

Abstract
Deer antlers are bony appendages that are annually cast and rapidly regrown in a seasonal process coupled to the reproductive cycle. Due to the uniqueness of this process among mammals, we reasoned that a fundamental characterization of antler progenitor cell behavior may provide insights that could lead to improved strategies for promoting bone repair. In this study, we investigated whether white-tailed deer antlerogenic progenitor cells (APC) conform to basic criteria defining mesenchymal stromal cells (MSC). In addition, we tested the effects of the artificial glucocorticoid dexamethasone (DEX) on osteogenic and chondrogenic differentiation as well as the degree of apoptosis during the latter. Comparisons were made to animal-matched marrow-derived MSC. APC and MSC generated similar numbers of colonies. APC cultures expanded less rapidly overall but experienced population recovery at later time points. In contrast to MSC, APC did not display adipogenic in vitro differentiation capacity. Under osteogenic culture conditions, APC and MSC exhibited different patterns of alkaline phosphatase activity over time. DEX increased APC alkaline phosphatase activity only initially but consistently led to decreased activity in MSC. APC and MSC in osteogenic culture underwent different time and DEX-dependent patterns of mineralization, yet APC and MSC achieved similar levels of mineral accrual in an ectopic ossicle model. During chondrogenic differentiation, APC exhibited high levels of apoptosis without a reduction in cell density. DEX decreased proteoglycan production and increased apoptosis in chondrogenic APC cultures but had the opposite effects in MSC. Our results suggest that APC and MSC proliferation and differentiation differ in their dependence on time, factors, and milieu. Antler tip APC may be more lineage-restricted osteo/chondroprogenitors with distinctly different responses to apoptotic and glucocorticoid stimuli.
Introduction
Fracture healing is a multistage regenerative process that under optimal conditions can restore bone function without generating permanent scar tissue.1,2 Successful repair depends on the type and extent of injury; the body's bone repair program often cannot restore function after large segmental losses due to disease or trauma.3
While bone repair and regeneration in most mammals follows this bounded paradigm, a rare exception is the deer antler, the only example of complete, repeated organ regeneration in an adult mammal.4 Antlers are bony appendages that in most species regrow, attain a fully mineralized, largely devitalized state, and are then cast off after the rutting season.4 This seasonal process is coupled to the reproductive cycle and associated with fluctuations in levels of circulating androgens.5 Due to their size, nutritional requirements, and role in sparring and fighting contests between rival males, antlers serve as outward indications of mate quality.6,7 Annual regeneration requires some of the fastest rates of bone growth in nature, exceeding 2 cm/day in some species.6
Antlers elongate through endochondral ossification occurring in growth centers at the distal end of each antler tine.8 Within each growth center, antlerogenic progenitor cells (APC) reside in a niche called the reserve mesenchyme, where undifferentiated APC undergo rapid proliferation as well as robust apoptosis.9,10 More proximally, APC undergo chondrogenic differentiation while osteoblasts are derived from cells in the perivascular niches that intercalate cartilage trabeculae.
Although the antler transcends barriers that limit our ability to promote regeneration in our own species, little is known of APC. As antler regrowth is thought to be due to the de novo generation of progenitor cells (rather than through de- or transdifferentiation of existing cells), it is widely believed that APC are one or more populations of multipotent cells.11,12 Whether APC can definitively be considered “stem” or “progenitor” cells awaits further characterization.
Cells derived from antler tip APC generate bone and cartilage in vivo, yet we are aware of only a few peer-reviewed studies that have examined the mesenchymal lineage differentiation potential of APC.9,13 We reasoned that a fundamental characterization of APC behavior may provide insights that could lead to improved strategies for promoting bone repair.
In this study, we set out to test whether heterogeneous populations of white-tailed deer antler tip APC conform to basic criteria defining mesenchymal stromal cells (MSC), namely self-renewal and multipotency. We compared colony formation, cell expansion rates, and in vitro and in vivo differentiation capacities of reserve mesenchyme APC to animal-matched phalangeal marrow-derived MSC. We also investigated the effects of the glucocorticoid dexamethasone (DEX) on osteogenesis in vitro. In chondrogenic cultures, we explored the effects of DEX on cell number, apoptosis, and matrix production.
Materials and Methods
Specimen acquisition
Cells were obtained from three wild, ∼2- to 4-year-old white-tailed bucks (Odocoileus virginianus) in early August, toward the end of the rapid regrowth phase in Southeast Michigan. Specimen acquisition conformed to institutional animal care and use standards and state wildlife policies.
