

Abstract
In an unbiased genetic screen designed to isolate mutations that affect synaptic transmission, we have isolated homozygous lethal mutations in Drosophila importin 13 (imp13). Imp13 is expressed in and around nuclei of both neurons and muscles. At the larval neuromuscular junction (NMJ), imp13 affects muscle growth and formation of the subsynaptic reticulum without influencing any presynaptic structural features. In the absence of imp13, the probability of release of neurotransmitter and quantal content is increased, yet the abundance of the postsynaptic receptors and the amplitude of miniature excitatory junctional potentials are not affected. Interestingly, imp13 is required in the muscles to control presynaptic release. Thus, imp13 is a novel factor that affects neurotransmitter release at the fly NMJ. Its role in the context of synaptic homeostasis is discussed.
Introduction
Synaptic homeostasis originates when synaptic activity is altered, for example, when key components required for proper synaptic transmission are disrupted or altered in the postsynaptic terminal. These disturbances trigger a postsynaptic response that signals to the presynaptic terminal to reset release parameters, thereby restoring synaptic output to previous levels. Hence, synaptic homeostasis enables overall neuronal output to remain stable and can be viewed as a specialized form of synaptic plasticity, which limits the risk of unbalanced synaptic output by activity-dependent alterations of neuronal excitation (Burrone and Murthy, 2003; Davis, 2006; Turrigiano, 2007).
The neuromuscular junction (NMJ) of the Drosophila larva has provided one of the best-characterized examples of synaptic homeostasis. For example, absence or reduction of the DGluRIIA subunits of the postsynaptic glutamate receptors (GluRs), or inhibition of their activity, leads to a significant decrease of quantal size (the depolarization induced by the release of a single synaptic vesicle) and an increase in the quantal content (the number of vesicles that are released presynaptically during invasion of an action potential) (Petersen et al., 1997; Davis et al., 1998; DiAntonio et al., 1999; Frank et al., 2006). However, inappropriate composition of the subunits of GluRs can also result in homeostatic compensation, although quantal size is not affected in this paradigm (DiAntonio et al., 1999).
The initial step by which postsynaptic receptors control presynaptic release is mediated by the influx of calcium (Ca2+) ions, which regulate the activity of postsynaptic calmodulin kinase II (CaMKII) (Haghighi et al., 2003). However, chronic hyperpolarization of the muscle by overexpression of Kir2.1 potassium channel also induces synaptic homeostasis (Paradis et al., 2001), and this may also be attributable to an increased postsynaptic Ca2+ influx (Haghighi et al., 2003; Frank et al., 2006). Activation of CaMKII may impinge on the retrograde signal that controls presynaptic features.
The nature of the signal from the larval muscles that triggers synaptic homeostasis has remained elusive. It has been shown that a bone morphogenetic protein pathway is necessary for retrograde signaling (Haghighi et al., 2003; van der Plas et al., 2006), because it renders the neurons competent to respond to the signal (Goold and Davis, 2007). In addition, synaptic homeostasis is abolished in the absence of functional presynaptic Cav2.1 calcium channels, emphasizing the fact that regulated calcium entry in the presynaptic terminal may adjust the probability of release of synaptic vesicles in a rapid and reliable manner during homeostasis (Frank et al., 2006).
Here, we report the isolation of mutations in the Drosophila homolog of importin 13 (imp13) in a screen designed to isolate genes that affect synaptic transmission. We find that imp13 affects synaptic transmission in the visual system and the larval NMJ. At the NMJ, imp13 functions postsynaptically to control presynaptic release in the context of synaptic homeostasis.
Materials and Methods
Drosophila melanogaster strains and genetics.
y w;; P{ry+t7.2 neoFRT}82Biso (FRT82Biso) isogenized flies were used for mutagenesis. Mutagenesis was performed as described previously (Mehta et al., 2005). Briefly, we fed male FRT82Biso flies, previously starved for 12–16 h, with 15 mm ethyl methane sulfonate (EMS) (in 1% aqueous sucrose solution, dispersed with repeated aspiration with a 10 ml syringe). After a 12 h feeding, we transferred the mutagenized flies in vials with food, in which they were left for additional 12 h to “clean” themselves from any traces of EMS on their bodies. We then crossed them with virgin females y w, P{ey-FLP.N}2, P{GMR-lacZ.C(38.1)}TPN1; P{ry+t7.2 neo-FRT}82B, P{w+ ry+ white-un1}90E l(3)cl-R31/TM6B, Tb1 to generate F1 flies that are >95% homozygous mutant in the cells of the visual system (Newsome et al., 2000). Hereafter, P{ry+t7.2 neo-FRT}82B, P{w+ ry+ = white-un1}90E l(3)cl-R31 will be referred to as FRT82B cl, and y w, P{ey-FLP.N}2, P{GMR-lacZ.C(38.1)}TPN1 will be referred to as eyFLP. Of 210,000 male flies screened, we established 423 stocks that showed defective postsynaptic responses to light stimuli in electroretinogram (ERG) assays (see Results). On the basis of lethality, we categorized the mutants in 40 lethal complementation groups.
We also used the ey3.5FLP driver, which is only active in the photoreceptors (Mehta et al., 2005; Bazigou et al., 2007). Non-mutagenized eyFLP; FRT82B cl/TM3, Sb and ey3.5FLP; FRT82B cl/TM3, Sb were used as control animals in the experiments for visual system defects. For R7 MARCM (mosaic analysis with a repressible cell marker) analysis, we used the GMR–FLP/MARCM system, as described previously (Lee et al., 2001). For marking the R8 photoreceptor axons, we introduced the Rh6–green fluorescent protein (GFP) transgene (Tahayato et al., 2003) into the background of mutant strains and performed clonal analysis by crossing to ey3.5FLP; FRT82B cl.
