STEP: For this and the following steps, make sure the pellet is thoroughly homogenized (a great deal of insoluble material will still remain, however). Use a spatula to chop the pellet finely.
-
15
Sonicate for 30 seconds on maximum power, keeping the samples on ice.
-
16
Centrifuge at 25400 ×g for 20 minutes at 4 °C in a JA-20 rotor.
-
17
Resuspend the pellet in each tube in 25mL TW buffer.
-
18
Repeat Step 16, resuspend the pellet in each tube in 25mL W buffer and then repeat Step 18.
-
19
Repeat Step 16 and add 175μL DMSO to the pellet in each tube, then combine both portions and mince the pellets finely with a spatula.
-
20
Add 13.2mL Unfolding buffer and soak for 1 h at RT with gentle shaking.
-
21
Homogenize in a dounce homogenizer, completely pulling the plunger through the entire volume several times, until no more large pieces are visible.
-
22
Centrifuge at 17640 xg for 20 minutes at 4°C in a JA-20 rotor.
-
23
Remove the supernatant but save in a fresh tube.
-
24
Resuspend the pellet again in 13.2mL Unfolding buffer the supernatant with the supernatant from Step 23.
-
25
Dialyze this supernatant overnight into 2L of urea buffer A stirring at 4°C; briefly, cut a strip of dialysis tubing sufficiently long to hold the entire supernatant plus a few cm extra at each end. Soak the tubing in urea buffer A for 10 min, and then wash the inside of the tubing with urea buffer A. Clip one end of the tubing, pipette the supernatant into the tubing, and clip the other end closed. Change the dialysis buffer to 2L of fresh buffer A, and dialyze for at least 3h at 4°C. (A white precipitate may form during the dialysis.)
-
26
Pipette the protein solution out of the dialysis bag and centrifuge it at 7840 ×g for 20 min at 4°C in a JA-20 rotor.
-
27
Measure the conductivity of the supernatant and that of the urea buffer. Ensure that the conductivity of the supernatant is less than 1.25 times that of the buffer alone. If it is not, dilute the protein with additional fresh urea buffer until this is the case. (If the ionic concentration of the protein solution is too high, it may not bind to the cation exchange column).
-
28
Assemble a 5mL HiTrap Q column and a 5mL HiTrap S column in series such that the flow will pass through the Q column first and then directly onto the S column.
-
29
Wash the assembled columns in 3 column volumes (CV) water (30 mL total), and then 3CV urea buffer A.
-
30
Load the supernatant from Step 27 onto the columns using a peristaltic pump or FPLC.
-
31
Wash with 2CV urea buffer A.
-
32
Remove the Q column, and attach the S column to the FPLC. (The Q column can be reused after elution with Urea buffer B)
-
33
Elute the S-column with a linear gradient from 0–37.5% urea buffer B (remainder urea buffer A) over 3CV, then 100% urea buffer B for 3CV, collecting 1mL fractions. Wash the column in 100% urea buffer B until the UV absorbance returns to baseline, and then for an additional 5CV.
-
34
Run peak fractions on a 20% SDS-PAGE gel to determine which fractions contain the histone and have the minimum number of other nonhistone bands It can be determined which band corresponds to the histone by comparing the analytical gel samples taken earlier before induction of protein expression and from the crude lysate.
-
35
Pool the histone-containing fractions, and dialyze against three changes of 4L distilled water containing 0.1mM PMSF and 5mM 2-Mercaptoethanol: first for > 2h, then overnight, then > 2h. A white precipitate will likely form during the dialysis.
-
36
Centrifuge the dialyzed protein at 25400 ×g for 20 min at 4 °C in a JA-20 rotor.
-
37
Collect the supernatant and measure the concentration by Bradford.
-
38
Lyophilize supernatant (for tetramer/dimer assembly it is useful to do this in aliquots of ~5mg protein). To lyophilize, measure out 5 mg aliquots of protein solution in 15 mL tubes. Flash freeze in liquid nitrogen. Uncap tubes and place in a sample chamber of a freeze-drying lyophilizer, and lyophilize samples according to manufacturer’s instructions.
PAUSE POINT: Lyophilized protein can be stored at −80 °C indefinitely.
H3/H4 tetramer or H2A/H2B dimer assembly TIMING: ~ 2 days
-
39
If proceeding directly from supernatant (from Step 37), dilute with 7 volumes of Unfolding buffer. If proceeding from lyophilized histones (Step 38), resuspend directly into this buffer at 10mg/mL (resuspending at this high concentration helps to avoid having to concentrate the protein before gel filtration, or having protein that is too dilute after gel filtration).
-
40
Mix ~10mg H3 and an equimolar amount of H4, or ~10mg H2A and an equimolar amount of H2B.
-
41
Dialyze against Refolding buffer, three changes of 4 L: for >2h, then overnight, then >2h.
-
42
If the volume has increased to more than 5mL, concentrate this in an amicon conical concentrator, 10k MWCO, to a final volume of ≤5mL.
-
43
Inject onto a Superdex 200 16/60, run at 1mL/min in Refolding buffer, collect 1mL fractions.
-
44
Run fractions on a 20% denaturing polyacrylamide gel, pool the fractions that have approximately equal amounts of H3 and H4 (or H2A and H2B), aliquot, and freeze in liquid nitrogen.
PAUSE POINT: Store indefinitely at −80 °C.
CENP-A/H4 expression TIMING: ~ 4–5 days
-
45
Repeat Steps 1–2 using the CENP-A/H4 bicistronic expression plasmid.
-
46
Pick a single bacterial colony, inoculate into 5mL LB/chloramphenicol/carbenicillin and grow at 37°C for 8–10 h.
-
47
Dilute the culture into 2× 50ml cultures in LB/chloramphenicol/carbenicillin and grow at 37°C overnight.
-
48
Dilute cultures 1:50 – 1:100 in 3× 2L 2xYT media.
-
49
Grow at 37 °C, shaking, to an OD600 = 0.2 and then switch to RT until OD600 = 0.5–0.6
-
50
Add IPTG to a final concentration of 0.2mM from a 200mM stock in water to induce protein expression.
-
51
Grow for ~6 hours shaking.
-
52
Repeat Step 8, wash pellets in PBS and repeat Step 8 again.
-
53
Freeze cell pellets in liquid nitrogen.
PAUSE POINT: store frozen cells at −80 °C indefinitely.
-
54
The HA column is stored in 500mM potassium phosphate, pH 6.8. Before cleaning, pre-equilibrate the column by washing with 1CV (~60ml) 20mM potassium phosphate, pH 6.8.
-
55
Clean column thoroughly with ~ 1CV (60 – 100ml) 0.5 M KOH.
-
56
Equilibrate HA column with 2 CV (~120ml) 20mM potassium phosphate, pH 6.8.
-
57
Prepare HA column buffers A and B without 2-Mercaptoethanol, filter and store overnight at 4°C.
-
58
Thaw cell pellets from Step 53 by resuspending in ice-cold lysis buffer. Use 60ml of lysis buffer for the cell pellets from 6L bacterial culture (see Step 48) and pool in a glass beaker.
-
59
Sonicate 6 times at 200W maximum power for 20sec separated by 1 min intervals. Keep on ice between the cycles. (Optional: take a ~1 μl sample for an analytical gel in Step 71)
-
60
Centrifuge at 110,000 g in a Ti70 rotor for 1h. (Optional: take a ~2 μl sample for an analytical gel in Step 71)
-
61
Load supernatant onto pre-equilibrated HA column from Step 56 at a flow rate of 1–2ml/min using a peristaltic pump. Save flow through. (Optional: take a ~2 μl sample for an analytical gel in Step 71)
-
62
Attach HA column to the FPLC.
-
63
Wash HA column with 6 CV (~ 400ml) of 28% HA column buffer B (corresponds to 1M NaCl). Save the wash. (Optional: take a ~4 μl sample for an analytical gel in Step 71)
-
64
Run linear gradient from 28% HA column buffer B (remainder HA buffer A) to 100% buffer B over 2 CV (120ml) and collect 6ml fractions.
-
65
Run fractions (~20μl) on a denaturing 20% polyacrylamide gel to determine which fractions contain soluble CENP-A/H4 tetramers. Relevant fractions will contain visible bands at approximately 18kDA (hsCENP-A) and 13 kDa (H4).
-
66
Pool relevant fractions and dialyze overnight against 4L dialysis buffer.
-
67
Change dialysis buffer and dialyze for another 2-4h.
-
68
Load sample onto 1ml HiTrap SP FF column at a flow rate of 1ml/min. Save flow through. (Optional: take a ~20 μl sample for an analytical gel in Step 71)
-
69
Wash with 10 CV (10ml) of 37% S column buffer B.