To obtain MSC, red marrow was flushed from phalanges and adherent cells cultured in the complete medium: Dulbecco's modified Eagle's medium (Gibco, Life Technologies, Grand Island, NY), 10% fetal bovine serum (FBS; Gibco), 100 units/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. APC were harvested from the antler tip reserve mesenchyme, which was dissected, minced, and plated in the complete medium.
As cellular antler tissue is only available on a seasonal basis, comparisons were made using both fresh and frozen cells. To freeze, confluent cells were detached using 0.25% trypsin-EDTA (Gibco), resuspended at 5×106 cells/mL in FBS, and 10% dimethyl sulfoxide. After 24 h at −80°C in a Mr Frosty cell freezing container (Nalge Nunc International, Rochester, NY), cells were transferred to liquid nitrogen. Thawed passage 1 cells were cultured in the complete medium supplemented with 6 mM l-glutamine and without amphotericin B.
Cell enumeration over time
First passage cells were seeded on 96-well plates (3200/cm2). Between 1 and 8 days, relative cell number was estimated by measuring the fluorescent intensity of a nuclear binding dye (CyQUANT; Life Technologies) at 485 nm excitation/530 nm emission. Results were corrected using CyQUANT-only control wells. Cell enumeration was conducted at the same time each day.
Colony formation
First passage cells were seeded on six-well plates at low density (17/cm2). After 14 days of culture in the complete medium, cells were fixed in 1% glutaraldehyde and stained with 0.1% Crystal Violet. Colonies visible to the naked eye were counted manually.
Osteogenesis
First passage fresh or thawed cells were plated at 5000/cm2 in the complete medium. At 70–80% confluence, the medium was supplemented with 0.1 μm DEX, 0.05 mM l-ascorbic acid 2-phosphate (Sigma-Aldrich Co. LLC, St. Louis, MO), and 10 mM β-glycerophosphate (Sigma, St. Louis, MO).14 In a subset of thawed cells, DEX was omitted. Unless stated otherwise, the osteogenic medium contained DEX.
To detect mineralized nodules, cultures were stained in 1% Alizarin Red-S and solubilized in 0.5 N HCl+5% SDS. Solubilized stain was transferred to 24-well or 96-well plates. Optical densities (OD) at 415 nm were normalized to those of the buffer. To correct for cell number, parallel wells were stained with Crystal Violet. Crystal Violet was solubilized in a 1:1 solution of ethanol and 1% Triton X-100, and the OD was measured at 562 nm.
Alkaline phosphatase activity was measured using Moreau's modification of Lowry's method.15 Briefly, cells were lysed in a Triton/glycine buffer and homogenized by repeated pipetting. Lysates were transferred to 96-well plates and a 12.5 mM pNPP substrate was added. Samples were incubated for 90 min at 37°C and 1 N NaOH was then added to stop the reaction. The resultant pNP content was quantified by measuring the OD at 405 nm and corrected using buffer-only control wells. Enzyme activity was expressed as pmol of pNP divided by incubation time and lysate protein content (BCA assay; Pierce/Thermo Fisher Scientific, Rockford, IL).
Adipogenesis
First passage fresh or thawed cells were plated at 5000/cm2 in the complete medium. At 95–100% confluence (designated day 0), the medium was replaced with the adipogenesis induction medium (AIM): high-glucose DMEM (Gibco), 10% FBS, 1% Pen-Strep, 1 μM DEX (Sigma), 0.2 mM indomethacin (Sigma), 1.7 μM recombinant human insulin (Sigma), and 0.5 mM 1-methyl-3-isobutylxanthine (Sigma). On day 3, the AIM was replaced with the adipogenesis maintenance medium (AMM): high-glucose DMEM (4.5 g/L), 10% FBS, 1% Pen-Strep, and 1.7 μM recombinant human insulin.16 For the first 12 days of culture, cells were exposed to a total of three 3-day AIM treatments separated by a 1-day AMM treatment. From day 13 to 21, AMM was used exclusively and replaced every 3–4 days. To detect lipid droplets, cells were fixed overnight in 10% neutral buffered formalin (NBF), incubated in 60% isopropanol, and stained in 0.5% Oil Red-O.
Chondrocyte induction
Chondrogenic differentiation experiments were performed in micromass cultures.17 250,000 cells were pelleted in 15-mL conical tubes and cultured in the chondrogenic medium: high-glucose DMEM, 1% Pen-Strep, 10 ng/mL, recombinant human TGFβ3 (Sigma), 0.1 μM DEX, 50 μg/mL l-ascorbic acid 2-phosphate, 40 μg/mL l-proline (Sigma), and 1% ITS+ (BD Biosciences, Sparks, MD).
