

Abstract
Objective:
To examine safety, tolerability, and efficacy of PF-04494700, an inhibitor of the receptor for advanced glycation end products (RAGE), in mild to moderate Alzheimer disease (AD).
Methods:
Double-blind, placebo-controlled trial at 40 academic centers (United States). Subjects with AD and Mini-Mental State Examination score 14–26 were randomized to PF-04494700 60 mg/day × 6 days, then 20 mg daily (high dose); 15 mg/day × 6 days, then 5 mg daily (low dose); or placebo, for 18 months. Clinical and laboratory measures were used to evaluate safety and tolerability. The primary efficacy measure was the Alzheimer's Disease Assessment Scale–cognitive (ADAS-cog). Secondary measures assessed clinical stage, function, behavior, MRI, and CSF biomarkers.
Results:
A total of 399 subjects were randomized. In a prespecified interim analysis, when 50% of subjects had completed the 6-month visit, the high dose was associated with confusion, falls, and greater ADAS-cog decline and was discontinued. A second prespecified analysis compared low-dose and placebo groups for futility and safety approximately 12 months after all subjects were randomized. This analysis met criteria for futility, and treatment was discontinued. There were no safety concerns in the low-dose group. Analyses including post-futility data showed decreased decline on the ADAS-cog in the low-dose group at month 18. Other clinical and biomarker measures showed no differences between low-dose treatment and placebo.
Conclusions:
PF-04494700 at 20 mg/d was associated with increased adverse events and cognitive decline. At 5 mg/d, PF-04494700 had a good safety profile. A potential benefit for this low dose on the ADAS-cog is not conclusive, because of high dropout and discontinuation rates subsequent to the interim analyses.
Classification of evidence:
This study provides Class I evidence that in patients with AD high-dose PF-04494700 increased cognitive decline at 6 months and Class IV evidence that low-dose PF-04494700 slowed cognitive decline at 18 months.
The receptor for advanced glycation end products (RAGE) is widely expressed in the brain and has been implicated in Alzheimer disease (AD). RAGE may be involved in AD pathogenesis through interactions with amyloid β protein (Aβ) and advanced glycation end products.1,2 RAGE is upregulated in the brain in AD, colocalizes with plaques, induces oxidative stress, and contributes to inflammation and neurodegeneration.3 RAGE has been implicated in the transport of Aβ across the blood–brain barrier.4 Inhibition of RAGE activation may decrease pathogenic events in AD. TransTech Pharma, Inc. discovered that PF-04494700, which inhibits in vitro interactions of RAGE with Aβ42 at nanomolar concentration, is orally bioavailable and crosses the blood–brain barrier. Chronic oral dosing of PF-04494700 in AD transgenic mice led to a reduction of amyloid load in the brain, improved performance on tests of spatial memory, and normalization of electrophysiologic recordings from hippocampal slices (data on file, Trans Tech Pharma, unpublished).
After phase 1 clinical development in healthy normal subjects, a placebo-controlled clinical trial examined 2 doses of PF-04494700 in subjects with mild to moderate AD over 3 months.6 This study did not reveal any major safety problems. Pfizer, Inc. licensed the drug from TransTech Pharma and sponsored a phase 2 clinical trial together with the Alzheimer's Disease Cooperative Study (ADCS), an academic consortium funded by the National Institute on Aging to conduct therapeutic research in AD.
METHODS
Study design.
A parallel 3-arm phase 2 study was conducted between January 2007 and December 2010 at 40 study sites across the United States. The primary research question was to evaluate the safety, tolerability, and efficacy of 2 doses of PF-04494700 compared to placebo in subjects with mild to moderate AD. The enrollment target was 399 subjects (133 per group) randomized to placebo or to PF-04494700 at 20 mg daily (after a loading dose of 60 mg daily for 6 days) or 5 mg daily (after a loading dose of 15 mg daily for 6 days) for 18 months. The loading dose was required because of the long half-life of PF-04494700.
Standard protocol approvals, registrations, and patient consents.
Subjects provided informed consent; if they had impaired decisional capacity, caregivers provided consent and subjects assented to participate. The study was conducted under local institutional review board supervision and under an investigational new drug application from the US Food and Drug Administration. It is listed on ClinicalTrials.gov (NCT00566397).
Study visits.