The semi-lethal P element line 5-SZ-3929 (DrosDel collection) (Ryder et al., 2004) was used for generation of both precise and imprecise excisions. Of 270 dysgenic crosses, five viable precise excision lines and seven lethal imprecise excision lines (that failed to complement any of the alleles of the 3R23 complementation group) were kept. Two imprecise excisions, imp13164 and imp13177, cause late third-instar lethality. Homozygous larvae were screened by PCR to delimit the deletion breakpoints within the genomic region of imp13. Both strains were balanced over TM6B, Tb1 balancer, so that mutant larvae could be readily identified for electrophysiology experiments. As controls in electrophysiology experiments, we used animals from the precise excision line imp13Ex2, as well as rescued animals of the genotype y w;P{w+ HA–FLAG–imp13–Genomic Rescue1}; imp13164 (for Genomic Rescue1 and Molecular Biology Section for tagging the construct, see Fig. 2). For rescue experiments in the muscle or neuron of the mutant animals, we used the myosin heavy chain (MHC)–GAL4 (Schuster et al., 1996) and C155–GAL4 (Lin and Goodman, 1994) drivers, respectively, to overexpress UAS–imp13 in imp13164/Df(3R)DG2 animals. We also used dpp–GAL4 (G. Mardon, Houston, TX) and BG57–GAL4 (Budnik et al., 1996) lines for overexpression of upstream activating sequence (UAS)–FLAG–Imp13 or UAS–Imp13–red fluorescent protein (RFP).
Figure 2.

Complementation group 3R23 maps within CG7212, the Drosophila homolog of imp13. A, Mutations in complementation group 3R23 map in the region 90E7–91A5, because they fail to complement the deficiencies Df(3R)DG2 and Df(3R)Exel6178 (red bars) but not Df(3R)RD31 and Df(3R)Cha7 (green bars). Gray bars indicate regions that are possibly uncovered by the corresponding deficiencies. B, Meiotic recombination mapping with an array of molecularly mapped P elements indicates that the mutations lie near P element EY6955. C, Genomic organization of the region around EY6955. Two genomic rescue constructs, Rescue1 and Rescue2 (red bars), rescue the corresponding mutations (H). D, Exon–intron structure of CG7212. Numbered boxes depict the exons, and the black segments of the boxes correspond to the coding sequence. The semi-lethal P element 5-SZ-3929 was imprecisely excised (see Materials and Methods). The breakpoints of excision lines imp13 177 and imp13164 are symbolized by arrows. E, Protein structure of Imp13. Imp13 contains the importin β N terminal domain (IMBN, red box), characteristic of all members of the importin β family. The position of the identified point mutations is symbolized by a white asterisk, and the molecular lesion is indicated. F, Phylogenetic comparison of various Imp13 homologs from model organisms (worm, fly, zebrafish, mouse, and human) generated by MatGat (Campanella et al., 2003); accession numbers are included in parentheses. Percentages of similarity (black) and identity (red) are given. H.s., Homo sapiens; M.m., Mus musculus; D.r., Danio rerio; D.m., D. melanogaster; and C.e., Caenorhabditis elegans. G, Clonal analysis in the eye shows that imp13177 excision leads to absence of ON and OFF transients and a dramatically reduced hyperpolarization curve, similar to imp13DC775 and imp13FY561 point mutations shown in Figure 1, A and B. H, Lethal phase analysis of mutations in imp13 (L2, second-instar larval stage; L3, third-instar larval stage; ND, not determined; MGL, maternal germ-line clones; asterisks indicate the genotypes assayed for germ-line clones). Red boxes indicate that the corresponding allelic combinations could be rescued to adulthood in the presence of Genomic Rescue1 (C) in the genetic background of the mutant fly strains.
To examine the effect of the deletions in the visual system, imp13164 and imp13177 were recombined onto the FRT82Biso parental chromosome. To assess the lethal phase of different allelic combinations, we balanced the mutants over a TM3, P{GAL4-Kr.C}DC2, P{UAS-GFP.S65T}DC10, Sb1 (Casso et al., 2000), available from the Bloomington Stock Center, and hereafter referred to as TM3, KrGFP. To assess the lethal phase of mutants derived from maternal germ line clones, we crossed FRT82B imp13177 with hsFLP. Virgin female progeny of the above cross were crossed to males w[*]; P{neoFRT}82B P{ovoD1–18}3R/st[1] betaTub85D[D] ss[1] e[s]/TM3, Sb[1]1] (Chou et al., 1993). To induce recombination in the germ line, larvae were heat shocked at 38°C twice, each time for 1 h, during 2 consecutive days. Virgin female progeny were then crossed to males (http://flybase.org/cgi-bin/uniq.html?FBst0004431%3Efbst; Df(3R)DG2/TM3, KrGFP). To avoid competition between mutants and wild-type larvae in lethal phase assays, non-GFP larvae were selected and transferred to new grape juice plates, in which they were monitored daily for survival. Flies were reared at room temperature.
Molecular biology.