-
70
Run a linear gradient from 37% S column buffer B (remainder S column buffer A) to 100 % S column buffer B to elute soluble CENP-A/H4 tetramer.
-
71
Run fractions (~20μl) on a denaturing 20% polyacrylamide gel to determine which fractions contain soluble CENP-A/H4 tetramers of high purity (no contaminating bands are detectable on the gel) aliquot, and snap-freeze in liquid nitrogen.
PAUSE POINT. Store indefinitely at −80 °C.
Preparation of biotinylated DNA TIMING: ~ 3–4 days (excluding plasmid preparation)
-
72
Transform the 19×601 plasmid 3,13,38 into competent SURE2 bacteria (use SURE2 cells only as a control) using standard procedures: mix 1μl of a mini-prep plasmid DNA with 40 μl competent bacteria on ice and incubate for 20 min. Heat shock for 45 sec at 42°C, transfer for 2min back on ice before adding 500 μl LB medium. Incubate shaking at ~200rpm for 1h at 37°C. Plate ~ 100μl of the transformation and of the cells-only-control, respectively, each onto a LB/chloramphenicol/carbenicillin plate and incubate at 37°C overnight. Next day, pick a single colony and inoculate a 5ml starter culture in LB medium with chloramphenicol/carbenicillin for 8h at 37°C. Dilute the starter culture 1/1000 into 2L LB medium supplemented with chloramphenicol and carbenicillin and grow overnight at 37°C.
-
73
Purify plasmid DNA with a Qiagen Giga Kit Tip-10000 (or any other comparable method), following the manufacturer’s instructions exactly, but scale the amounts of buffers specified in the manual to 2L of bacterial culture (instead of 2.5L). Typically, we get 7–10mg purified plasmid DNA from each preparation.
PAUSE POINT Store plasmid DNA at −20°C indefinitely.
-
74
Digest 2× 500–750μg plasmid DNA with EcoRI, XbaI, DraI, HaeII (10μl of each enzyme at 20000U/ml) in a final volume of 1ml (per reaction) at 37°C overnight. Check for efficient digestion by running a 0.7% agarose gel using standard procedures: load 0.5–1μl of each digest and run at 100V for approximately 20min (the exact conditions for elecrophoresis have to be determined individually). The 19×601 fragment runs at 3.8kb, while the digested backbone appears as 4 major bands < 1kb (Figure 3a).
-
75
Add 5M NaCl to the digest to reach a final concentration of 0.5M NaCl (e.g. add 111μl 5M NaCl to a 1ml reaction).
-
76
Add 20% PEG, 0.5M NaCl to a final of 5% PEG, 0.5M NaCl (e.g. 370μl to a 1111μl reaction).
-
77
Centrifuge for 10 min at ~5000 ×g at RT in a tabletop microcentrifuge.
-
78
Transfer supernatant into a fresh tube (take 1μl sample for analysis on an agarose gel in Step 81) and keep original tube in a rack at RT.
-
79
Add 20% PEG, 0.5M NaCl to the transferred supernatant to a final concentration of 5.5% PEG, 0.5M NaCl.
-
80
Repeat Steps 77–79, adding additional PEG (in 0.5% final concentration increments), and centrifuging, until the final PEG concentration reaches 10%.
-
81
Load collected 1μl samples onto a 0.7% agarose gel to identify fractions containing the 19×601 fragment. Typically, the desired 19×601 DNA fragment precipitates between 8–9% final PEG concentration (Figure 3b).
-
82
Resuspend precipitated DNA (from appropriate tubes stored in Step 78) in TE pH 8.0 and pool fractions of comparable quality. The volume of TE for resuspension depends on the efficiency of PEG precipitation and has to be determined empirically. As a guideline, assume that the 19×601 fragment spans approximately half the length of the plasmid and so there is ~ 250μg precipitated 19×601 DNA/500μg plasmid DNA input. If two fractions contain about equal amounts of precipitated 19×601 DNA it is reasonable to assume that each of these tube contains ~125μg 19×601 DNA. To resuspend precipitated DNA at 1–2μg/μl, use ~100μl TE for each tube. If 2× 500μg plasmid DNA was digested, a total of 4 tubes with ~100μl would be the result that can be dialyzed in 2× 200μl dialysis buttons (see Step 83).
-
83
Dialyse resuspended DNA in 50–200μl dialysis buttons into TE pH 8.0 overnight to eliminate residual PEG.
-
84
Change dialysis buffer one more time and dialyse for another 3–4h.
-
85
Harvest 19×601 DNA using a Hamilton syringe and determine the concentration at OD260 using a spectrophotometer. Typically, the digestion of 1mg 19×601 plasmid DNA yields 300–400μg digested and purified 19×601 DNA fragment.
-
86
Optional: Analyze 1μl of a 1:10 dilution on a 0.7 % agarose gel to verify purity.
-
87
A concentration of ~2.5μg/μl or higher is ideal. If required, NaOAc/EtOH precipitate DNA using standard protocols: add 1/10th volume 3M NaOAc and 2 volumes ice cold 95% EtOH. Incubate at −20°C for at least 20 min (or overnight). Centrifuge for 10 min at 10000 ×g and decant supernatant. Wash with ~ 500μl 70% EtOH, centrifuge again for 10 min at 10000 ×g, decant supernatant. Air-dry DNA pellet and resuspend in TE pH 8.0 in an appropriate volume to adjust the concentration.
PAUSE POINT: Store DNA at −20°C until use.
-
88
Set up a biotinylation reaction by mixing 500μg digested and purified 19×601 DNA in a final volume of 400μl with biotin-14-dATP, α-thio-dGTP, α-thio-dTTP and dCTP at 35μM final concentration for each nucleotide and 24μl Klenow fragment. CRITICAL STEP: Thiolated nucleotides are resistant to removal by some nucleases and help prevent enzymatic removal of the biotin label in cell extracts.
-
89
Incubate for 3h at 37°C.
-
90
Purify biotinylated 19×601 DNA using appropriate numbers of Qiagen PCR purification columns (considering a DNA binding capacity of 10μg DNA per column e.g. use 25–30 columns for 300μg DNA as a starting material), following the manufacturer’s instructions. Elute with 30μl EB buffer (from the Qiagen kit) or water per column, pool eluted fractions and precipitate using NaOAc/EtOH precipitation, as described in Step 87. Alternatively, purify large biotinylation reactions using G-50 gel filtration resin to remove the unincorporated nucleotides 10.
-
91
Resuspend in 100μl TE pH 8.0 and measure the concentration at OD260 using a spectrophotometer. A final concentration of 3–5μg/μl is ideal. Typically, the input of 500μg digested 19×601 DNA into the biotinylation reaction yields ~300μg of biotinylated 19×601 DNA.
-
92
Optional: To analyze the biotinylation efficiency, incubate 500ng biotinlyated 19×601 DNA with and without 0.5μl Streptavidin-FITC as well as non-biotinylated 19×601 DNA with 0.5μl Streptavidin-FITC in TE pH 8.0, 2.5mM NaCl for 20 min at RT.
-
93
Run reactions on a 0.7% agarose gel for 1–2h and analyze by imaging the DNA by EtBr stain and the biotinylation by FITC fluorescence (e.g. using a Typhoon 9400 Variable Mode Imager). Efficient biotinylation causes the 19×601 DNA to migrate a single band more slowly in the presence of streptavidin (Figure 3c).
PAUSE POINT: Store biotinylated DNA at −20°C until use.
Figure 3. Generation and analysis of biotinylated DNA arrays for chromatin reconstitution.

(a) Schematic map of the 19×601 nucleosome positioning sequence plasmid (left panel) indicating the position of the 601 sequence repeats and restriction sites (RE) of interest. Result of a virtual RE digest with EcoRI, HaeII, DraI and XbaI (right panel) to facilitate the purification of the 19×601 sequence (~3800bp) from the digest plasmid backbone (fragments < 700bp) by PEG precipitation. (b) Representative agarose gel of the PEG precipitation procedure. At ~ 8.5–9% PEG the fragment of interest precipitates while DNA fragments <700bp remain soluble in the supernatant. (c) To asses the biotinylation efficiency of the array DNA (Biotin-DNA), and of non-biotinylated DNA as a control (Non-Biotin DNA), the DNA was incubated for 15 min with (+) or without (−) Streptavidin-FITC. Subsequently, the reactions were separated by agarose gel electrophoreses and the DNA was visualized by Ethidium Bromide (EtBr) stain. Bound Streptavidin-FITC (FITC) caused a gel mobility-shift and is detected by fluorescence imaging (lower panel). Figure is adapted from 2.