After 14 days, hypertrophic induction medium: high-glucose DMEM, 1% Pen-Strep, 50 ng/mL thyroxine (Sigma), 1 nM DEX, 20 mM β-glycerophosphate, 50 μg/mL l-ascorbic acid 2-phosphate, 40 μg/mL l-proline, and 1% ITS+. After 28 days, micromasses were fixed in 10% NBF, paraffin embedded, and 6-μm-thick sections were cut and mounted onto glass slides. In a subset of thawed cells, DEX was omitted from both chondrogenic and hypertrophic media.
Micromass collagen content was evaluated using Van Gieson's Stain. Proteoglycan content was visualized with Safranin-O and verified using Alcian Blue (pH 2.5) on separate sections. Alizarin Red staining was also performed for micromasses in the DEX effects portion of the study.
To measure apoptosis and cell number within micromasses, slides were dual-labeled using a TUNEL-Hoechst kit (Click-iT TUNEL assay; Molecular Probes/Life Technologies, Eugene, OR). Microscopy and image capture were conducted using a Zeiss Axiovert 200M (Carl Zeiss Microscopy, LLC, Thornwood, NY). For each section, three images were taken in the same field of view. First, an auto-exposed brightfield image was taken to estimate the tissue area. Next, Hoeschst-stained and TUNEL image were captured sequentially using standardized exposure times. Image analysis was carried out using ImageJ software (National Institutes of Health, Bethesda, MD). Mean TUNEL image grayscale intensities were normalized to cells per brightfield image area.
Ectopic ossicle growth model
Passage 1 APC and MSC from three deer were thawed and expanded in culture. To validate the model, MSC were also harvested from the long bone marrow of 5-week-old C57BL/6 mice as described previously.18 Deer cells were cultured in the complete medium and C57/BL6 MSC in α-MEM (Gibco) containing 100 U/mL penicillin, 100 mg/mL streptomycin, 20% FBS, and 10 nM DEX.19
Cells were seeded at 18000/mm3 on Type I collagen sponges (Gelfoam; Pharmacia&Upjohn Co., New York, NY). “Blank” sponges were soaked in the complete medium. Sponges were implanted subcutaneously in 4-week-old immunodeficient mice (Athymic Nude; Charles River Laboratories International, Inc., Wilmington, MA). After 6 weeks, implants were removed, fixed for 48 h in 10% NBF, and kept in 70% ethanol preparatory to histology.
Using microcomputed tomography (μCT), implants were scanned in air and reconstructed on 18 μm voxels (GE Healthcare Preclinical Imaging, London, ON, Canada). Analysis was performed using GE Microview software on three-dimensional regions of interest (ROI) interpolated from two-dimensional splines. After generating image histograms to estimate mineralized tissue grayscales, a global threshold was applied to each ROI, and mineral content and density were calculated.
After μCT, implants were demineralized in 10% EDTA, paraffin embedded, and 7-μm sections were mounted onto glass slides. Proteoglycan content was visualized using Alcian Blue-Periodic Acid-Schiff stain (Poly Scientific R&D Corp., Bay Shore, NY). Putative osteoclasts were identified using a TRAP detection kit (Sigma).
Statistical analysis
As each experiment involved a small sample size (2–3 biological replicates), multiple experimental replicates (3–12) were performed for each assay. Generalized linear mixed models, which take into account nested data structures and repeated measures, were used for comparisons (SPSS; IBM Corp., Armonk, NY). For fresh and thawed cell models, “celltype” was a fixed effect and “animal” was a random effect. For the DEX study, “DEX treatment,” “celltype,” and a “DEX×celltype” interaction were fixed effects with “animal” again as a random effect. As ossicle μCT involved APC, MSC, and blanks, “celltype” was used as a fixed effect; no random effect was considered. Statistical comparisons were limited to implants with detectable mineralized tissue. Values more than 2.5 standard deviations from the mean were omitted. Figures show least squared means and standard deviations.
Results
Cell enumeration over time
Rapid antler growth rates in vivo prompted us to compare APC and MSC expansion in culture. While MSC numbers increased before reaching a plateau at 6 days, APC numbers declined between 1 and 4 days and then increased between 5 and 8 days (Fig. 1A). Contrary to expectations, APC did not exhibit greater in vitro proliferative capacity compared to MSC.
FIG. 1.