Visits occurred at screening, baseline (within 4 weeks after screening), then at 4 weeks, 3, 6, 9, 12, 15, and 18 months, and a safety follow-up visit at 21 months. Visits included clinical and safety evaluations, blood draw for plasma biomarker and pharmacokinetic analysis, and pill counts to assess compliance. Primary clinical outcome measures were obtained at baseline and at subsequent 3-monthly visits, and secondary clinical outcome measures at baseline and at 6-monthly intervals. Brain MRIs were obtained at baseline and at 12 and 18 months. Lumbar punctures for CSF biomarkers were performed at baseline and 12 months on a subgroup of subjects. Amyloid imaging was implemented late during the study and was obtained on too few subjects to yield meaningful results.
Subjects.
Key eligibility criteria included age ≥50 years, a diagnosis of probable AD,7 Mini-Mental State Examination (MMSE)8 score between 14 and 26, and good general health. There could be no evidence of stroke contributing to dementia (modified Hachinski [Rosen] score ≤4 and no stroke in cognitively significant areas on brain imaging). Further inclusion criteria included treatment with a stable dose of an acetylcholinesterase inhibitor or memantine for ≥4 months prior to randomization and an available caregiver to act as informant and supervise study medications. Exclusion criteria included uncontrolled hypertension, unstable cardiac or pulmonary disease, diabetes (or hemoglobin A1c at screening >6%), weight less than 40 kg or greater than 100 kg within the past 2 years, chronic use of nonsteroidal anti-inflammatory drugs or immunosuppressive agents, drugs that increase QTc or inhibit CYP 3A4, and markedly abnormal ECG or QTc (QTcB or QTcF) on any screening 12-lead ECG >450 ms (women) or >430 ms (men). There could be no history of treatment for cancer (past 5 years), drug or alcohol abuse, or major psychiatric illness. Women could not be of childbearing potential. Subjects could not have taken another investigational drug for 3 months before screening.
Randomization and masking.
Subjects were assigned with equal probability to one of 2 doses of PF-04494700 or placebo, using a permuted block randomization scheme stratified by site, generated by the ADCS Data Core. After subjects signed informed consent and eligibility was confirmed, study sites obtained randomization numbers from the ADCS Data Core. Adequacy of masking was assessed by questionnaires completed by subjects, caregivers, study coordinators, and site investigators.
Study medication.
For the first 6 days, subjects received a loading dose of 60 mg (three 20-mg capsules), 15 mg (three 5-mg tablets), or 3 placebo capsules. The maintenance dose was decreased to 20 mg (high dose), 5 mg (low dose), or placebo, taken once daily, for the remainder of the study.
Outcome measures.
The primary efficacy measure was the 70-point Alzheimer's Disease Assessment Scale–cognitive (ADAS-cog),9 a psychometric test that assesses memory, attention, reasoning, language, orientation, and praxis. Primary safety measures included reports of adverse events, blood and urine tests, and ECG measures.
Secondary clinical measures (table e-1 on the Neurology® Web site at Neurology.org) included Clinical Dementia Rating–sum of boxes (CDR-sb),10 ADCS Activities of Daily Living Scale (ADCS-ADL),11 Neuropsychiatric Inventory (NPI),12 and MMSE. Subjects also received a neuropsychological test battery, including Digit Symbol Substitution Test,13 Forward and Backward Digit Span Test,14 Controlled Oral Word Association Test,15 Stroop Color Word Interference Test,16 and Trail Making Test (parts A and B).17 Caregivers received a Quality of Life questionnaire18 and a Resource Utilization Schedule.19
Laboratory and biomarker measurements.
Brain MRI was performed at baseline and 12 and 18 months on 1.5T scanners, with standardized acquisition parameters based on those in the Alzheimer's Disease Neuroimaging Initiative study (www.adni-info.org), and images were used for volumetric analysis20–22 (supplemental text). CSF obtained by lumbar puncture was used to analyze AD-related biomarkers (supplemental text). APOE genotyping was performed and DNA was banked for pharmacogenomic studies on subjects who consented. Plasma was assayed for study drug levels at each visit and was stored for biomarker studies.
Safety assessments.
Subjects received complete physical and neurologic examinations at baseline and vital signs and brief examinations at subsequent visits. Clinical laboratory studies, urinalysis, and ECGs (centrally read, including QTc analysis by a cardiologist) were obtained at all visits.
Adverse events were classified according to severity and causality by site investigators and reported to the ADCS and sponsors using standard methods.
If subjects decided to withdraw from the study or were discontinued by site investigators, an early termination visit was scheduled within 14 days, including clinical and safety evaluations similar to the baseline visit.