Determination of the protein concentration of histone preparations TIMING: ~ 1h
-
94
Before assembling chromatin arrays, precisely determine the protein concentration for each histone preparation using a 280nm absorption reading. First, centrifuge a sample of the protein solutions (from Step 44 or 71) for 10 min at 10000 ×g at 4°C.
-
95
Mix 5μl of the supernatant with 5μl 8M Guanidine hydrochloride to denature the protein and use a spectrophotometer to obtain the 280nm absorption.
-
96
Use the theoretical extinction coefficient ε (the specific absorbance of a (pure) protein in solution over a 1 cm path length at 1 M concentration at 280nm) and the calculated molecular weight (MW) of each histone to determine the protein concentration in mg/ml. Both parameters can be calculated from the amino acid sequence (http://web.expasy.org/protparam/). The protein concentration in mg/mL can be determined by dividing the protein absorbance at 280 nm by the extinction coefficient and then multiplying by the protein’s molecular weight.
Chromatin Array Assembly TIMING: ~ 5 days
-
97
Estimate the required ratios of H3/H4 and CENP-A/H4 tetramers to DNA arrays for equal stoichiometry and use a slight excess of H2A/H2B dimers (2.2x the DNA amount), as tetramers can assemble on the DNA without dimers, and a slight excess of dimer will help avoid this.
CRITICAL STEP: A slight molar excess of H2A/H2B dimers is fine, but the exact ratio of histone tetramers (CENP-A/H4 and H3/H4) should be determined for each histone preparation by titration. For example, try ratios of DNA:tetramer of 1:0.8, 1:0.9, 1:1.0, 1:1.1 and 1:1.2 to determine the ideal ratio for high nucleosome saturation and to avoid under- or oversaturation. Undersaturation means that not all 19 nucleosome positioning sequences are occupied by a histone octamer, while oversaturation means that an amount of histone greater than that contained in 19 nucleosomes is associated with an array. Oversaturation might occur through nonspecific interaction of basic histone complexes with the negatively charged DNA and potentially leads to enhanced aggregation of chromatin arrays (as observed by microscopy) while undersaturation reduces aggregation when compared to fully saturated chromatin arrays (see TROUBLESHOOTING).
-
98
Assemble chromatin arrays by salt dialysis. A schematic of the general set up is shown in Figure 4a. First, prepare 500ml cold HS assembly buffer and 2L cold LS assembly buffer, adjust pH to 7.5 and keep at 4°C.
-
99
Set up chromatin arrays assembly reactions in 1.5ml tubes on ice. Typically, we use 50μl reactions or 200μl reactions at a final DNA concentration of 0.2μg/μl or 0.15μg/μl, respectively. Mix appropriate volumes of H2O, 10xTE and 5M NaCl to achieve a final concentration of 1xTE and 2M NaCl. Add, in the following order, biotinylated DNA, H2A/H2B dimers and CENP-A/H4 or H3/H4 tetramers at a molar stoichiometry of 1:2.2:1.
-
100
Moisten the membrane in each “Slide-a-lyzer mini dialysis unit” on the surface of the 500 ml cold HS assembly buffer. Pipette each assembly mixture (either 50 or 200μl reactions) into one dialysis unit.
-
101
Cap the dialysis units and adjust the units in a floating tube rack such that they are approximately the same depth into the buffer; this will minimize (though not eliminate) differences in post-dialysis volume between samples. Alternately, if changes in the dialysis volume become problematic, use 50μl (or 200μl) fixed volume dialysis buttons (Figure 4b), as described in Box 1.
-
102
Transfer the beaker to a magnetic stir plate at 4 °C. Set stirring speed to very low. If using the dialysis buttons (Box 1), adjust length of dental floss to not have the dialysis buttons too close to the surface and to avoid entanglement of individual buttons if multiple are used at the same time.
-
102
Prepare tubing for the array assembly as shown in Figure 4a, by connecting a 1ml plastic pipette to one end of two pieces of peristaltic pump tubing, and clip both pieces of tubing with the plastic pipettes at the same end into the peristaltic pump. Set flow speed to 0.5ml/min. Put one pipette into the 2L beaker containing low salt assembly buffer and attach the corresponding loose end of tubing with a binder clip to the rim of the dialysis beaker. Attach the second pipette, using a binder clip, to the rim of the dialysis beaker and put the loose end into a liquid waste container or the sink.
-
105
When the buffer exchange is complete (~67h later), exchange dialysis buffer one more time with fresh LS assembly buffer and dialyze for an additional 4h.
-
106
Harvest assembled arrays by pipetting out of the dialysis unit, taking care not to puncture the dialysis tubing, and transfer the assembly reaction into a 1.5ml tube on ice. The volume may differ for each assembly, so assess the final volume of each array and adjust experimental volumes accordingly. If using the dialysis buttons (Box 1), pierce the dialysis tubing with a clean Hamilton syringe to transfer the array assembly to a 1.5ml tube on ice.
PAUSE POINT Assembled arrays can be stored for up to ~3 months at 4°C.
-
107
To assess the level of saturation of the assembled arrays, use 500ng assembled array DNA and digest with 1μl Ava1in AD Buffer supplemented with 1mM DTT, 1xBSA in a final volume of 20μl at RT overnight. Use 500ng biotinylated DNA (no nucleosomes) as a control (Figure 5, panel a).
CRITICAL STEP: In the 19×601 array DNA, each positioning sequence is separated by an AvaI restriction site, thus digestion with AvaI generates 19x ~ 200bp fragments.
-
108
Add Glycerol (80% vol/vol) to a final concentration of 20% (6.7μl/20μl reaction).
-
109
Run the whole reaction (~27μl) of digested arrays and DNA on a 5% native acrylamide/bisacrylamide gel based on 0.5x TBE in 0.5x TBE as a running buffer. (The appropriate run time depends on the gel system used.)
-
110
Mix 200ml 0.5xTBE with 15μl EtBr and pour into a gel-staining container.
-
111
Disassemble the gel system, remove spacers and one glass-plate leaving the gel stuck to the second glass-plate. It is easy to tear the gel when detatching it from the plate, so turn the gel plate upside down, hold it over the gel-staining container, and use a spatula to slowly detach one corner/side of the gel from the remaining glass-plate. Using the spatula, detach as much of the gel from glass-plate so that it rolls slowly into the container. Stain gel, gently rocking or shaking for 20min at RT.
-
112
Wash gel 3x with H2O to remove residual EtBr.
-
113
Transfer gel to a UV transmittable glass plate.
-
114
Analyze gel by fluorescence imaging (e.g. using a Typhoon 9400 Variable Mode Imager) (Figure 5a–b). Efficient array assembly causes a band shift from ~200bp (DNA control) to ~700bp (mononucleosomes).
TROUBLESHOOTING
Figure 4. Experimental apparatus for assembling chromatin arrays by salt dialysis.

(a) To assemble arrays, use a 500ml and a 2Lbeaker as a reconstitution vessel and as a low salt buffer reservoir, respectively, and a peristaltic pump to drive the buffer exchange from high salt to low salt buffer over ~ 67h. The dialysis buttons are attached with binder-clips to the rim of the beaker and the speed of the magnetic stir bar is set to very low to avoid entanglement if multiple dialysis buttons are present. Usually, we cover the beakers with plastic wrap or tinfoil to prevent dirt from falling into the beakers (not depicted). (b) The individual parts that are required to assemble the fixed volume (50 or 200μl) dialysis buttons for chromatin assembly: dialysis button, dental floss, rubber gasket, dialysis tubing, golf tee or 12mm head of a socket wrench (left panel). The dialysis button assembly is set up as described in Box 1; the rubber gasket (with the dental floss threaded through the gasket) is slid down either a golf tee or a 12mm head of a socket wrench, which is holding the dialysis tubing in place after the sample has been pipetted into the buttons and covered with the dialysis tubing (middle panel). The dialysis tubing is then held in place by the rubber gasket, tightly sealing the dialysis button, and subsequently the dental floss can be used to attach the buttons to the rim of a beaker using binder clips (right panel).
BOX 1. Using fixed volume dialysis buttons.
Cut a square of dialysis tubing about four times the diameter of a dialysis button, cutting off the edges of the dialysis tubing to obtain single-layer pieces.
Wet dialysis tubing in HS assembly buffer.
Pipette the chromatin assembly mixture into the button. If the mixture does not completely fill the button to the top, add additional HS assembly buffer as incomplete filling of the button can trap air bubbles, preventing efficient buffer exchange (small bubbles are fine).