Comparison of antlerogenic progenitor cells (APC) and mesenchymal stromal cell (MSC) cell number and colony formation. (A) Relative cell number over time as measured by optical density (OD). *p≤0.05 APC versus MSC at each time point. Eight to 16 wells per group per time point, n=3 bucks. (B) Colony formation at day 14 (n=3, p=0.89).
Colony-forming units
The ability of single cells to form colonies is a measure of self-renewal, a key progenitor cell trait.20 The mean numbers of visible colonies generated by APC and MSC were similar, but APC demonstrated more interanimal variability (Fig. 1B).
Adipogenic differentiation
The capacity to differentiate into adipocytes, osteoblasts, and chondrocytes is another characteristic of MSCs.21 After 21 days of exposure to adipogenic media, both fresh and thawed MSC displayed widespread intracellular Oil Red-O-positive lipid droplets. No lipid droplets were observed for either fresh or thawed APC at the same time point (Fig. 2).
FIG. 2.
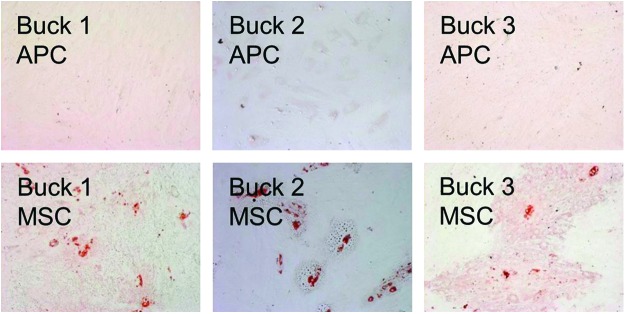
Adipogenic differentiation of thawed cells. MSC exhibited Oil Red-O-positive lipid droplets, whereas none were apparent for APC under these conditions. Color images available online at www.liebertpub.com/tea
Osteogenic differentiation
We performed osteogenic differentiation of APC and MSC in culture using both fresh and thawed cells. In a separate experiment, we measured the effects of the glucocorticoid DEX on osteogenic differentiation in thawed cells.
APC and MSC displayed different patterns of alkaline phosphatase activity over time. In both fresh and thawed cells, APC activity was greater than MSC at earlier time points while the reverse was true by day 14 in thawed cells (Fig. 3). In fresh cells, the APC activity was greater than MSC at day 16 (Fig. 3A). However, MSC activity increased over time in the same stepwise manner seen in thawed cells.
FIG. 3.

Alkaline phosphatase activity in fresh and thawed cells. (A) Fresh cell alkaline phosphatase activity (n=2 bucks). (B) Thawed cell activity (n=3). (C) Alkaline phosphatase activity in thawed cells cultured with or without dexamethasone (DEX) (n=2). * or line: p≤0.05, #: p≤0.1.
DEX affected APC and MSC alkaline phosphatase activity differently (Fig. 3C). DEX increased the APC activity at day 7 but had no significant effect at days 10 or 14. In MSC, however, DEX consistently led to decreased alkaline phosphatase activity.
APC and MSC in the osteogenic culture underwent different patterns of mineralization as well. In thawed cells cultured with DEX, by days 21/22, MSC had greater cell number-corrected mineralization, while no significant difference was seen by days 28/30 (Fig. 4E, F). On the other hand, MSC maintained greater mineralization at day 30 in fresh cells (Fig. 4D). APC consistently had fewer cells in fresh and thawed cultures compared to MSC (Fig. 4A–C).
FIG. 4.

Relative cell number and mineralization during osteogenic differentiation. (A) Fresh cell number (Crystal Violet OD, n=2 bucks). (B) Thawed cell numbers (n=3). (C) Thawed cell numbers with and without DEX (n=2). (D) Fresh cell mineral (Alizarin Red-S OD normalized by Crystal Violet, 24-well plate). (E) Thawed cell mineral (96-well plate). (F) Thawed cell mineral, with and without DEX (96-well plate). * or line: p≤0.05. Line+#: p≤0.1.
DEX influenced APC and MSC mineralization in different ways (Fig. 4F). At day 21, DEX reduced mineralization in both APC and MSC. By day 28, DEX led to increased mineralization in APC (APC+DEX: 0.232±0.08, APC−DEX: 0.142±0.06, p=0.02) but did not significantly alter Alizarin Red content in MSC at this time point (p=0.23) (Fig. 4F).
DEX significantly lowered cell number in MSC at day 28, but the absolute difference was small (Fig. 4C). DEX also lowered APC cell number at day 28, but the difference was not statistically significant (p=0.09).