Data and safety monitoring board.
A data and safety monitoring board (DSMB) with membership independent of the study sponsors received and reviewed data every 3 months. They could request unblinding of data in relation to prespecified safety and futility analyses or in response to safety concerns.
Statistical analysis.
A detailed statistical analysis plan (SAP) was developed and finalized by the ADCS and Pfizer. An ADCS Biostatistician (R.G.T.) supervised data analyses, independent of Pfizer. The Full Analysis Dataset was based on a modified intent-to-treat approach, consisting of all subjects who received at least one dose of study medication and had a baseline and postbaseline observation for the measurement of interest. The primary analysis compared differences in mean treatment effect using an analysis of covariance model applied to the full analysis dataset. Covariates considered for inclusion in the model (besides baseline MMSE), if warranted by the data (i.e., based on imbalance across groups and association with outcome measure), included age, sex, years of education, and APOE genotype. Missing values were imputed via the multiple imputation method. Predictor variables used in the imputation model included changes from baseline at 3, 6, 9, 12, and 15 months. Point estimates, standard errors, confidence intervals, and p values were computed using the multiple imputation method. Similar approaches were used to analyze changes in secondary endpoints.
A prespecified interim safety analysis, conducted when 50% of subjects had completed a month 6 visit, examined safety data, mean changes on ADAS-cog, and the percentages of subjects who declined by ≥10 points on the ADAS-cog. A prespecified interim futility and safety analysis was conducted 12 months after all subjects were randomized. A futility rule was prespecified to assess the probability of rejecting the null hypothesis of a treatment difference vs placebo at 18 months (i.e., conditional power) using a threshold of 10%.
After the futility conclusion was reached based on the second interim analysis, an amended SAP was developed to allow for postfutility exploratory analyses and was approved by the DSMB.
Power.
With 133 subjects per group, the study had 80% power, at a significance level of 0.05, to detect a 3-point difference in change from baseline to 18 months in ADAS-cog scores (assuming a SD of 6.5) between a PF-04494700 dose group and placebo, allowing for 25% missing data and 2 interim analyses.
RESULTS
Figure 1 (CONSORT diagram) shows the flow of subjects: 701 subjects were screened for eligibility; 302 were excluded and 399 randomized. The interim safety analysis, conducted in August 2009 after 50% of subjects had a 6-month visit, revealed an increased frequency of serious adverse events (SAE), in particular falls and confusion, in the high-dose group relative to the low-dose and placebo groups. A higher percentage of subjects in the high-dose group declined by ≥10 points on the ADAS-cog (relative to baseline scores) at months 3 and 6. Safety concerns or accelerated cognitive decline were not evident in the low-dose group. On the recommendation of the DSMB, subjects randomized to the high dose discontinued study drug, received a safety evaluation, and were asked for consent to be followed for safety, clinical, and laboratory assessments. Subjects in the low-dose and placebo groups were re-consented before continuing in the study.
Figure 1. CONSORT flow diagram: RAGE inhibitor clinical trial.

ADAS-cog = Alzheimer's Disease Assessment Scale–cognitive; RAGE = receptor for advanced glycation end products.
The second prespecified interim analysis, comparing the low-dose and placebo groups for futility, was conducted in August 2010. This analysis showed no major safety concerns; however, the futility criteria for ADAS-cog were met. The DSMB reviewed these data and recommended discontinuation of the study. Subjects were informed of these findings, and, whenever possible, continued to be followed off study drug for up to 3 months. Discontinuation rates before month 18 were higher in the high-dose arm (87/135 [64%]) compared to the low-dose (77/132 [58%]) or the placebo group (74/132 [57%]) (figure e-1). Figure 1 lists reasons for discontinuation. Forty-six subjects in the high-dose group, 69 in the low-dose group, and 68 in the placebo group completed month 18 visits.
Clinical data.