Place the dialysis tubing on the button, being careful not to trap any air and hold in place using a golf tee (or a head of a socket wrench for 200μl dialysis buttons).
Thread a length of flat dental floss through the rubber gasket, and slide the gasket down over the tee and into the groove on the dialysis button. See Figure 4b for illustration of dialysis button assembly.
Use the dental floss to suspend the button in the dialysis beaker.
Figure 5. Analysis and quality control of the chromatin array assemblies.

(a) Representative CENP-A and H3 chromatin arrays at >90% saturation of the nucleosome positioning sites (H3 and CENP-A lanes, respectively) and biotinylated control DNA (DNA) were separated on a native gel after AvaI digestion to generate ~ 200 bp DNA fragments and/or H3 and CENP-A mononucleosomes (which run at the size of ~ 700 bp DNA). The chromatin arrays assemble with equal histone protein stoichiometry as indicated by the lower panel showing the stochiometry of histones in undigested chromatin arrays by Coomassie stain (as described in ref. 2). Figure is adapted from 2. (b) Two sets of chromatin arrays were digested with AvaI and subsequently resolved on a native gel. Oversaturated arrays display an additional higher molecular weight band (oversaturation band) and undersaturated arrays display a lower molecular weight band (undersaturation band) corresponding to the ~200bp fragment of digested control DNA when compared to the mononucleosome band (mononucleosome). (c) Chromatin arrays that are >90% saturated at their nucleosome positioning sequences typically cause the beads to form small to medium sized (10–50 beads) aggregates and display an even DNA stain when assessed microscopically (left panel). Heavily oversaturated chromatin arrays might form larger aggregates and the DNA staining sometimes appears rather patchy (right panel). Oversaturated chromatin arrays may cause centromere and kinetochore assembly to occur less efficiently when compared to >90% saturated chromatin arrays.
Preparation of Chromatin Beads TIMING: ~ 1h
-
115
Prewash 45μl Dynabeads three times in ~300μl 1x bead buffer using a magnetic particle concentrator to collect beads between individual washes allowing the supernatant to be aspirated. To wash beads, pipet them up and down or flick by hand. The appropriate volume of beads to start with depends on the number of samples that will be analyzed in each experiment. 45μl Dynabeads are sufficient to analyze the binding of four different centromere or kinetochore proteins to CENP-A arrays versus H3 arrays as described in Step 119A and 119B. It will also include a coverslip for the H4 staining and for the bead only control (no chromatin arrays coupled), both of which are required for the subsequent quantification by fluorescence microscopy (Figure 6a). Split beads into three tubes (15μl/tube; one for CENP-A arrays, one for H3 arrays and one for the bead only control), place onto magnetic particle concentrator and aspirate the supernatant.
-
116
To each tube, add 10μl chromatin arrays (CENP-A or H3 chromatin) from Step 106 (or 10μl of array buffer for the bead only control) in bead buffer supplemented with 2.5% Polyvinyl alcohol (PVA) to make a final volume of 50μl (the volume of this reaction should be scaled up or down according to the amount of beads used in a particular experiment).
CRITICAL STEP: Chromatin arrays are typically assembled at a final DNA concentration of 0.15μg/μl or 0.2μg/μl (see Step 99). Typically, for immunofluorescence analysis of centromere and kinetochore proteins, analysis of microtubule polymerization and the mitotic checkpoint assay we couple 2μl chromatin arrays (at 0.15μg/μl) or 1.5μl chromatin arrays (0.2μg/μl) to 3μl Dynabeads for each sample.
-
117
Couple chromatin arrays to beads using a vortex adaptor for 1.5ml tubes at low shaking speed (the exact setting has to be determined for each vortex model used) for at least 30 min at RT. As a guideline, make sure that the speed is fast enough to keep the beads in solution preventing the beads from settling down in the tube but not so fast that the bead/buffer mixture spills all over the tube. Typically, good coupling causes the beads to clump slightly.
CRITICAL STEP: In our hands, clumps of 10–30 beads are optimal for centromere and kinetochore assembly. Presumably, a certain density of CENP-A chromatin is required to efficiently promote centromere and kinetochore assembly as well as microtubule polymerization. Similarly, clumps of 12–20 beads have been reported to be optimal for spindle assembly around DNA coated beads 10. The size of the bead clumps can be assessed by microscopy (Figure 5c).
TROUBLESHOOTING
-
118
ash the chromatin coupled beads 1x in ~200–300μl bead buffer.
Figure 6. Set up of an immunofluorescence experiment and of spin down tubes for the analysis of microtubule polymerization.
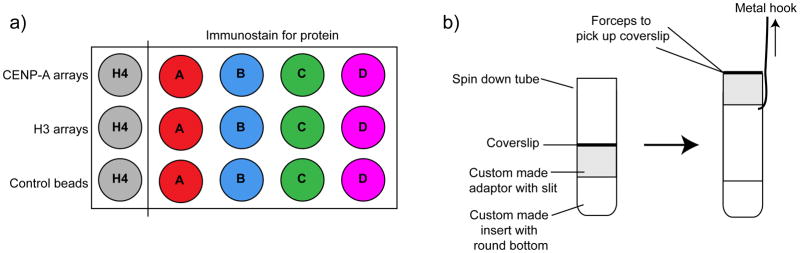
(a) Schematic showing the number of coverslips that are required to compare the binding efficiency of four different proteins (A–D) to CENP-A and H3 chromatin arrays. Each individual antibody staining is accompanied by a bead only control that is used to subtract the background fluorescence of the beads during quantification. To normalize the amount of bound protein to the amount chromatin arrays, a separate H4 staining is used to quantify the amount of chromatin arrays coupled to beads. (b) Two custom-made plastic adaptors are placed into Corex spin down tubes, one with a round bottom to fit the tube and a second with a slit on one site for retrieval with a metal hook. The coverslip is placed on top of the adaptors and, after spindown, the adaptor with the slit is pulled upwards by inserting the metal hook. Once accessible, the coverslip can be handled with forceps for further processing. Image adapted from 10.
Functional Assays to investigate centromere and kinetochore assembly and function
-
119
For functional assays to investigate centromere and kinetochore assembly and function, use option A to analyze centromere (and kinetochore) assembly in CSF extract; option B to analyze kinetochore (and centromere) assembly in cycled extract; option C to analyze microtubule polymerization and stabilization in CSF extracts; or option D to analyze mitotic checkpoints in cycled extracts.
A. Centromere (and kinetochore) assembly in CSF extracts. TIMING: ~ 90min (without chromatin coupling, extract making and immunofluorescence)
-
Wash chromatin beads from Step118 two times with ~200–300μl CSF-XB supplemented with 0.05% Triton-X-100.
TROUBLESHOOTING
Aspirate supernatant and mix chromatin coupled beads with 100μl freshly prepared CSF extract (see Box 2) per tube on ice by pipetting the extract-bead mixture 2–3 times up and down and transfer to 18–20°C waterbath.
To allow centromere and kinetochore protein assembly, keep at 18–20°C for 60 min and flick tube every 15 min to keep chromatin beads in solution. Optional: If you want to analyze the assembly of kinetochore or mitotic checkpoint proteins, add Nocodazole (or DMSO as a control) at 10μg/ml final concentration after 5–10 min of incubation.
Transfer the reactions to the magnet to collect the beads. Optional: If the extract is very viscous, it might take some time to efficiently collect the beads. To speed up the process, the quick spin option of a tabletop centrifuge can be used to lightly pellet the beads before using the magnet. This should take a few seconds; determine the exact time empirically. Avoid spinning too hard, since this will make it harder to resuspend the bead pellet afterwards.
Aspirate extract and quickly wash chromatin beads three times in ~200–300μl cold CSF-XB buffer supplemented with 0.05% Triton-X-100.
To fix the samples, resuspend chromatin beads in 100μl CSF-XB + 0.05% Triton-X-100 by pipetting up and down and add Formaldehyde to a final concentration of 2%. Incubate for 5 min at RT.
-
Wash three times with ~200–300μl AbDil and adjust concentration to 1μl chromatin on beads per 10μl AbDil.
PAUSE POINT: Fixed samples can be stored in AbDil for a 1–2 days at 4°C and processed later.
Box 2. Xenopus CSF extract preparation TIMING: ~ 2h (without priming frogs).
To induce maturation of oocytes, prime female frogs with 0.5ml PMSG using a 1ml disposable syringe and a 25-gauge needle. Optional: For optimal egg laying prime frogs a second time two days after the first priming injection with 0.25ml PMSG. Primed frogs can be used for egg laying for 2 weeks after priming.