For construction of the genomic rescue constructs, BACR27G04 (GenBank accession number AC009462) was digested with BglII, and a 7.2 kb fragment spanning the genomic region of both CG7212 and CG7208 was cloned into pBluescript KSII (Stratagene) at BamHI. Subsequently, a SmaI digestion and religation removed the region corresponding to CG7208. For examination of the protein expression, we tagged the smaller genomic rescue construct containing CG7212 by subcloning a 2xHA (hemagglutinin tag-3xFLAG composite tag at a BglII site, inserted just before the stop codon in the coding sequence of importin 13. For insertion of the BglII site, we performed PCR-based site-directed mutagenesis of the corresponding plasmid, using iProof high-fidelity polymerase (Bio-Rad) and appropriately designed mutagenic oligos, phosphorylated by T4 polynucleotide kinase (New England Biolabs). All genomic fragments were subcloned into pP{CaSpeR-4} as NotI/XhoI fragments. For cDNA rescue experiments, we PCR amplified the full coding sequence of cDNA from LD35896 (Drosophila Gene Collection) and cloned it into pP{UAST} and pP{UAST-FLAG} as a NotI fragment by conventional cloning and pP{UAST-RFP} using Gateway technology (Invitrogen). All constructs were verified by sequencing before injection for generation of transgenic lines.
Immunohistochemistry, image acquisition, and processing.
Third-instar larval fillets and adult brains were fixed in HL3 (Stewart et al., 1994) or PBS with 3.5% formaldehyde for 15 min, respectively, and washed in PBS with 0.2% Triton X-100. For stainings with anti-DGluRIIA, we used Bouin's fixative (picric acid/formaldehyde/acetic acid mixed in a 15:5:1 ratio) for 20 min at room temperature, followed by multiple 30 min washes before application of the antibody. Antibody dilutions used were as follows: mouse anti-actin (cloneC4; MP Biomedicals); mouse anti-Bruchpilot (nc82), 1:100 (Wagh et al., 2006); mouse anti-Chaoptin, 1:100 [24B10; Developmental Studies Hybridoma Bank (DSHB), University of Iowa, Iowa City, IA] (Fujita et al., 1982); mouse anti-Dlg, 1:500 (4F3; DSHB) (Parnas et al., 2001); mouse anti-FasII, 1:100 (1D4; DSHB); anti-DGluRIIA, 1:100 (8B4D2; DSHB) (Schuster et al., 1991); rabbit anti-GluRIIB, 1:500 (Marrus et al., 2004); guinea pig anti-Eps15, 1:1000 (Koh et al., 2007); rabbit anti-HRP, 1:1500 (Jackson ImmunoResearch); mouse anti-hemagglutinin, 1:500 (Sigma-Aldrich); mouse anti-Flag, 1:500 (Sigma-Aldrich); and rabbit anti-Synaptotagmin (Syt), 1:1000 (Littleton et al., 1993). Secondary antibodies conjugated to cyanine 3 (Cy3), Cy5, or Alexa 488 (Jackson ImmunoResearch and Invitrogen) were used at 1:250. All antibody incubations were performed at 4–8°C overnight in the presence of 5% normal goat serum.
Images from fluorescently labeled specimens were acquired with a Zeiss LSM510 confocal microscope and processed using NIH ImageJ software and Adobe Photoshop 7.0 (Adobe Systems).
For quantification of the intensity of immunostaining, larvae of different genotypes were dissected and fixed simultaneously and processed in the same microcentrifuge tubes. After completion of the procedure, they were mounted on the same slide and scanned by confocal microscopy under identical settings. The intensity of fluorescence was quantified with NIH Image J as follows: a two-dimensional projection of the maximum fluorescence at the NMJ was created from a series of confocal sections. Then, we delineated manually the area of interest, included by HRP staining for GluRIIA or included by Dlg staining for GluRIIB (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). The average pixel intensity within the delineated area was then calculated in the channel of interest (GluRIIA, GluRIIB).
Retina sections and transmission electron microscopy.
Retina sections, transmission electron microscopy (TEM) of photoreceptors and NMJ boutons were performed as described previously (Zhai et al., 2006; Ly et al., 2008). Laminas were prepared and analyzed from at least five different animals. NMJ samples were prepared from three to five animals. Thick sections were prepared for inspection of the integrity of sample. Sections were acquired from boutons at muscles 6 and 7 from abdominal segments A2 to A4. For the quantification of clustered vesicles, we included vesicles located within the range of 250 nm away from a T-bar (active zone), as performed previously (Aravamudan et al., 1999). For the quantification of docked vesicles, we included vesicles that surround the T-bar (250 nm) and are <30 nm away from the plasma membrane. For the quantification of vesicle diameter, ∼50 vesicles were randomly chosen from each bouton. Images were analyzed using NIH ImageJ software.
Electrophysiology.
ERGs were recorded as described previously (Mehta et al., 2005). Third-instar larval fillets were prepared in HL3 (Stewart et al., 1994), and recordings were performed at various extracellular Ca2+ concentrations. Ca2+ was provided as a chloride salt at the indicated concentrations. Larval motor axons were severed and excitatory junctional potentials (EJPs) were recorded from muscle 6 of abdominal segments A2 and A3 at room temperature. Quantal content (QC) was estimated by including failures and correcting for nonlinear summation of EJPs as indicated previously (Stevens, 1976) (also see McLachlan and Martin, 1981). Cooperativity coefficients were then assessed by determining the slope of log-transformed measurements for quantal content for Ca2+ concentrations of 0.15, 0.25, and 0.35 mm. Spontaneous miniature EJPs (mEJPs) were recorded in the presence of 0.5 mm extracellular Ca2+ and 10 μm tetrodotoxin (Sigma-Aldrich). EJPs and mEJPs were analyzed using pClamp6 (Molecular Devices) and Mini Analysis Program (Synaptosoft) software, respectively.