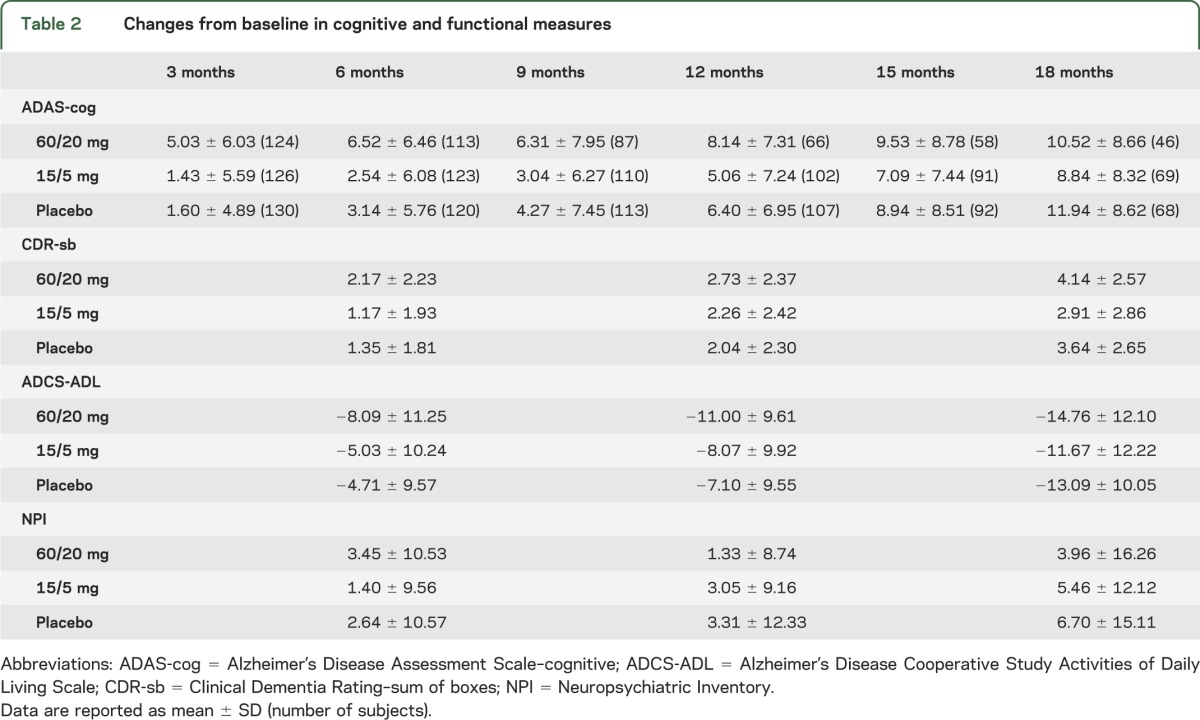
Across treatment groups, subjects were well-matched for demographic and clinical data at baseline (table 1). At baseline, almost all subjects were taking an acetylcholinesterase inhibitor, and about two-thirds of subjects in each group were taking memantine. Changes over time in clinical outcome measures are shown in table 2.
Table 1.
Demographic and clinical characteristics of subjects at baseline
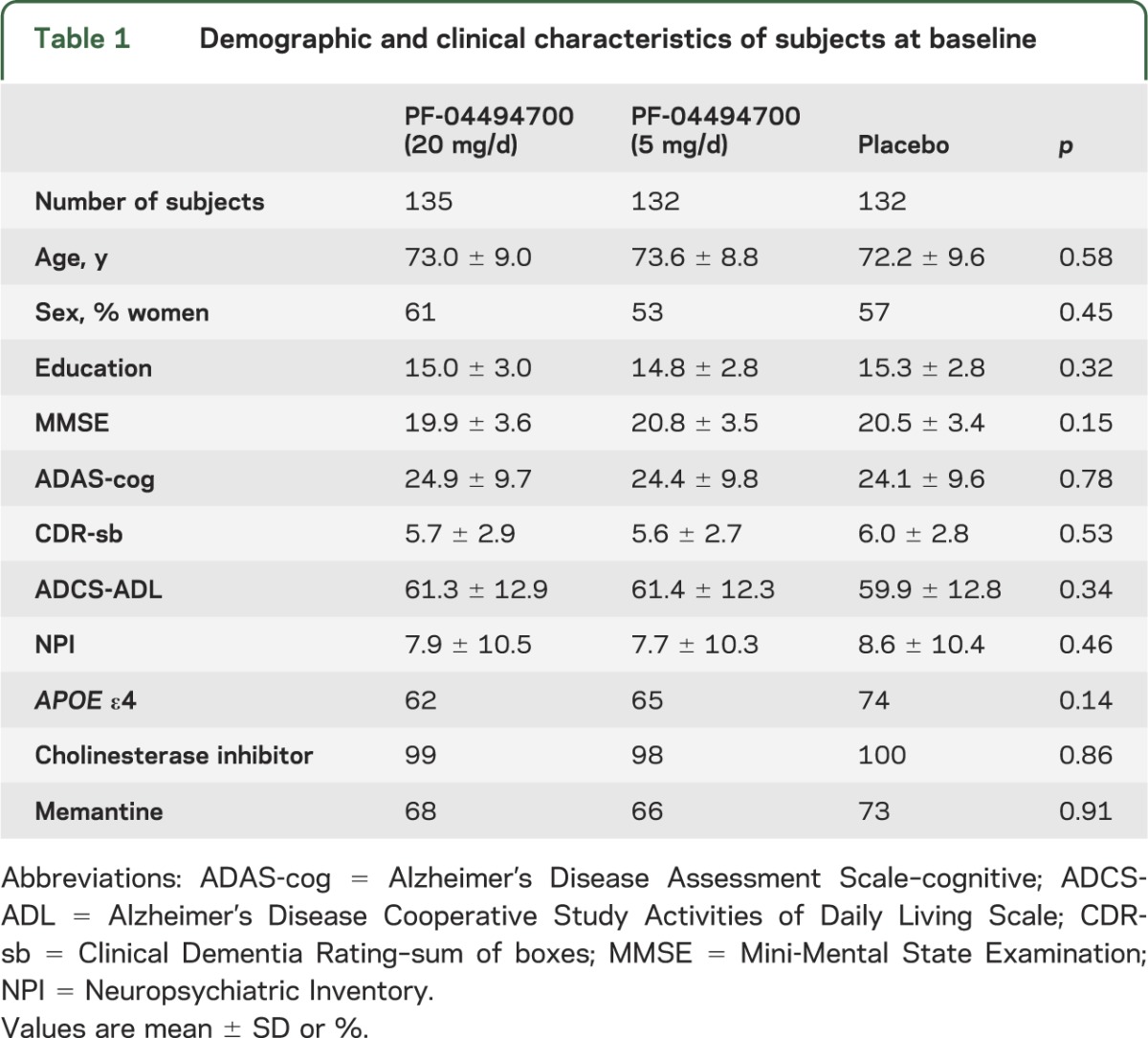
Table 2.
Changes from baseline in cognitive and functional measures
Low-dose treatment vs placebo: Primary outcome.
In the post-futility exploratory analysis of ADAS-cog, changes favored low dose compared to placebo at month 18 (p = 0.008, analysis of covariance [ANCOVA] with multiple imputation) (table 2 and figure 2). ADAS-cog changes at month 18 also favored the low dose when other statistical models were applied: complete case (p = 0.02), ANCOVA with last observation carried forward (p = 0.03), mixed model repeated measures (p = 0.04), and generalized estimating equations (p = 0.03).
Figure 2. Changes in ADAS-cog scores.
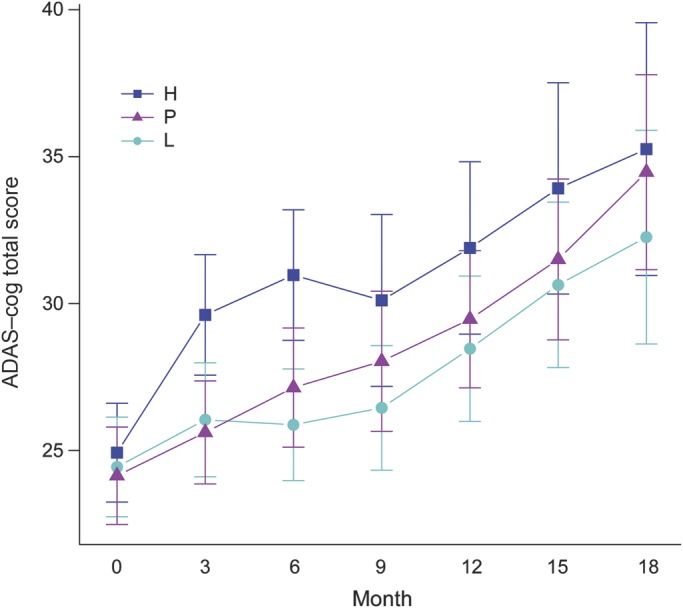
Alzheimer's Disease Assessment Scale–cognitive (ADAS-cog) scores are shown for each treatment group as mean and 95% confidence interval. The figure shows a post-futility data analysis, using analysis of covariance with modified intent-to-treat. H = high-dose; L = low-dose; P = placebo.
Low-dose treatment vs placebo: Secondary outcome measures.