Inject 3 frogs with 0.5ml hCG (500U) to induce egg laying 16–18h before the extract will be prepared.
After injection, separate frogs into individual plastic containers with a minimum of 2L 1x MMR made with deionized water and keep at 16°C overnight.
Before making buffers for the extract preparation, rinse all glassware with deionized water before use. Prepare required buffers (2L 1xMMR, 200ml dejellying buffer (the cysteine is added immediately before use), 500ml CSF-XB buffer) and keep at 16°C.
Coat a Pyrex glass dish and a 100ml glass beaker with gelatin to prevent eggs sticking to the glass by pouring ~ 10–20ml and ~ 5ml, respectively, hot, liquid gelatin solution into the water filled containers and swirling the container to coat the sides. Keep containers with water/gelatin solution on your bench until further use.
-
Remove frogs from the plastic container, rinse frog eggs 1–2x with ~500ml 1xMMR and garden eggs using a glass pasteur pipette to remove strings of eggs, puffy eggs, and activated eggs. Do not use the batch of eggs if bad eggs comprise more than 10% of the whole batch.
TROUBLESHOOTING
Prepare 50ml CSF-XB + protease inhibitors. Transfer 2ml of this solution into a separate tube, add CytochalasinB, and pipet 1ml each into two SW-55 centrifuge tubes.
-
Remove water/gelatin solution from the Pyrex glass dish and wash 1–2x with ~200ml 1x MMR. Pour gardened eggs from the plastic container into the Pyrex dish and wash eggs again 1–2x with ~200–300ml 1xMMR. Add the cysteine to the dejellying buffer (see REAGENT SETUP) and mix until the cysteine is dissolved. Immediately, remove 1x MMR from the Pyrex dish containing the eggs and pour in ~50ml of the dejellying buffer. Swirl gently, remove the dejellying buffer and pour in the remaining 150ml of the dejellying buffer. Swirl again gently, remove eggs (if necessary) that activate (the metaphase arrest of meiosis II is released) as indicated by the contraction of the dark pigments. Observe the progression of the dejellying process by eye. When eggs are fully dejellied they become closely packed when collected at one site of the Pyrex dish when the dish is tilted. This process takes 5–10 min.
CRITICAL STEP: Eggs should not be left in dejellying solution for too long as they will start to lyse.
When dejellying is complete, quickly pour off dejellying buffer and wash thoroughly 3–4 times with ~ 200–300ml of CSF-XB.
Pour eggs into the 100ml beaker from step 5 above, containing a few ml of CSF-XB buffer (eggs should not be poured into a dry glass container as this could cause lysis).
Pour off the CSF-XB buffer and exchange with ~10 ml of CSX-XB + protease inhibitors. Remove buffer and pour in remaining 40ml of CSF-XB + protease inhibitors.
Using a fire-polished pipette, transfer eggs into the SW-55 centrifuge tubes containing 1ml CSF-XB + protease inhibitors + CytochalasinB from step 7, above. To avoid exposing eggs to air or crushing them, gently suck up eggs with pipettes and only release eggs after the tip of the pipette is submerged in the buffer without touching the sides of the tube. Depending on the amount of eggs, fill up one tube (in this case an appropriate amount of water has to be added to the second tube, which will serve as a balance for the centrifugation step later) or both tubes equally.
Remove excess buffer on top of the eggs with an aspirator and use forceps to gently slide tubes into the adaptor tubes (13ml Falcon tubes) without caps.
Pack the eggs using a clinical centrifuge by centrifuging for ~30 sec at ~300 xg and then for ~30 sec at ~500 xg (ref. 9).
Remove tubes from adaptor tubes, aspirate excess liquid (some excess buffer will be visible on top of the packed eggs) and crush eggs by centrifuging for 15min at 9480 xg in SW-55 swinging bucket rotor].
After the spin, carefully put tubes on ice.
-
Clean tubes by wiping with ethanol and pierce, bevel up, with a 1ml syringe and a 16-gauge needle to collect the cytoplasmic fraction (straw colored middle layer).
CRITICAL STEP: Avoid taking black particles from the bottom layer containing yolk granules and nuclei as well as contamination from the yellow top layer containing lipid droplets.
Carefully measure the amount of extract and expel into a fresh tube on ice.
Add appropriate amount of protease inhibitors (1000x), CytochalsinD (1000x), cold Sucrose (1:40) and energy mix (1:20) to the extract and mix well. Use extract within 1–2 h as the extract quality declines over time.
B. Kinetochore and (centromere) assembly in cycled extracts. TIMING: ~ 4 h (without chromatin coupling, extract making and immunofluorescence)
Repeat Steps 119Ai–ii, using 50μl freshly prepared CSF extract.
After 5–10 min, add CaCl2 (final concentration 0.6mM) and incubate for 80 min at 18–20°C to drive the CSF arrested extract into interphase. Flick tubes every 15 min to keep chromatin beads in solution. Optional: Take a sample for a western blot to monitor the cell cycle stage (as described in ref. 2).
Add 1 volume (50μl per tube) fresh CSF extract and incubate for 90 min to drive the extract into mitosis. Flick tubes every 15 min to keep chromatin beads in solution. Optional: If analyzing assembly of spindle assembly checkpoint proteins, premix CSF extract with Nocodazole (or DMSO as a control) at 10μg/ml final concentration.
After 90 min, wash and fix samples as described in Steps 119Avi–vii.
C. Microtubule polymerization and stabilization in CSF extracts. TIMING: ~ 5 h (excluding chromatin coupling and extract preparation)
Repeat Step 119Ai
Aspirate supernatant and mix 2μl chromatin coupled beads with 20–50μl freshly prepared CSF extract per spindle assembly reaction in a 0.5ml Eppendorf tube on ice.
Mix reactions by pipetting 2–3 times up and down. As a control for efficient spindle assembly, prepare a sample with 250 sperm nuclei/μl extract.
Transfer samples to an 18–20°C waterbath (empty 1000ul pipette tip boxes left open to the air work well; adjust temperature with a bit of ice as needed) and incubate at 18–20°C for 75–90 min. Flick tubes gently every 15min to keep chromatin beads in solution. Optional: Add 100ng/μl Nocodazole to the reaction or transfer reaction to 4°C for 10min to induce microtubule depolymerization.
Pre-fix reactions for 10min at RT by pipetting the reaction into 1ml dilution buffer supplemented with 2.5% Formaldehyde. Use a cut-off tip to avoid shearing of the assembled spindles and mix samples by inverting the tubes.
Place custom made adaptors into Corex tubes, place a poly-L-lysine coated coverslip on top and add 5ml of cushion (Figure 6b) 9,10.
Carefully layer prefixed samples onto cushion using a plastic transfer pipette.
Centrifuge reactions for 20min at 3290 ×g in a JS13.1 rotor onto coverslips.
Using a metal hook and forceps, take the coverslips out of Corex spin down tubes and postfix in ice cold MeOH for 5 min 9,10.
After MeOH fixation, place coverslips into a humidified chamber and wash 3 times with AbDil (by pipetting AbDil with a plastic transfer pipet onto the coverslip until it is completely covered) to remove the MeOH.
Block in AbDil for 30 min at RT.
for immunofluoresce analysis, incubate with mouse-anti-tubulin antibodies (DM1α) at 1:2000 for 1h at RT or at 4°C overnight. Optional: Co-incubate mouse-anti-tubulin antibodies with antibodies recognizing another protein of interest that was raised in a different species (e.g. rabbit-anti-Mad2 antibodies).
Wash coverslips 3 times for 5 min in AbDil.
Incubate with ~40–50μl (or so that the coverslip is completely covered) Hoechst (10μg/ml in AbDil) for 10 min at RT to stain DNA. For mounting and subsequent microscopic analysis and quantification proceed to Step 128.
D. Mitotic Checkpoint Assay. TIMING: ~ 2 days
Repeat Step 119Ai. For each sample use 2μl chromatin arrays (0.15μg/μl) coupled to 3μl of Dynabeads (see Step 116).
Optional: take 5μl of CSF extract, mix with 45μl SDS-PAGE sample buffer and leave at RT for the subsequent western blot analysis of the cell cycle stage of the extract used in that particular experiment.
Add 20μl of fresh CSF extract to each sample from Step 119Di in a 0.7ml Eppendorf tube, mix by pipetting up and down and incubate 5–10 min at 18–20°C in a waterbath (empty 1000 ul pipette tip boxes left open to the air work well; adjust temperature with a bit of ice as needed).
Add calcium to a final concentration of 0.6mM and mix by flicking the tubes.
Incubate for 80 min at 18–20°C, flicking the tubes every 15 min.