Chondrogenic differentiation
We used a micromass model to induce chondrogenic differentiation in thawed APC and MSC. We also explored whether the robust apoptosis seen in the reserve mesenchyme could be recapitulated in these high-density cultures. In a separate experiment, chondrogenic differentiation and apoptosis were investigated in thawed APC and MSC with and without DEX.
Evidence of a chondrogenic phenotype was found in both APC and MSC micromasses (Fig. 5). However, matrix composition and the effect of DEX depended on cell type.
FIG. 5.

Chondrogenic micromass histology (buck 1 cells, 200× magnification). Color images available online at www.liebertpub.com/tea
Safranin-O staining showed a widespread distribution in MSC micromasses, but staining was faint in APC micromasses (data not shown). Alcian Blue staining verified the presence of proteoglycans in the latter (Fig. 5, middle panel). DEX qualitatively increased the intensity and distribution of Alcian Blue and Safranin-O (data not shown) in MSC micromasses while having the opposite effect in APC micromasses. Van Gieson's staining confirmed collagen production throughout APC and MSC micromasses and was not affected by DEX.
Alizarin Red-S staining was performed to investigate matrix mineralization during chondrocytic differentiation (Fig. 5, right panels). Again, DEX affected APC and MSC in seemingly opposite ways. MSC Alizarin Red staining was qualitatively more robust with DEX, whereas the reverse was apparent in APC.
Cellularity of APC and MSC micromasses also differed. In thawed cell micromasses, APC displayed significantly more cells per matrix area than MSC (Fig. 6A, B). DEX did not affect the number of APC cells per area (p=0.58) but increased cellularity in MSC (MSC+DEX 11.4±0.39 vs. 7.1±0.35 in untreated MSC, p<0.001).
FIG. 6.

Chondrogenic micromass cellularity and apoptosis. (A, B) Cells per area for thawed cells (n=3 bucks) and thawed cells with or without DEX (n=2). (C, D) TUNEL grayscale intensity normalized by cells per area for thawed cell and thawed cells with or without DEX. Different scales due to lower permeabilization of tissue in latter study. (E, F) Another measure of apoptosis: percent of pixels with nonzero grayscale, thawed cell and thawed cells with or without DEX. (G, H) Images of TUNEL stain, buck 1 APC and MSC, 200× magnification. Six sections per mass. * or line: p≤0.05.
Compared to MSC, APC micromasses were more apoptotic. In TUNEL-stained images, APC micromasses had greater mean grayscale intensities and proportion of nonzero grayscale pixels compared to MSC (Fig. 6C–F). In APC micromasses, DEX increased both measures of apoptosis while reducing or having no effect on MSC.
Ectopic ossicle formation
Ectopic ossicle formation assesses cell osteogenic differentiation in a murine model assumed to be more representative of the native cell milieu than cell culture plastic.22 As described in the previous literature, C57BL/6 cell-seeded implants generated a mineralized shell surrounding a marrow-like region containing erythrocytes, likely host-derived (histological data not shown). Although APC- and MSC-seeded scaffolds did not generate an apparent marrow region, histological analysis revealed proteoglycan-positive regions containing buried osteocyte-like cells (Fig. 7A–C), as well as the presence of multinucleated TRAP-positive cells (Fig. 7D–F). No TRAP-positive cells were apparent in blank ossicles.
FIG. 7.

Histology and microcomputed tomography (μCT) of ectopic ossicles (n=3 bucks). (A–C) Alcian Blue-Periodic Acid-Schiff (400×): APC, MSC, blank. Arrows show embedded cells. (D–F) TRAP staining (400×): APC, MSC, blank. Arrows show TRAP-positive cells. (G, H) μCT data: tissue mineral content and tissue mineral density. Line: p≤0.05. Color images available online at www.liebertpub.com/tea
Although lower than in ossicles formed by BL/6 MSC (data not shown), mineral content was similar in APC and cervid MSC ossicles (p=0.63) (Fig. 7G, H). MSC had a slightly greater mineral density compared to either APC or blanks (Fig. 7G, H).
Discussion
We initiated this study as an exploration of white-tailed deer APC in terms of basic criteria defining MSCs. Our results indicate that in addition to self-renewal and multiple lineage differentiation, reserve mesenchyme-derived APCs exhibit a phenotype distinct from animal-matched marrow-derived progenitor cells.
Both APC and MSC demonstrated clonogenicity or the ability to generate colonies in a density-independent manner in culture.20 The lack of difference in mean APC and MSC colony numbers suggests that overall self-renewal capacity is conserved across different connective tissue progenitors in a regionally restricted white-tail population. On the other hand, the greater APC standard deviation indicates a greater variation in self-renewal of antlerogenic cells from deer to deer compared to marrow-derived MSC or could reflect differences in antler growth stages in individual animals.