To assess the passive properties of the membrane (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), a 1 nA depolarizing current was injected in the muscle. The input resistance of the muscle was calculated by the alteration in potential difference across the membrane divided by the amplitude of the injected current, according to Ohm's law. As far as capacitance is concerned, we measured the time constant τ (tau) of the depolarization curve by fitting a single-exponential equation to the curve, as performed previously (Paradis et al., 2001). For quantification of the resting membrane potential, we calculated the baseline from different recordings from different genotypes at various [Ca2+]o concentrations. There were no differences regarding all three parameters (input resistance, time constant τ, and resting membrane potential) among different genotypes (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Calcium imaging.
Calcium imaging was performed as described previously (Macleod et al., 2002, 2003; Rossano and Macleod, 2007) (also see http://www.jove.com/index/details.stp?ID=250), with the following modifications. More specifically, a forward filling pipette was prepared by standard procedures (pulling and fire polishing). Subsequently, the pipette was mounted on a slide with the support of two small pieces of wax of unequal height, so that the pipette lies at an angle of ∼30°, with the tip of the pipette pointing downward and toward a Sylgard plate, onto which the larva fillets have been dissected. Larvae were dissected carefully in Schneider's cell culture medium, so that muscle fibers were not damaged. The nerve axons were severed near their contact points with the larval brain. After aligning the pipette and the larva fillet, the end of a severed nerve axon was suctioned into the forward filling pipette by application of negative pressure with the aid of plastic tubing and a syringe adjusted at the pipette's back end. The whole procedure was completed within 5 min after severing the nerves, so that “sealing” of the nerve end is avoided. At this stage, the tubing was removed. The loading of the nerve was then performed by inserting a plastic filament containing 3 mm fura-2 dextran into the pipette and bringing it into close proximity with the cut nerve. The dye was then released.
The preparation was left in the dark for ∼45 min, for the active transport of the dye to the nerve terminal to occur. During that period, Schneider's medium was changed twice. After incubation, we removed the remaining dye from the pipette and washed extensively with Schneider's medium. We then left the Ca2+ dye to equilibrate for another 60 min, during which Schneider's medium was change twice and eventually exchanged with HL6 solution (Macleod et al., 2002, 2003), containing 1 mm [Ca2+]o and 7 mm l-glutamic acid. After 25 min, imaging was performed. Fura-2 fluorescent dye was excited using an X-Cite 120 fluorescence illumination system (EXFO Photonic Solutions), attached on an upright Zeiss Axioskop Optical microscope. To minimize photo damage, a 25% neutral density filter was always inserted in the excitation light path. All preceding procedures were performed in the dark. Images were acquired at 340 and 380 nm, with duration of exposure equal to 180 ms. Five paired frames were acquired from each NMJ for calculation of the resting levels of intracellular Ca2+. Fluorescent images were captured through a Zeiss Optical objective (model Plan-NeoFLUAR 2.5X) and an AxioCam MRm CCD camera and processed using AxioVision 4.6.3 (Zeiss). Fluorescence intensity from images was calculated by the average pixel intensity within a delineated area of nerve terminals, after subtracting the average “background” pixel intensity of the medium within an area of identical size, near and around the loaded synaptic boutons.
Statistical analysis.
Statistical analysis was performed using Excel (Microsoft) or GraphPad Prism (GraphPad Software). The numbers of the samples used in each quantification is depicted at the base of the bars in each graph. Hypothesis testing was based on one-way ANOVA, followed by post hoc pairwise comparisons among all groups by Tukey–Kramer test (statistically significant for p < 0.05) or nonparametric Kruskal–Wallis test, followed by Dunn's multiple pairwise comparisons (statistically significant for p < 0.05).
Results
Mutations in importin 13 cause defective synaptic transmission in the visual system
To identify novel genes that affect synaptic transmission, an F1 mosaic forward genetic screen was conducted using the eyFLP system (Newsome et al., 2000), as described previously (Mehta et al., 2005). In brief, flies carrying randomly induced mutations are made homozygous for the mutation exclusively in cells of the visual system and screened for defects in their ability to sense light. We then performed ERGs (Pak et al., 1969). These extracellular electrophysiological recordings typically consist of an upward spike (the ON transient), followed by a negative sustained potential (hyperpolarization curve) and a downward spike (the OFF transient) (Fig. 1A). The ON and OFF transients represent the relay of the signal from the photoreceptors to the postsynaptic neurons in the lamina (Coombe and Heisenberg, 1986), the first neuropil of the adult visual system. The hyperpolarization curve corresponds to the primary electrical response and the depolarization of the cell membrane (Pak et al., 1969).
Figure 1.