After 80 min, take 5μl of sample and mix with 45μl SDS-PAGE sample buffer for western blotting and leave at RT. CRITICAL STEP: Avoid taking beads by not flicking the tubes 15–20 min before taking the sample so that the chromatin beads will sink down.
To the remaining 15μl sample, add 15μl (1 volume) fresh CSF extract that has been kept on ice supplemented with either Nocodazole at 10μg/ml or DMSO as a control. Mix by carefully pipetting up and down to avoid bubbles.
Incubate for 90 min at 18–20°C, flicking the tubes every 15 min.
After 90 min, take 5μl of sample (t=0′) and mix with 45μl SDS-PAGE sample buffer for western blotting and leave at RT. CRITICAL STEP: Again, avoid taking beads, as described in Step 119Dvi.
Add calcium to the remaining 25μl of sample to a final concentration of 0.6mM and mix by flicking the tubes.
Incubate for 10 min at 18–20°C, take 5μl of sample (t=10′) and mix with 45μl SDS-PAGE sample buffer for western blotting and leave at RT. CRITICAL STEP: Again, avoid taking beads, as described in Step 119Dvi.
-
Repeat Step 119Dxi for t=20′, t=30′ and t=40′ samples.
PAUSE POINT: Store samples in SDS sample buffer at −20°C for a few weeks.
Heat western blot samples for 10 min at 95°C.
Load 20μl of each sample (containing ~2μl of extract) on 12.5% SDS-PAGE. The exact run time depends on the gel system used and has to be determined empirically.
Transfer separated proteins onto PVDF membrane in CAPS transfer buffer for 1 h 45 min at 400mA.
Block membrane for 30 min in 5% milk in PBS-T.
Incubate with ~30–40μl primary antibodies (rabbit-anti-phosho-Wee1, 1:1000 (ref. 16); mouse-anti-tubulin (DM1α), 1:2000) at 4°C overnight.
Wash three times for 5 min in PBS-T.
Incubate for 30 min at RT with ~30–40μl fluorescently labeled secondary antibodies at a dilution of 1:500 (goat-anti-rabbit-Alexa 647; goat-anti-mouse-Alexa 488).
Wash three times for 5 min in PBS-T.
-
Wash 1–2 times with PBS and analyze the western blot via fluorescence detection (e.g. using a Typhoon 9400 Variable Mode Imager).
TROUBLESHOOTING
Immunofluorescence and data analysis TIMING: ~ 2–3 days
-
120
To allow immunofluorescence analysis of samples prepared in Step 119A and B, put the appropriate number of poly-L-lysine coated coverslips in a humidified chamber and let them dry (do not close the lid of the chamber). The number of coverslips depends on how many different proteins are analyzed. For example, if 4 different proteins are analyzed, 15 coverslips are needed (see Figure 6a).
-
121
Pipette 20μl of chromatin beads in AbDil (equals 2μl chromatin beads) from Step 119Avii or 119Biv onto each coverslip and let the beads attach to the coverslip for 15min at RT without letting them dry out.
-
122
Block the coverslips in AbDil for 30 minutes by pipetting 30–50μl on top of the attached beads (if beads had been stored in AbDil before for more than 30 min, this step can be omitted).
-
123
Remove the blocking AbDil by aspiration and incubate each coverslip using antibodies to your proteins of interest diluted in AbDil (the concentration for each specific antibody has to be determined individually) for 1 hour at RT or overnight at 4 °C.
CRITICAL STEP: Efficient histone H4 staining at 1:100 could only be obtained with incubation at 4°C overnight.
-
124
Wash coverslips 3 times for 5 min each in AbDil by pipetting AbDil on top of the coverslip using a plastic transfer pipet to fully cover the coverslip.
-
125
Stain the coverslips using appropriate fluorophore-conjugated secondary antibodies for 30 min at RT.
-
126
Wash coverslips 3 times for 5 min in AbDil. For H4 coverslips proceed to Step 130.
-
127
When co-staining for HsCENP-A using an antibody raised in the same animal as one of the other primary antibodies, it needs to be directly conjugated to a fluorophore (we use rabbit-anti-HsCENP-A conjugated to Alexa 647). Block with whole IgG of that species for 45 minutes before incubation with a directly labeled anti-HsCENP-A for 1h at RT. (The primary antibodies used are typically raised in rabbits; in this case block with whole rabbit IgG.)
-
128
Wash coverslips 3 times for 5 min in AbDil.
-
129
Incubate with 30–40μl propidium iodide (1:1000 in AbDil) for 10 min at RT to stain DNA.
-
130
Wash coverslips 3 times for 5 min in AbDil.
-
131
Wash 1–2 times in PBS.
-
132
Add 5–7μl mounting media to an appropriately labeled slide, aspirate PBS from coverslips, and put, using forceps and a pipette tip, the appropriate coverslips upside-down onto the microscope slide. If required, carefully remove the excess mounting media using Kimwipes. Seal slides with nail polish.
PAUSE POINT: Store slides at −20°C until analysis.
-
133
Mount each slide on the microscope and image at least three fields of beads per slide, axially sectioning at 0.2μm intervals through the beads. Typically, a total of 60–600 beads should be imaged. Image each relevant wavelength at each section. The wavelength to use depends on the fluorescent label of the secondary antibodies, directly conjugated primary antibodies and on whether Propidium iodide or Hoechst stain has been used.
CRITICAL STEP: Determine exposure times for each wavelength empirically, such that the camera is brought to ~75% of saturation to maximize dynamic range; ensure that the camera does not saturate. Keep exposure times the same for different samples stained with the same marker.
-
134
If images are not in a format readable by the imread function in MATLAB (e.g. TIFF), convert them to such a format. ImageJ (http://rsbweb.nih.gov/ij/) with the bio-formats plugin (LOCI, University of Wisconsin Madison, http://loci.wisc.edu/software/bio-formats) can convert most common image formats to TIFF. Ensure that the bit-depth of images remains constant during the conversion so as not to lose information during the conversion. Do not convert to a format that uses lossy compression (e.g. JPEG-compressed image formats).
-
135
Quantify each image using MATLAB. We have provided a MATLAB script (see Supplementary Methods) that is freely available at http://straightlab.stanford.edu/software, which we use to quantify our images. It reads in TIFF images where each color channel is in a separate file, finds beads using the DNA-stained images, and quantifies the intensity of each bead in each color channel. Detailed instructions for using the script are included in the documentation in the script file, and see Box 3 for a description of the functions that are carried out by the script (or could be carried out using a similar method).
Box 3. Image analysis with MATLAB. TIMING: ~1–3h.
The following steps are carried out by the script we have provided in Supplementary Methods, which is also available at http://straightlab.stanford.edu/software, or could be carried out using a similar method:
First, construct a bead-like structuring element that when convolved with the image will find the centers of beads. In our images, the beads appeared as circles with a radius of 17 pixels that had a bright ring of in-focus fluorescence about 2 pixels wide at the edge of the bead, dim fluorescence interior to the bead, and low signal outside the bead. Therefore, we made our structuring element a circle of radius 17 pixels, with zeros on the interior, a value of 10 at the two pixels at the border of the circle, and a value of −29.5 for 2 pixels outside the circle. (The exact numerical values used were chosen empirically based on manual inspection of the quality of bead finding on a test set of images.)
Next, convolve the bead image with the structuring element. This should produce a set of points that are near centers of the identified beads. Note that this will leave out a large number of beads in the image that may be out of focus, or part of aggregates, or that otherwise do not appear like this regular pattern. (We could detect no bias associated with this subset of beads identified, as assayed by manually identifying a different but not necessarily disjoint subset of beads in the same images and quantifying using both methods.)
Construct an image mask by drawing a 17-pixel-radius circle around each point found as a bead center. Where beads overlap, assign each pixel to the bead to which it is nearest.
For each channel, compute the integrated intensity of each bead by summing the values of all pixels within its region of the mask.
Normalize by the number of pixels in each bead (which will be constant for nonoverlapping beads, but is necessary for the case where there are any partially overlapping beads).
Background-correct the images by choosing three bead-sized areas not near any beads as an average background measurement, and subtracting this off the measurements for the beads.
To correct for the autofluorescence of the Dynabeads for each individual channel and given exposure time as well as the background signal for each individual antibody staining, use non-chromatin coated Dynabeads as described in Steps 115–116 that were incubated egg extract and processed similar to the samples of each particular experiment.