There were no differences at a threshold of p < 0.05 between low dose and placebo at 18 months on the CDR-sb, MMSE, ADCS-ADL, or NPI (table 2), or on neuropsychological test scores.
High-dose treatment vs placebo.
The 6-month interim safety analysis identified higher frequencies in the high-dose compared to the placebo group of all SAEs (13.2% vs 8.3%), falls (10.3% vs 6.1%), and confusion (8.1% vs 4.5%). Mean 6-month change on the ADAS-cog in this analysis was 8.0 (±6.6) points for the high-dose group, 3.2 (±5.4) for the low-dose, and 3.1 (±5.9) for the placebo group (Kruskal-Wallis, p < 0.001). Changes in vital signs (blood pressure, pulse rate, temperature), weight, or body mass index from baseline until August 2009 did not differ among the 3 treatment groups. The incidence of worsening on the ADAS-cog (≥10 points) trended higher in subjects with lower baseline MMSE scores (53% for subjects with baseline MMSE <20 vs 39% for those with baseline MMSE ≥20; p = 0.13). The APOE ε4 allele was not associated with faster progression on the ADAS-cog with treatment.
To assess whether the accelerated cognitive decline observed in the high-dose treatment arm persisted after treatment discontinuation, we examined trajectories of change on the ADAS-cog during active treatment and after follow-up. The rate of decline on the ADAS-cog for subjects who continued to be followed after stopping the high dose of PF-04494700 was significantly slower than the rate of decline while on treatment.
Neuroimaging findings.
Sixty-two subjects in the placebo group and 62 in the low-dose group had serial MRIs. At baseline, these subjects did not differ significantly regarding age, sex, education, and MMSE; there was a trend for higher CDR-sb score in the low-dose group (5.0 ± 2.4) compared to the placebo group (5.6 ± 2.4) (p = 0.086, Wilcoxon). Volumetric analysis of MRI was carried out by the Center for Imaging and Neurodegeneration at UC San Francisco. There were no significant differences between the low-dose and placebo groups in changes in hippocampal (figure e-2) or whole brain measures from baseline to 12 or 18 months.
CSF biomarkers.
CSF levels of Aβ1-x, 1-40, and 1-42, and of total tau and phospho-tau-181, were measured by sandwich ELISA. There were relatively small changes in CSF concentrations of these analytes among subjects from baseline to month 12, and no significant differences were found among the 3 treatment groups. These analyses are limited by small numbers of subjects (figure e-3, table e-2).
Safety data.
Frequencies of adverse events are shown according to MedDRA coding in table e-3. Decreased psychiatric disorders and increased gastrointestinal disorders (diarrhea, constipation, and nausea) were observed in the active dose groups. There were no significant differences in laboratory blood or urine parameters or ECG findings among the 3 groups. There were no distinguishing abnormal MRI findings across the groups, and amyloid-related imaging abnormalities (ARIA)23 were not detected. In particular, changes of vasogenic edema or parenchymal hemorrhage were not identified.
DISCUSSION
This phase 2 clinical trial examined 2 doses of PF-04494700, compared to placebo, in a planned 18-month design. Due to adverse effects (not observed in a prior 3-month trial), the high-dose arm was discontinued prematurely. The occurrence of confusion, falls, and accelerated cognitive decline suggests CNS toxicity. The mechanism of this toxicity is not clear: there were no abrupt or acute changes among study subjects, and it was not related to increased brain atrophy or to ARIA.23 CSF and plasma levels of Aβ did not change in a distinctive way in the high-dose group. Follow-up data on subjects in the high-dose group suggest slowing of cognitive decline after the study drug was discontinued, although data are limited to those subjects who returned to complete follow-up testing. Findings therefore indicate a reversible adverse effect of the high dose of the study drug on brain function, but we cannot definitively conclude whether this was an on- or off-target effect.
The low dose of PF-04494700 did not raise safety concerns regarding either clinical or laboratory measures. The post-futility analysis found decreased decline in ADAS-cog in the low-dose group at 18 months compared to placebo. This finding of potential benefit must be treated with caution because of the modifications to the study as a result of the 2 interim analyses: attrition due to dropouts and discontinued dosing by month 18 and the collection of data points after subjects were requested to discontinue study drug may have influenced the results. The futility analyses conducted as the prespecified interim analysis included 84 subjects who had completed 18 months of treatment, whereas the post-futility analyses conducted after the trial was finished included 137 subjects with 18 months of ADAS-cog data. We do not know how long it takes for a cognitive response to an amyloid-targeted treatment to emerge and suggest that future anti-AD trials should be cautious about including futility analyses. Changes in secondary clinical outcome measures did not indicate significant benefits of low-dose treatment. Interpretation of drug effects is complicated by the lack of a biomarker that directly assesses target engagement by the study drug. MRI and CSF biomarkers related to AD did not provide evidence for benefits mediated by changes in neurodegeneration or amyloid clearance, although the number of subjects with longitudinal CSF biomarkers was relatively small.