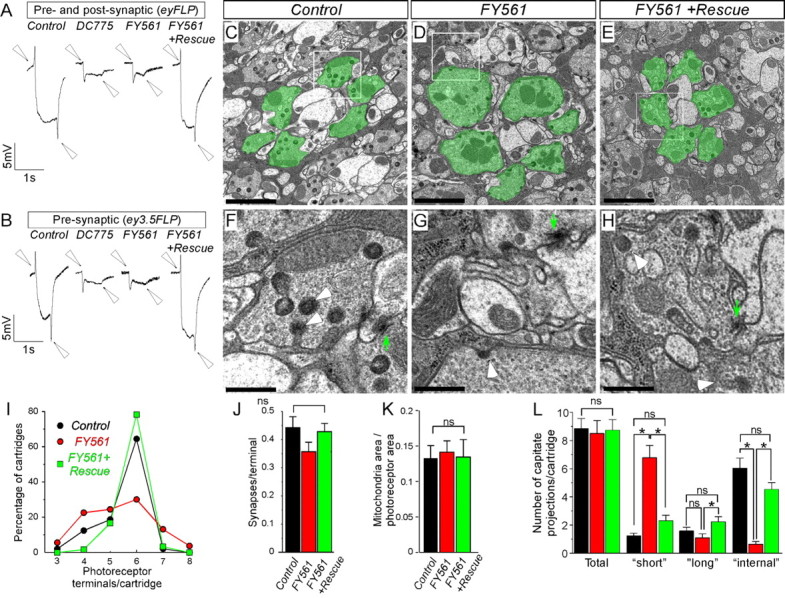
Neurotransmission in the adult visual system of Drosophila is impaired in mutants of the 3R23 complementation group. A, B, ERGs from control, mutant, and rescued animals. In A, both photoreceptors and postsynaptic cells are mutant because of the recombination mediated by the eyFLP transgene, whereas in B, only photoreceptors are mutant attributable to the activity of the ey3.5FLP transgene. Arrowheads indicate the ON and OFF transients. C–H, TEM of synaptic cartridges in the laminas of control (C, F: ey3.5FLP; FRT82B cl/TM3), mutant (D, G: ey3.5FLP; FRT82B cl/FRT82B FY561), and rescued (E, H: ey3.5FLP; Rescue; FRT82B cl/FRT82B FY561) animals. Photoreceptors of a single cartridge are pseudocolored green in C–E. F, G, and H are magnifications of the indicated regions within white frames in C, D, and E, respectively. White arrowheads point to glial capitate projections and green arrows indicate active zone T-bars. Scale bars: C–E, 2 μm; F–H, 0.5 μm. I–M, Bar graphs of ultrastructural characteristics of R1–R6 photoreceptors (control, n = 265 photoreceptors/48 cartridges; FY561, n = 284/53; and Rescue; FY561, n = 350/60). I, Diagram of percentage of synaptic cartridges (ordinate) according to the number of photoreceptors that they include (abscissa), for control (blue), mutant (red), and rescued (green) animals. J–L, Bar graphs for the number of synapses (J), ratio of mitochondrial area to photoreceptor terminal area (K), total number of capitate projections, and number of capitate projections of each morphological class (“short,” “long,” and “internal”) per cartridge for each genotype (L). ns, Not significant.
One of the lethal complementation groups, 3R23, consists of two alleles, DC775 and FY561. The ERG defects caused by these mutations are quite severe (Fig. 1A,B). Not only do these strains lack both ON and OFF transients, but they also exhibit extremely reduced hyperpolarization curves, indicating that multiple aspects of photoreceptor function are affected. When only the photoreceptors are made mutant using the ey3.5FLP driver (Mehta et al., 2005; Bazigou et al., 2007), the observed phenotype is identical to flies in which presynaptic and postsynaptic neurons are mutant (Fig. 1, compare A, B). We conclude that mutations of the complementation group 3R23 affect presynaptic aspects of synaptic transmission and phototransduction in the eye.
To assess whether there are potential defects in ommatidial development, axonal guidance, and synapse formation of the photoreceptors, we performed light microscopy and TEM studies of the visual system. Each ommatidium of the compound eye of the fly comprises eight photoreceptors. In thick sections of the retina, seven of eight photoreceptors can be observed at any given level. Photoreceptors develop normally in the absence of 3R23 (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). Analysis of the axonal projection profile of the R7 and R8 photoreceptors show that they target the appropriate layers in the medulla and do not display patterning defects (supplemental Fig. S1C–H, available at www.jneurosci.org as supplemental material). TEM analysis of the R1 and R6 photoreceptor terminals in the lamina show that synaptic cartridges are formed and that the number of synapses are normal, although the number of synaptic terminals per cartridge is more variable than controls (Fig. 1C–E,J). In addition, the number of synapses, the area of mitochondria, and the total number of capitate projections, which are the invaginations of the epithelial glia that surround the synaptic cartridges, are unaltered in the different genotypes (Fig. 1C–E,J–L). However, in mutant photoreceptor terminals, the glial capitate projections are shallower than in wild type (Fig. 1F—H,L). Capitate projections are the sites of endocytosis of synaptic vesicles (Fabian-Fine et al., 2003), and their morphology is altered in mutants that affect synaptic transmission (Fabian-Fine et al., 2003; Ohyama et al., 2007). Together, these data indicate that the 3R23 alleles affect neurotransmission in the visual system.
The identified mutations were mapped to cytological region 90E7–91A5 based on complementation tests with an array of deficiencies and fine mapped with P elements (Fig. 2A,B) (Zhai et al., 2003). Sequencing of the genes in this region revealed that, in the FY561 strain, CG7212 carries a Q811STOP mutation (Fig. 2E) (supplemental Fig.S2, available at www.jneurosci.org as supplemental material). However, we could not identify the lesion in DC775. CG7212 is the Drosophila homolog of importin 13, a member of the importin β protein family (Fig. 2F) (Mingot et al., 2001; Artero et al., 2003; Quan et al., 2008). To confirm that CG7212 is indeed the cause of the lethality and ERG phenotypes, we identified two other mutations in 3R23: imp13sd4 and imp13sd6. These alleles were isolated in a similar genetic screen for synaptic transmission mutants (Babcock et al., 2003) and failed to complement the lethality of imp13FY561. During sequencing of CG7212, we identified the lesions G875D in imp13sd4 and Q351STOP in imp13sd6 (Fig. 2E) (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We also generated two additional alleles of CG7212 by imprecise excision of the semi-lethal P element insertion (5-SZ-3929). Both alleles delete the majority of the predicted coding sequence. Both excision alleles imp13177 and imp13164 are strong loss of function or null alleles, because they exhibit the same phenotype as homozygotes as well as over a deficiency with respect to the lethal phase (Fig. 2G) and other phenotypes (see below). Examination of the phenotypes of the newly acquired alleles by clonal analysis and ERGs showed that they all confer the same phenotype as the initially identified mutations (compare Figs. 2G, 1A,B) (data not shown). Finally, we also constructed two genomic rescue constructs, the smallest of which spans only the genomic region of CG7212. This transgene rescues the lethality of most allelic combinations (Fig. 2H). Our complementation and rescue data suggest that there are additional lethal hits in the genetic background of some of the alleles, namely the mutations imp13sd4, imp13sd6, and imp13FY561 (Fig. 2H). However, every aspect of the eye phenotype is rescued by the genomic transgene, indicating that the second lethal hits in these mutants are not responsible for the phenotype observed in the visual system (Figs. 1, 2G). Note that the lethality of any mutant allele or allelic combination cannot be rescued by neuronal, muscle, or ubiquitous overexpression of imp13 cDNA.