For the normalization, first determine the average bead measurement for each protein of interest and for H4 by dividing the sum of all (background-subtracted) bead measurements by the number of beads analysed. (Typically, 60 – 600 beads were analysed for each antibody staining of chromatin samples and 50–100 for non-chromatin coated Dynabeads.) Similarly, for each protein of interest and for H4, determine the average bead measurement for the non-chromatin coated beads. Next, subtract off bead-specific background by subtracting the value for non-chromatin-coated control samples from the average bead measurements for each protein of interest and H4. Finally, normalize the resulting measurements for each protein of interest by dividing by the resulting H4 bead measurement for any given condition.
TIMING
Steps 1–38, H3/H4 tetramer or H2A/H2B dimer expression: ~5–6 days
Steps 39–44, H3/H4 tetramer or H2A/H2B dimer assembly: ~2 days
Steps 45–71, CENP-A/H4 expression: ~4–5 days in total
Steps 72–73, preparation of biotinylated DNA (large scale plasmid preparation): ~2 days
Steps 74–87, preparation of biotinylated DNA (digest, PEG precipitation and dialysis): ~1 day
Steps 88–93, preparation of biotinylated DNA (biotinylation and purification of biotinylated DNA): ~1 day
Steps 94–96, determination of protein concentration of histone preparations; ~1h
Steps 97–105, chromatin array assembly (setting up the chromatin assembly reactions and dialysis): ~4 days
Steps 106–114, chromatin array assembly (chromatin array harvesting, AvaI digest and analysis): ~ 1 day
Steps 115–118, preparation of chromatin beads: ~1h
Box 2, Xenopus CSF extract preparation: ~2h
Step 119Ai–vii, centromere (and kinetochore) assembly in CSF extracts (experimental setup, monitoring and fixation): ~90min
Step 119Bi–iv, kinetochore (and centromere) assembly in cycled extracts (experimental setup, monitoring and fixation): ~4h
Step 119Ci–xiv, microtubule polymerization and spindle assembly in CSF extracts (experimental setup, monitoring, fixation and immunofluorescence): ~5h
Step 119Di–xii, mitotic checkpoint assay (experimental setup, monitoring, and sampling): ~4–5h
Step 119Dxiii–xxi, mitotic checkpoint assay (running gels, and western blotting including antibody overnight incubation): ~1 day
Steps 120–132, immunofluorescence including antibody overnight incubation, mounting: ~1–2 days
Steps 133–135, imaging for ~15 slides of a typical centromere and kinetochore assembly experiment: ~3–5h
Box 3 image analysis with MATLAB: ~1–3h, depending on the quality and number of images
TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting.
| Step | Problem | Possible Reason | Possible Solution |
|---|---|---|---|
| 114 | Arrays are over- or undersaturated (Figure 5c) | Too much or not enough of the histones were used for chromatin assembly | Titrate amounts of histone proteins again to achieve 90–100 % array saturation |
| 117 | Large bead aggregates form during the coupling process (Figure 5d) | Arrays are oversaturated | Titrate amounts of histone proteins again to achieve 90–100 % array saturation |
| Box 2 (step 6) | Low quality eggs (strings of eggs, puffy eggs or activated eggs comprise more than 10% of the whole batch) |
|
|
| 119Ai | Chromatin beads stick to side of tubes | CSF-XB buffer was not supplemented with Triton-X-100 | Add 0.05% Triton X-100 to CSF-XB buffer before washing the chromatin beads |
| 119Dxxi | Extract did not exit from mitosis | Extract quality was low or not enough calcium was added |
|
| No mitotic arrest in appropriate samples detected |
|
|
|
| 135 | No kinetochore or spindle assembly checkpoint proteins were detected |
|
|
ANTICIPATED RESULTS
Centromere (and kinetochore) assembly in CSF extracts
When CENP-A and H3 chromatin beads are incubated in CSF extracts (or released from CSF arrest into interphase by the addition of calcium), CENP-A chromatin arrays will specifically assemble a subset of conserved centromere proteins including CENP-C, CENP-N and CENP-K that will not be recruited to H3 chromatin (Figure 2a and 7a) 2. This assays provides a quick and robust assay system to investigate the chromatin requirements for efficient centromere assembly as well as the mechanism of centromere assembly itself quantitatively using fluorescence microscopy.
Figure 7. Representative images of anticipated results using the cell free system to analyze centromere and kinetochore assembly and function.

(a) Representative immunofluorescence images comparing the binding of the centromere protein CENP-C to CENP-A and H3 chromatin arrays in CSF extracts. The experiment was carried out as described in Figure 2a. In the left hand column, the separate histone H4 staining is shown (separated by a black line), which can be used to assess the amount of bound chromatin arrays that is used for the normalization of the quantification. The next columns show DNA, CENP-A and CENP-C staining. A merge image of the DNA (red) and CENP-C (green) channels is shown in the right columnl. This shows that reconstituted CENP-A chromatin specifically induces efficient binding of CENP-C in egg extracts, but a comparable amount of H3 chromatin does not (as indicated by similar H4 levels). Scale bar, 5μm (b) Representative immunofluorescence images for a kinetochore assembly experiment in cycled extracts as described in Figure 2b. The recruitment of the kinetochore protein Ndc80 and the spindle assembly checkpoint protein Mad2 to CENP-A and H3 chromatin arrays in the presence and absence (+/−) of nocodazole is displayed. The left column shows the DNA. The next two columns to the right show CENP-A, followed by Ndc80 or Mad2, respectively. A merge of the DNA (red) and Ndc80 or Mad2 (green) channels is shown in the right column (DNA/Protein). This shows that the kinetochore protein Ndc80 gets specifically recruited to CENP-A chromatin arrays. Moreover, the spindle assembly checkpoint protein Mad2 is specifically increased in the presence of Nocodazole. This suggests that CENP-A chromatin promotes centromere and kinetochore assembly in egg extract and that spindle assembly checkpoint protein recruitment responds to microtubule depolymerization in a physiological manner. Scale bar, 5μm (c) Representative images of microtubule polymerization around reconstituted CENP-A, H3 and natural Xenopus sperm chromatin from a typical experiment as described in Figure 2c. After 90 min incubation time in CSF extracts, reactions were shifted to 4°C for 10 min (4°C) or treated with nocodazole (10μg/ml). Microtubules (green) polymerized around chromatin under control conditions (left column) and were stabilized by sperm and CENP-A chromatin, but not H3 chromatin, during a 10 min cold shock (middle column). The amount of mitotic checkpoint protein Mad2 (magenta) on CENP-A and sperm chromatin increased upon microtubule depolymerization, indicating the presence of functional kinetochores (right column). Scale bar, 10μm (d) Additional representative images of microtubule polymerization around CENP-A chromatin including a bipolar spindle (left column), a half spindle (middle column) and microtubule bundles (right column). Scale bar, 10μm (e) Representative western blot analysis of the mitotic checkpoint assays as described in Figure 2d. The cell cycle stage of the extract is assayed by phospho-Wee1 (P-Wee1) levels, and tubulin levels are shown as a loading control. Samples from different time points (t 0′, t 10′, t 20′, t 30′, t 40′) are shown for CENP-A and H3 chromatin on beads, either incubated with nocodazole (+) or with DMSO (−) as a control. These representative data show that CENP-A chromatin, but not H3 chromatin, delays the exit of mitosis in the presence of Nocodazole as indicated by persisting P-Wee1 levels. This indicates that reconstituted CENP-A chromatin is capable to activate a physiological mitotic checkpoint response in egg extracts when assembled kinetochores are detached from microtubules (through depolymerization by Nocodazole). Figure adapted from 2.
Kinetochore (and centromere) assembly in cycled extracts
CSF extracts are actually a meiotic system, but by adding calcium to release CSF extracts into interphase and the subsequent addition of fresh CSF extract containing mitotic cyclins, they can be cycled into metaphase of the first mitotic division (Figure 2b). In cycled egg extracts, CENP-A chromatin beads, but not H3 chromatin beads, will promote the assembly of functional kinetochores (Figure 7b). Moreover, when Nocodazole is added to the extract to depolymerize microtubules, a subset of mitotic checkpoint proteins (e.g. Mad2) is specifically recruited to CENP-A chromatin arrays while other general kinetochore components such as the microtubule binding protein Ndc80 are recruited independent of microtubule polymerization state (Figure 7b).
Microtubule polymerization and stabilization in CSF extracts
Typically, CENP-A chromatin beads will promote efficient microtubule polymerization in CSF extract and stabilize a subset of these, the so-called kinetochore fibers, when the reaction is shifted to 4°C for 10 min (Figure 7c). As previously reported, canonical chromatin is capable of promoting microtubule polymerization in a kinetochore independent manner 6,10, but the efficiency of reconstituted H3 chromatin to polymerize microtubules is low when compared to CENP-A chromatin, and stabilization of kinetochore fibers is never observed 2. The phenotypes for microtubule association observed for CENP-A chromatin arrays vary from spindle like structures to only small microtubule bundles that are associated with CENP-A chromatin arrays (Figure 7d). The phenotype variation most likely reflects again variation of kinetochore assembly efficiency depending on array saturation, coupling efficiency and Xenopus extract quality.