Preclinical data supported the ability of PF-04494700 to decrease amyloid pathology in transgenic mouse models related to AD, accompanied by improvements on behavioral testing and electrophysiology. Another compound that inhibits RAGE-Aβ interactions was shown to have similar efficacy in a transgenic mouse model of AD.24 The doses of PF-04494700 selected for this clinical trial result in blood levels in humans in a similar range to blood levels in mice associated with pathologic and behavioral improvement. Benefits of antiamyloid treatment in animal models have not directly extrapolated to human clinical trials in AD. It is possible that intervention at the stage of clinical AD dementia, as in this trial, may be too late to appreciate major clinical benefits.
Key aspects of trial design to identify disease-modifying effects in AD, including the optimal duration of treatment, remain uncertain. The interim analyses in this study, while leading to appropriate discontinuation of the high-dose arm and a conservative decision about futility, complicated the analysis and interpretation of the clinical data. In view of the high overall dropout and discontinuation rates over 18 months, and the collection of data points after subjects were requested to discontinue study drug, this study does not provide definitive conclusions about the clinical efficacy of treatment with the low dose of PF-04494700 in AD.
Supplementary Material
ACKNOWLEDGMENT
Organizational support was provided by Sarah Walter and Tamie Sather (ADCS Clinical Operations, UCSD) and Wendy Davison and Alison Hochbaum (Pfizer, Inc.). Data Safety Monitoring Board members were George Alexopoulos, MD, PhD (Chair), Karl Kieburtz, MD, Serge Gauthier, MD, Richard Kryscio, PhD, Bruce Miler, MD, and Eva Lonn, MD.
GLOSSARY
- Aβ
amyloid β protein
- AD
Alzheimer disease
- ADAS-cog
Alzheimer's Disease Assessment Scale–cognitive
- ADCS
Alzheimer's Disease Cooperative Study
- ADCS-ADL
Alzheimer's Disease Cooperative Study Activities of Daily Living Scale
- ANCOVA
analysis of covariance
- ARIA
amyloid-related imaging abnormalities
- CDR-sb
Clinical Dementia Rating–sum of boxes
- DSMB
data and safety monitoring board
- MMSE
Mini-Mental State Examination
- NPI
Neuropsychiatric Inventory
- RAGE
receptor for advanced glycation end products
- SAE
serious adverse events
- SAP
statistical analysis plan
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Galasko: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content. Dr. Bell: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content. Dr. Mancuso: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content. Dr. Kupiec: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content. Dr. Sabbagh: study concept and design, drafted and revised manuscript for intellectual content. Dr. van Dyck: drafted and revised manuscript for intellectual content. Dr. Thomas: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content. Dr. Aisen: study concept and design, analysis and interpretation, drafted and revised manuscript for intellectual content.
STUDY FUNDING
Funding provided by Pfizer, Inc., and by the National Institute on Aging (grant AG10483). The compound was discovered by TransTech Pharma. Pfizer licensed the compound for development for this trial but has returned the intellectual property to TransTech Pharma after the trial concluded. TransTech Pharma reviewed a draft of this manuscript but did not sponsor the trial or have input into the data analyses presented here.