All allelic combinations are lethal at the second-instar or in the third-instar larval stages (abbreviated as L2 and L3, respectively) (Fig. 2H). To further examine whether the lethal phase of the mutations is attributable to maternal contribution of imp13, we induced germ-line clones (Chou et al., 1993) and examined the lethal phase of animals devoid of both maternal and zygotic imp13 function. Animals derived from germ-line clones that lack the gene and are zygotic ally null still develop until the third-instar stage. Therefore, we conclude that imp13 is required during late larval stages, and the lethal stage of other allelic combinations is not attributable to perdurance of maternally provided product.
In summary, we have identified mutations that indicate a role for imp13 in synaptic transmission in the visual system of Drosophila. We therefore decided to examine the possible function of imp13 in synaptic transmission at the larval NMJ, because this synapse is much better characterized.
Imp13 is expressed in neurons and muscles
Members of the importin β protein family are considered to be “housekeeping” genes, expressed in a wide variety of tissues at varying levels. In human tissues, Imp13 is enriched in fetal lung and heart (Zhang et al., 2000), brain, and spinal cord (Quan et al., 2008). To examine whether Imp13 is localized at the NMJ, we attempted to generate antibodies against Imp13. Although the sera recognize Imp13 when overexpressed (supplemental Fig. S3A–C, available at www.jneurosci.org as supplemental material), we were not able to assess the endogenous expression of imp13 by immunofluorescent stainings or Western blot analysis. When overexpressed in neurons or muscles, an Imp13–RFP fusion protein is localized to synapse (supplemental Fig. S3D–I, available at www.jneurosci.org as supplemental material). To follow the endogenous expression of imp13, we tagged the genomic rescue construct and confirmed that it encodes a protein of the expected length (supplemental Fig. S3J–L, available at www.jneurosci.org as supplemental material) and that it rescues imp13 null mutants. In the larval brain, Imp13 is enriched in neurons, but it is also expressed in other cell types, probably glia, mainly localizing in perinuclear structures (Fig. 3A–C). At the neuromuscular junction, we were able to detect Imp13 expression only in the muscle nuclei but not at the synapse (Fig. 3D–F), in contrast to when we use overexpression conditions (supplemental Fig. S3G–I, available at www.jneurosci.org as supplemental material). In summary, imp13 is expressed widely and is localized in and around the nucleus in neurons and muscles.
Figure 3.

Imp13 is expressed in the nervous system and somatic muscles but not the synapse. A–C, Analysis of expression of imp13–HA–FLAG in a part of the ventral nerve cord of third-instar larvae. Single-channel representation of A for Elav, nuclear marker for neurons (B), and Imp13–HA (C). Arrowheads in A–C point to nuclei, whereas the arrow indicate non-neuronal cells. D, Analysis of expression of imp13–HA–FLAG genomic rescue construct at third-instar NMJ. Single-channel representation of D for Eps15, a presynaptic endocytic protein (E), and Imp13–HA (F). Arrowhead points to the nucleus of muscles. Scale bars: A–F, 10 μm.
Imp13 mutants exhibit postsynaptic defects at third-instar NMJs
We first examined the structural features of third-instar NMJs at the level of confocal light microscopy (Fig. 4A,B). The number of synaptic boutons per muscle surface is not altered in mutants when compared with control animals (Fig. 4D), although there is a small decrease (∼15%) of the total surface area of muscles 6 and 7 (Fig. 4C). TEM reveals that the density and size of synaptic vesicles, as well as the number of clustered and docked synaptic vesicles are not affected in the mutants (Fig. 4E–H) (supplemental Fig. S4A,B, available at www.jneurosci.org as supplemental material). Furthermore, there are no changes in mitochondrial area, as indicated by the relative area they occupy within the terminals (supplemental Fig. S4E, available at www.jneurosci.org as supplemental material). The number of active zones (Fig. 4K) and the length of the postsynaptic density (Fig. 4L) are also not altered. However, we find defects in the subsynaptic reticulum (SSR) in terms of thickness (the total area that SSR occupies) (Fig. 4M) and density (the average number of layers of SSR that surround each bouton) (Fig. 4N). Interestingly, muscle overexpression of imp13 cDNA is sufficient to rescue the muscle size (Fig. 4C), the thickness of SSR (Fig. 4M) and SSR density (Fig. 4N), suggesting that imp13 functions in the muscle for controlling certain aspects of synapse development or function.
Figure 4.