White-tailed deer antlers regrow rapidly, about 0.25 inches (6.35 mm) per day.23 In vitro, however, APCs from these deer expanded less rapidly than MSC overall. Interestingly, after 5 days of decline in a monolayer culture, APC numbers began to recover.
Cells were cultured from late summer antlers when rapid growth had begun to abate. However, proliferation of red deer (Cervus elaphus) APC in vitro is independent on the stage of antler regrowth during which the cells were harvested.24 It is conceivable that the situation is similar in the white-tailed deer as well.
Slower APC expansion in the monolayer culture may instead have been due to one or more disadvantageous components of the culture system—initial plating density, oxygen tension, the lack of APC-specific mitogenic factors in the medium, and so on. For example, APC in a micromass culture consistently exhibited a greater cell number per matrix area compared to MSC. These data raise the possibility that APC mitogenesis may be more sensitive to milieu compared to MSC (i.e., monolayer versus three-dimensional micromass).
In terms of cell potency, our results suggest that, compared to MSC, antler tip reserve mesenchyme APC are more lineage-restricted osteo/chondrogenic progenitors, with little adipogenic capacity. This result may reflect that adipose tissue is not a primary antler constituent while it is present in the phalangeal marrow space. In the case of stem cells, differentiation is generally limited to the lineages present in the tissue of origin.25 However, as heterogeneous cell cultures were used here, further investigation will be needed to determine whether differences in APC and MSC adipogenesis were exaggerated by the presence of committed adipocytic precursors in the latter culture. These findings contrast with those of Rolf et al. and Berg et al., who reported positive Oil Red-O staining in fallow deer (Dama dama) and red deer (C. elaphus) APC, respectively, after exposure to the adipogenic medium.9,13 Besides the use of different deer species, possible sources of discrepancy between our results and those of other workers include the differentiation of a STRO-1+ cell subpopulation by Rolf et al. (rather than a heterogeneous population) and the harvest of cells from the cranial frontal crest (instead of the antler tip reserve mesenchyme) by Berg et al. The contrast in adipogenic capacity between our results and the latter study suggests that APC multipotency may depend on the anatomical location of the niche in which these cells reside.
Under osteogenic conditions, APC alkaline phosphatase activity was higher than that of MSC for the first 10 days in culture. This suggests higher responsiveness of APC to osteogenic factors, perhaps due to a greater degree of intrinsic osteo/chondrogenic commitment compared to MSC. Despite higher initial alkaline phosphatase activity, APC mineralization was delayed until later time points or reduced relative to MSC, perhaps reminiscent of the delay in complete mineralization that occurs in the antler after full regrowth is achieved.
DEX consistently inhibited mineralization in MSC cultures, but its effects on APC mineralization were time dependent. This suggests that DEX affects APC-derived osteoblasts in a manner dependent on differentiation stage, consistent with the studies of glucocorticoid action on osteoblasts.26 In contrast, DEX consistently reduced markers of early and late osteoblast differentiation in cervid MSC cultures.
In the murine ectopic ossicle formation model, APC-seeded scaffolds generated a similar amount of mineralized matrix compared to MSC after 6 weeks of implantation. This contrasts with our in vitro results in which thawed APC were only able to achieve statistically similar mineralization compared to MSC on a per-cell basis at later time points and in the presence of DEX. Fresh APC were not able to match MSC mineralization under the same conditions.
As with osteogenesis, APC and MSC undergoing chondrogenesis responded differently to DEX. While DEX led to increased proteoglycan content and mineralization in MSC-derived micromasses, the opposite was true for those from APC. These results demonstrate that, in vitro, DEX can play a role in supporting chondrocytic differentiation in MSC while reducing it in APC.
In addition to influencing matrix production in APC and MSC micromass, DEX treatment led to differences in cellularity and apoptosis. For APC, DEX fostered more robust apoptosis but had no significant impact on cellularity. In MSC micromass, DEX led to greater matrix production, increased cellularity, and lower apoptosis over untreated micromasses. This suggests that, in MSC undergoing chondrogenesis, DEX enhances both proliferation and differentiation, although these effects are likely dependent on the stage of cell lineage progression.