Spindle assembly checkpoint assay
To assess the ability of reconstituted CENP-A and H3 chromatin to activate the mitotic checkpoint in cycled Xenopus egg extracts, chromatin arrays are mixed with CSF extract, released into interphase, cycled, in the presence of Nocodazole, into metaphase of mitosis and then again released into anaphase (Figure 2d). During the course of the experiment, the cell cycle state of the extract is monitored by the cell cycle specific phosphorylation of Wee-1 using western blot analysis (Figure 7e) 2,16. Typically, phospho-Wee-1 is readily detectable in metaphase extracts (CSF and cycled) and phosphorylation levels decrease significantly within 30–40 min after release from metaphase arrest (Figure 7e). CENP-A chromatin that has assembled functional kinetochores will delay the dephosphorylation of Wee-1 in the presence of Nocodazole indicating a mitotic checkpoint dependent mitotic arrest (Figure 7e).
Supplementary Material
Acknowledgments
A.G. was supported by a postdoctoral fellowship from the German Research Foundation (DFG), C.J.F by a Stanford Lieberman Fellowship and this work was supported by NIH R01GM074728 to A.F.S.
Footnotes
Author Contribution Statement
A.G., C.J.F. and A.F.S. wrote the manuscript.
Competing financial interests
The authors declare no financial interests.
Supplementary Methods. beadMeasurements3.m. MATLAB script for quantifying fluorescence associated with beads in three channels, which implements the analysis described in Box 3. (Documentation on using the script is contained in the comments in the script file.)
References
- 1.Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nature reviews Molecular cell biology. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- 2.Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF. In vitro centromere and kinetochore assembly on defined chromatin templates. Nature. 2011;477:354–358. doi: 10.1038/nature10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huynh V, Robinson P, Rhodes D. A Method for the In Vitro Reconstitution of a Defined. Journal of molecular biology. 2005;345:957–968. doi: 10.1016/j.jmb.2004.10.075. [DOI] [PubMed] [Google Scholar]
- 4.Luger K, Rechsteiner TJ, Flaus AJ, Waye MM, Richmond TJ. Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol. 1997;272:301–311. doi: 10.1006/jmbi.1997.1235. [DOI] [PubMed] [Google Scholar]
- 5.Desai A, Deacon HW, Walczak CE, Mitchison TJ. A method that allows the assembly of kinetochore components onto chromosomes condensed in clarified Xenopus egg extracts. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12378–12383. doi: 10.1073/pnas.94.23.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heald R, et al. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature. 1996;382:420–425. doi: 10.1038/382420a0. [DOI] [PubMed] [Google Scholar]
- 7.Maresca TJ, Heald R. Methods for studying spindle assembly and chromosome condensation in Xenopus egg extracts. Methods Mol Biol. 2006;322:459–474. doi: 10.1007/978-1-59745-000-3_33. [DOI] [PubMed] [Google Scholar]
- 8.Murray AW, Kirschner MW. Cyclin synthesis drives the early embryonic cell cycle. Nature. 1989;339:275–280. doi: 10.1038/339275a0. [DOI] [PubMed] [Google Scholar]
- 9.Desai A, Murray A, Mitchison TJ, Walczak CE. The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods in cell biology. 1999;61:385–412. doi: 10.1016/s0091-679x(08)61991-3. [DOI] [PubMed] [Google Scholar]
- 10.Hannak E, Heald R. Investigating mitotic spindle assembly and function in vitro using Xenopus laevis egg extracts. Nature protocols. 2006;1:2305–2314. doi: 10.1038/nprot.2006.396. [DOI] [PubMed] [Google Scholar]
- 11.Carroll CW, Milks KJ, Straight AF. Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol. 2010;189:1143–1155. doi: 10.1083/jcb.201001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carroll CW, Silva MC, Godek KM, Jansen LE, Straight AF. Centromere assembly requires the direct recognition of CENP-A nucleosomes by CENP-N. Nature cell biology. 2009;11:896–902. doi: 10.1038/ncb1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowary P, Widom J. New DNA Sequence Rules for High Affinity Binding to Histone Octamer and Sequence-directed Nucleosome Positioning. JMB. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 14.Luger K, Rechsteiner T, Richmond T. Preparation of nucleosome core particle from recombinant histones. Methods in enzymology. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 15.Minshull J, Sun H, Tonks NK, Murray AW. A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell. 1994;79:475–486. doi: 10.1016/0092-8674(94)90256-9. [DOI] [PubMed] [Google Scholar]
- 16.Kim S, Song E, Lee K, Ferrell J., Jr Multisite M-phase phosphorylation of Xenopus Wee1A. Molecular and cellular biology. 2005;25:10580. doi: 10.1128/MCB.25.23.10580-10590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blow JJ, Laskey RA. Initiation of DNA replication in nuclei and purified DNA by a cell-free extract of Xenopus eggs. Cell. 1986;47:577–587. doi: 10.1016/0092-8674(86)90622-7. [DOI] [PubMed] [Google Scholar]
- 18.Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, Willard HF. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat Genet. 1997;15:345–355. doi: 10.1038/ng0497-345. [DOI] [PubMed] [Google Scholar]
- 19.Ikeno M, et al. Construction of YAC-based mammalian artificial chromosomes. Nat Biotechnol. 1998;16:431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- 20.Nakashima H, et al. Assembly of additional heterochromatin distinct from centromere-kinetochore chromatin is required for de novo formation of human artificial chromosome. J Cell Sci. 2005;118:5885–5898. doi: 10.1242/jcs.02702. [DOI] [PubMed] [Google Scholar]
- 21.Okada T, et al. CENP-B controls centromere formation depending on the chromatin context. Cell. 2007;131:1287–1300. doi: 10.1016/j.cell.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 22.Nakano M, et al. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev Cell. 2008;14:507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohzeki J, et al. Breaking the HAC Barrier: Histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 2012;31:2391–2402. doi: 10.1038/emboj.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnhart MC, et al. HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol. 2011;194:229–243. doi: 10.1083/jcb.201012017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gascoigne KE, et al. Induced ectopic kinetochore assembly bypasses the requirement for CENP-A nucleosomes. Cell. 2011;145:410–422. doi: 10.1016/j.cell.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P. Drosophila CENH3 is sufficient for centromere formation. Science. 2011;334:686–690. doi: 10.1126/science.1206880. [DOI] [PubMed] [Google Scholar]
- 27.Hyman AA, Middleton K, Centola M, Mitchison TJ, Carbon J. Microtubule-motor activity of a yeast centromere-binding protein complex. Nature. 1992;359:533–536. doi: 10.1038/359533a0. [DOI] [PubMed] [Google Scholar]
- 28.Kingsbury J, Koshland D. Centromere-dependent binding of yeast minichromosomes to microtubules in vitro. Cell. 1991;66:483–495. doi: 10.1016/0092-8674(81)90012-x. [DOI] [PubMed] [Google Scholar]
- 29.Sandall S, et al. A Bir1-Sli15 complex connects centromeres to microtubules and is required to sense kinetochore tension. Cell. 2006;127:1179–1191. doi: 10.1016/j.cell.2006.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akiyoshi B, Nelson CR, Ranish JA, Biggins S. Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev. 2009;23:2887–2899. doi: 10.1101/gad.1865909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akiyoshi B, et al. Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature. 2010;468:576–579. doi: 10.1038/nature09594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 34.Vary JC, Jr, Fazzio TG, Tsukiyama T. Assembly of yeast chromatin using ISWI complexes. Methods in enzymology. 2004;375:88–102. doi: 10.1016/s0076-6879(03)75006-x. [DOI] [PubMed] [Google Scholar]
- 35.Selleck W, Tan S. Recombinant protein complex expression in E. coli. Curr Protoc Protein Sci. 2008;Chapter 5(Unit 5):21. doi: 10.1002/0471140864.ps0521s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan S. A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- 37.Black BE, et al. Structural determinants for generating centromeric chromatin. Nature. 2004;430:578–582. doi: 10.1038/nature02766. [DOI] [PubMed] [Google Scholar]
- 38.Thastrom A, et al. Sequence Motifs and Free Energies of Selected Natural and Non-natural Nucleosome Positioning DNA Sequences. JMB. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.