imp13 mutants exhibit subtle structural defects at the NMJ. A–D, Examination of NMJ structure, muscle surface, and total number of boutons per unit of muscle surface area in control (A) and imp13164 (B) mutants. Boutons were visualized by anti-Eps15 (blue), anti-FasII (red), and anti-Syt (green) in A and B. Scale bars: A, B, 10 μm. C, D, Bar graphs of total surface of muscles 6 and 7 of abdominal segment A2 (C) and number of synaptic boutons per muscle surface (D) in imp13Ex2, imp13164, imp13177, and rescued animals by either a genomic rescue construct or overexpression of imp13 cDNA in neurons or muscles. The total size of the muscles in the mutants is statistically significantly smaller than in control and rescued animals, but the number of synaptic boutons is similar among the different genotypes. E–J, TEM in imp13Ex2 (control) (E, F), imp13164 (mutant) (G, H), neuronal rescue (I), and muscle rescue (J) animals. F and H are magnifications of the active zones in E and G, respectively, that clearly show the similar distribution of the clustered and docked vesicles in control and mutant animals. AZ, Active zones; m, mitochondria; SSR, subsynaptic reticulum; SV, synaptic vesicles; PSD, postsynaptic density. Arrows in E, G, K, and L span the SSR structure and indicate its thickness. SSR thickness and density were quantified as in the study of Budnik et al. (1996). K–N, Bar graphs of quantification of ultrastructural features in imp13Ex2 (E, F), imp13164 (G, H), imp13177 (not depicted), neuronal rescued (I), and muscle rescued (J) animals. The numbers at the bases of the bars in the bar graphs (C, D, K–N) represent the numbers of samples included in our analysis. For the confocal pictures, the numbers correspond to number of animals (1 set of muscle 6/7 per animal was quantified). For TEM studies, boutons were prepared from three to five animals from each genotype. In K, M, and N, the numbers within the bars correspond to the numbers of boutons examined. In L, the numbers within the bars correspond to the numbers of PSDs analyzed. Ns, Not significant; *p < 0.05.
The basic properties of synaptic transmission at the NMJ are normal at elevated Ca2+ but not at low Ca2+
To examine whether there are any defects in basal neurotransmission in imp13 mutants, we performed electrophysiological recordings at the larval NMJ. As control animals, we used precise excisions of the P-element insertion (imp13Ex2) and animals rescued by the tagged genomic rescue construct (Rescue; imp13164). As mutant animals, we used the severe loss-of-function (or null) excision alleles imp13164 imp13177, either as homozygotes, or in trans to Df(3R)DG2, to avoid background effects.
The passive properties of the muscle membrane, namely input resistance, time constant τ (representative of capacitance), and membrane resting potential, are similar among the different genotypes (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). We analyzed the amplitude and the frequency of mEJPs (“minis”). We did not observe any difference in the amplitude or frequency of the miniature events between control and mutant animals (Fig. 5A–C). Furthermore, the cumulative probability of the mEJP amplitude is identical among all the genotypes tested (Fig. 5D, and data not shown), consistent with the observation that there are no defects in synaptic vesicle size based on the TEM. These data also suggest that there are no defects in loading of synaptic vesicles with neurotransmitters or in postsynaptic receptor clustering or function (supplemental Fig. S6, available at www.jneurosci.org as supplemental material).
Figure 5.

Altered synaptic transmission in imp13 mutants. A, B, Bar graphs of frequency (A) and amplitude (B) of mEJPs in imp13 mutant and control animals. C, Representative traces of mEJP recordings from control imp13Ex2, mutant imp13164, and rescued animals. D, Cumulative probability distribution of the mEJP amplitude (bin size equals 0.5 mV). E, Bar graphs of EJP amplitude at 2 mm [Ca2+]o. EJP amplitude is similar for all genotypes analyzed. F, G, Summary of EJP amplitudes at 0.35 mm [Ca2+]o (F) and 0.5 mm [Ca2+]o (G) showing that neurotransmitter is statistically significantly increased in mutants (red) compared with control and rescued animals (black) (*p < 0.05). The number of the animals used for quantification is depicted at the bases of each bar in the bar graphs. H, Representative traces from control and mutant animals at 2 mm [Ca2+]o. I, Sample traces from control (imp13Ex2), mutant (imp13164), neuronal rescue, and muscle rescue animals at 0.35 mm [Ca2+]o.
To assess whether exocytosis is compromised in the absence of imp13, we first recorded evoked EJP at 2 mm extracellular Ca2+ ([Ca2+]o). We did not observe a difference between mutant and control animals in the amplitude of EJPs under these conditions (Fig. 5E,H), indicating that propagation of the action potentials and exocytosis are not defective in the mutant animals. To estimate basal neurotransmission properties more accurately, we compared the amplitude of EJPs in control and mutant animals at limiting concentrations of extracellular [Ca2+]o. At low [Ca2+]o, i.e., 0.35 and 0.5 mm, we observed that the EJP amplitude is significantly larger in mutant animals compared with controls (Fig. 5F,G,I). This suggests that QC is increased in the mutants at low Ca2+. Interestingly, these defects were rescued during overexpression of imp13 cDNA in muscles but not in neurons (Fig. 5F,G,I), suggesting that imp13 is required in the muscle for controlling neurotransmitter release presynaptically.
Importin 13 functions postsynaptically to control the presynaptic probability of release
The EJP amplitude is significantly larger in mutants when compared with different controls at 0.35 and 0.50 mm [Ca2+]o, suggesting that QC is increased (Fig. 6A,B). To determine whether elevated neurotransmitter release in the mutants is attributable to an altered cooperativity of release (Dodge and Rahamimoff, 1967; Jan and Jan, 1976; Stewart et al., 1994), we compared QC in mutants and control animals. Calcium cooperativity plots show that the slope of calcium sensitivity is very similar in mutant and control animals and close to the values reported previously (Stewart et al., 1994) (Fig. 6B), suggesting that there are no alterations in the properties of the presynaptic Ca2+ sensing machinery (Koh and Bellen, 2003).