Antler regrowth is associated with hormone levels changing throughout the mating cycle. However, it has been difficult to determine which factors directly regulate APC proliferation and differentiation. Factors such as testosterone, whose fluctuations are closely associated with the antler cycle,27 failed to promote mitogenesis in antler cell cultures.28 On the other hand, it is unclear whether IGF-1, which stimulates antler cell proliferation in vitro,28,29 is the key stimulatory factor in antler growth.24
DEX can promote or suppress bone cell proliferation and differentiation directly and through interactions with hormones such as IGF-1.26,30,31,32 DEX can also reduce antler cell proliferation in vitro.29 In APC cultures, we found that DEX reduced markers of chondrogenic differentiation while promoting apoptosis. High levels of apoptosis have been reported in the reserve mesenchyme, adjacent to regions of active chondrogenesis.10 This suggests that factors such as glucocorticoids might contribute to the shifting balance between progenitor proliferation and differentiation in the antler as it undergoes regeneration. Additionally, an inverse relationship between apoptosis and chondrogenesis raises the possibility that apoptosis may help maintain a population of nondifferentiating APC within the reserve mesenchyme. Compensatory proliferation, in which caspases released from apoptotic bodies act as mitogenic factors, is one means by which apoptosis can contribute to tissue homeostasis.33
The observed pattern of time, factor, and milieu dependence of APC proliferation and differentiation may reflect a system of regulation required to confine antler growth to a specific anatomical and temporal range. Antler regrowth does not proceed automatically in response to antler casting, it is initiated by seasonally determined signaling.34 Complete mineralization of the antler is delayed until circulating androgens peak during the fall rutting season, after it is fully regrown.5 Moreover, antler regrowth occurs concurrently with bone resorption elsewhere in body, indicating a differential responsiveness to circulating factors in the antler compared to other bone tissue.7
We have demonstrated that APC and cervid marrow-derived MSC differ in terms of proliferative capacity, differentiation, apoptotic potential, and hormone responsiveness. As knowledge of the APC phenotype improves, a compelling avenue of future research would be the elucidation of the mechanisms behind the properties and behavior of these cells. A thorough understanding of the antler's unique process of regeneration would be of great value in guiding the development of novel skeletal regenerative therapies.
Acknowledgments
The authors thank Karl Malcolm, previously of the University of Wisconsin, for his crucial work in obtaining bucks for cell culture. We also thank Earl Werner and Christopher Davis, of the University of Michigan Department of Ecology and Evolutionary Biology, for providing access to the Edwin S. George Reserve. Charles Roehm, of the Orthopaedics Research Laboratory at the University of Michigan, gave much needed help with habituating and tracking bucks. Laurie McCauley and Amy Koh-Paige, of the University of Michigan Department of Periodontics and Oral Medicine, were instrumental in helping us learn how to generate ectopic ossicles. This study was supported by the National Institutes of Health through grant R01-AR051504.
Disclosure Statement
No competing financial interests exist.
References
- 1.Alman B.A., Kelley S.P., and Nam D.Heal thyself: using endogenous regeneration to repair bone. Tissue Eng 17, 431, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Colnot C.Cell sources for bone tissue engineering: insights from basic science. Tissue Eng 17, 449, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimitriou R., Jones E., McGonagle D., and Giannoudis P.V.Bone regeneration: current concepts and future directions. BMC Med 9, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kierdorf U., Li C., and Price J.S.Improbable appendages: deer antler renewal as a unique case of mammalian regeneration. Semin Cell Dev Bio 20, 535, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Price J.S., and Allen S.Exploring the mechanisms regulating regeneration of deer antlers. Philos Trans R Soc Lond B 359, 809, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price J.S., Allen S., Faucheux C., Althnaian T., and Mount J.G.Deer antlers: a zoological curiosity or the key to understanding organ regeneration in mammals? J Anat 207, 603, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landete-Castillejos T., Estevez J.A., Martinez A., Ceacero F., Garcia A., and Gallego L.Does chemical composition of antler bone reflect the physiological effort made to grow it? Bone 40, 1095, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Price J.S., Oyajobi B.O., Oreffo R.O.C., and Russel R.G.G.Cells cultured from the growing tip of red deer antler express alkaline phosphatase and proliferate in response to insulin-like growth factor-I. J Endocrinol 143, R9, 1994 [DOI] [PubMed] [Google Scholar]