DISCLOSURE
D. Galasko serves as Editor of Alzheimer's Disease Research and Treatment, for which he receives payment; serves on Data Safety Monitoring Boards for Elan, Janssen, and Balance Pharmaceuticals; and is a consultant for Elan Pharmaceuticals, Inc., and Genentech, Inc. He receives research support from the NIH (AGO5131, AG036528, AG020206, AG10483), the Michael J Fox Foundation, and the Alzheimer's Drug Discovery Foundation. J. Bell is an employee of Pfizer, Inc. J. Mancuso is an employee of Pfizer, Inc. J. Kupiec is an employee of Pfizer, Inc. M. Sabbagh serves as a consultant to Eisai, Elan/Wyeth, Pramal, Takeda, and TransTech Pharma. He receives research support from NIH (AGO19610, AG024904, AGO10483) and is a site investigator in clinical trials sponsored by Avanir, Avid, Baxter, Bayer, Celgene, Elan, GE, Genentech, Lilly, and Pfizer. C. van Dyck serves as consultant to Bristol-Myers Squibb, Janssen, and Pfizer. He holds intellectual property rights with Shire and Marinus Pharmaceuticals. He receives research support from the NIH (AG10483, AGO24904, AGO34953, UH2TR000967) and is a site investigator in clinical trials sponsored by Baxter, Biogen Idec, Bristol-Myers Squibb, Eisai, Eli Lilly, Genentech, Janssen, Merck, Pfizer, and Roche. R. Thomas serves as a consultant to AC Immune, and receives research support from the NIH (AG10483). P. Aisen serves as a consultant for Anavex, Astellas, Astra-Zeneca, Biogen Idec, Bristol Myers Squibb, Ichor, iPerian, KKA, Merck, Neurophage, Novartis, Somaxon, and Toyama. He is an unpaid consultant to Lilly, Pfizer, and Roche. He receives research support from the NIH (AG10483, AGO24904). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Yan SD, Bierhaus A, Nawroth PP, Stern DM. RAGE and Alzheimer's disease: a progression factor for amyloid-beta-induced cellular perturbation? J Alzheimers Dis 2009;16:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt AM, Sahagan B, Nelson RB, et al. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer's disease. Curr Opin Investig Drugs 2009;10:672–680 [PubMed] [Google Scholar]
- 3.Fang F, Lue LF, Yan S, et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, A-beta accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. FASEB J 2010;24:1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003;9:907–913 [DOI] [PubMed] [Google Scholar]
- 5. Deleted in proof.
- 6.Sabbagh MN, Agro A, Bell J, et al. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord 2011;25:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 8.Folstein MF, Folstein SE, McHugh PH. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198 [DOI] [PubMed] [Google Scholar]
- 9.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer's Disease. Am J Psychiatry 1984;141:1356–1364 [DOI] [PubMed] [Google Scholar]
- 10.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414 [DOI] [PubMed] [Google Scholar]
- 11.Galasko D, Kershaw PR, Schneider L, et al. Galantamine maintains ability to perform activities of daily living in patients with Alzheimer's disease. J Am Geriatr Soc 2004;52:1070–1076 [DOI] [PubMed] [Google Scholar]
- 12.Cummings LM, Mega M, Gray K, et al. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2304–2314 [DOI] [PubMed] [Google Scholar]
- 13.Gilmore GC, Royer FL, Gruhn JJ. Age differences in symbol-digit substitution task performance. J Clin Psychol 1983;39:114–124 [DOI] [PubMed] [Google Scholar]
- 14.Wechsler D. Wechsler Adult Intelligence Scale–Revised Manual. New York: Psychological Corporation; 1981 [Google Scholar]
- 15.COWAT metanorms across age, education and gender. Appl Neuropsychol 2001;8:161–166 [DOI] [PubMed] [Google Scholar]
- 16.Golden GC. Stroop Color and Word Test: A Manual for Clinical and Experimental Uses. Wood Dale, IL: Stoelting; 1978 [Google Scholar]
- 17.Reitan R. Validity of the trail making test as an indicator of organic brain disease. Percept Mot Skills 1958;8:271–276 [Google Scholar]
- 18.Sano M, Zhu CW, Whitehouse PJ, et al. ; The Alzheimer's Disease Cooperative Study Group. ADCS Prevention Instrument Project: pharmacoeconomics: assessing health-related resource use among healthy elderly. Alzheimer Dis Assoc Disord 2006;20:S191–S202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith SC, Lamping DL, Banerjee S, et al. Development of a new measure of health-related quality of life for people with dementia: DEMQOL. Psychol Med 2007;37:737–746 [DOI] [PubMed] [Google Scholar]
- 20.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp 1999;8:272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–355 [DOI] [PubMed] [Google Scholar]
- 22.Fischl B, van der Kouwe A, Destrieux C, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex 2004;14:11–22 [DOI] [PubMed] [Google Scholar]
- 23.Sperling RA, Jack CR, Black SE, et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trails: recommendations form the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement 2011;7:367–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deane R, Singhe I, Sagare AP, et al. A multimodal RAGE-specific inhibitor reduces amyloid [beta]-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012;122:1377–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.