- 9.Rolf H.J., Kierdorf U., Kierdorf H., Schulz J., Seymour N., Schliephake H., Napp J., Niebert S., Wolfel H., and Wiese K.G.Localization and characterization of STRO-1+ cells in the deer pedicle and regenerating antler. PLoS One 3, e2064, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colitti M., Allen S.P., and Price J.S.Programmed cell death in the regenerating deer antler. J Anat 207, 339, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li C.Deer antler regeneration: a stem cell-based epimorphic process. Birth Defects Res C Embryo Today 96, 51, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Li C., Yang F., and Sheppard A.Adult stem cells and mammalian epimorphic regeneration—insights from studying annual renewal of deer antlers. Curr Stem Cell Res Ther 4, 237, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Berg D.K., Li C., Asher G., Wells D.N., and Oback B.Red deer cloned from antler stem cells and their differentiated progeny. Biol Reprod 77, 384, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Jaiswal N., Haynesworth S.E., Caplan A.I., and Bruder S.P.Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J Cell Biochem 64, 295, 1997 [PubMed] [Google Scholar]
- 15.Moreau R., Aubin R., Lapointe J.Y., and Lajeunsse D.Pharmacological and biochemical evidence for the regulation of osteocalcin secretion by potassium channels in human osteoblast-like MG-63 cells. J Bone Miner Res 12, 1984, 1997 [DOI] [PubMed] [Google Scholar]
- 16.Janderova L., McNeil M., Murrell A.N., Mynatt R.L., and Smith S.R.Human mesechymal stem cells as an in vitro model for human adipogenesis. Obes Res 11, 65, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Mackay A.M., Beck S.C., Murphy J.M., Barry F.P., Chichester C.O., and Pittenger M.F.Chondrogenic differentiation of cultured human mesenchymal stems cells from marrow. Tissue Eng 4, 415, 1998 [DOI] [PubMed] [Google Scholar]
- 18.Pettway G.J., Schneider A., Koh A.J., Widjaja E., Morris M.D., Meganck J.A., Goldstein S.A., and McCauley L.K.Anabolic actions of PTH (1–34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone 36, 959, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Pettway G.J., Meganck J.A., Koh A.J., Keller E.T., Goldstein S.A., and McCauley L.K.Parathyroid hormone mediates bone growth though the regulation of osteoblast proliferation and differentiation. Bone 42, 806, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianco P., Robey P.G., and Simmons P.J.Mesenchymal stem cells: revisting history, concepts, and assays. Cell Stem Cell 2, 313, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dominici M., Le Blanc K., Mueller I., Slaper-Cortenbach I., Marini F., Krause D., Deans R., Keating A., Prockop D.J., and Horwitz E.Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Krebsbach P.H., Kuznetsov S.A., Satomura K., Emmons R.V.B., Rowe D.W., and Robey P.G.Bone formation in vivo: comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblast. Transplantation 63, 1059, 1997 [DOI] [PubMed] [Google Scholar]
- 23.Ozoga John J.Whitetail Spring: Season of the Whitetail, Willow Creek Press, 1996, p. 82 [Google Scholar]
- 24.Kuzmova E., Bartos L., Kotrba R., and Bubenik G.A.Effects of different factors on proliferation of antler cells, culture in vitro. PLoS One 6, e18053, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orford K.W., and Scadden D.T.Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet 9, 115, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Delany A.M., Dong Y., and Canalis E.Mechanisms of glucocorticoid action in bone cells. J Cell Biochem 56, 295, 1994 [DOI] [PubMed] [Google Scholar]
- 27.Suttie J.M., Fennessy P.F., Lapwood K.R., and Corson I.D.Role of steroids in antler growth of red deer stags. J Exp Zool 271, 120, 1995 [DOI] [PubMed] [Google Scholar]
- 28.Li C., Littlejohn R.P., and Suttie J.M.Effects of insulin-like growth factor 1 and testosterone on the proliferation of antlerogenic cells in vitro. J Exp Zool 284, 82, 1999 [PubMed] [Google Scholar]
- 29.Li C., Wang W., Manley T., and Suttie J.M.No direct mitogenic effects of sex hormones on antlerogenic cells detected in vitro. Gen Comp Endocrinol 142, 75, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Delany A.M., Durant D., and Canalis E.Glucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol 15, 1781, 2001 [DOI] [PubMed] [Google Scholar]
- 31.Tuan R.S., Boland G., and Tuli R.Adult mesenchymal stem cells and cell-based tissue engineering. Arthritis Res Ther 5, 32, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoch A.I., Binder B.Y., Genetos D., and Leach J.K.Differentiation-dependent secretion of proangiogenic factors by mesenchymal stem cells. PLoS One 7, e35579, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fan Y., and Bergmann A.Apoptosis-induced compensatory proliferation. The cell is dead. Long live the Cell! Trends Cell Biol 18, 467, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kierdorf U., Kierdorf H., and Szuwart T.Deer antler regeneration: cells, concepts, and controversies. J Morphol 268, 726, 2007 [DOI] [PubMed] [Google Scholar]