Figure 6.
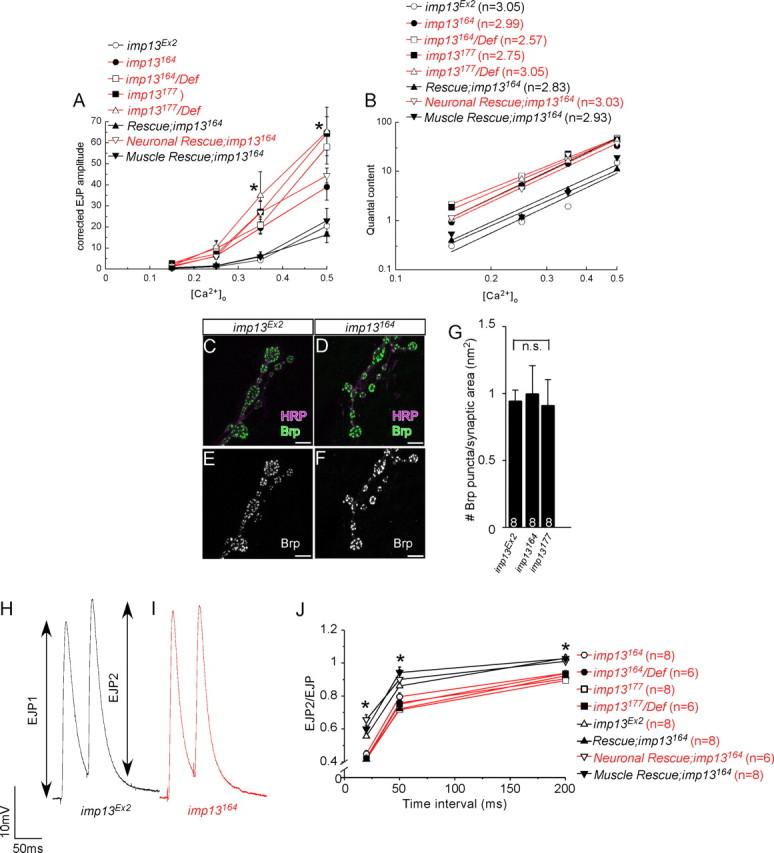
imp13 mutations trigger a retrograde signal that increases presynaptic neurotransmitter release. A, Graph of EJP amplitudes, corrected for nonlinear summation according to Stevens (1976), in mutant (red) and control (black) animals at various concentrations of [Ca2+]o (*p < 0.05, statistically significant difference between mutant and control animals,). For the 0.35 and 0.5 mm concentrations of [Ca2+]o, the number of the animals used in our quantification is the same with the one shown at the bases of the bars of the bar graphs in Figure 5, F and G. For the 0.15 and 0.25 mm concentrations of [Ca2+]o, we did not find any differences among genotypes (6–8 animals assessed from each genotype). B, Linear plot of log(QC) versus log[Ca2+] shows that QC is increased in imp13 mutants (red) compared with control animals (black), without affecting sensitivity to Ca2+ (cooperativity values n are shown on the top left of the graph). Quantal content was calculated from recordings from animals shown in Figures 5, F and G, and 6A. C–F, Visualization of the number of active zones by anti-Brp in control (C, D) and mutant (E, F) animals. G, Quantification of Brp-positive punctae in D–G suggests that the increase in presynaptic release in the mutants is not attributable to additional sites of release. The number of the animals used for quantification is depicted at the bases of each bar in the bar graphs. H, I, Sample traces from imp13Ex2 (control, E, black) and imp13164 (mutant, G, red) animals from paired-pulse stimulation at 200 ms intervals. J, Diagram of the paired-pulse ratio for time intervals 20, 50, and 200 ms at 2 mm extracellular Ca2+ reveals that synaptic depression is more profound in mutants (red) compared with control animals (black), confirming that the probability of release is higher in the absence of imp13. Depression of the EJP amplitude is rescued by overexpression of Imp13 in muscles, but not neurons, of the mutants, suggesting that Imp13 functions in the muscle to control presynaptic probability of release. The number of the animals used for quantification is depicted next to each genotype.
The QC is proportional to the number of release sites and the probability of release. Based on the observation that the number of active zones, visualized as punctae positive for Bruchpilot (Brp), an active zone marker (Kittel et al., 2006; Wagh et al., 2006), are similar between mutants and controls (Figs. 4K, 6C–G), we conclude that the probability of release is elevated in imp13 mutants.
If the probability of release is higher in imp13 mutants when compared with control animals, then paired-pulse stimulation (PPS) at saturating levels of [Ca2+]o (2 mm) should lead to a more extensive depletion of the synaptic vesicle pool and a greater synaptic depression of the EJP amplitude in mutant animals compared with controls. We therefore performed PPS at three different time intervals (20, 50, and 200 ms) and observed a significant depression in mutant animals when compared with controls (Fig. 6H–J). Because PPS represents a measure of presynaptic release (Zucker and Regehr, 2002; Futai et al., 2007), we conclude imp13 mutants exhibit a higher release of neurotransmitter than wild-type animals without affecting the number of release sites.
We then asked whether the PPS phenotype can be rescued by overexpression of Imp13 presynaptically or postsynaptically. Overexpression of Imp13 in the muscles, but not in the neurons, of mutant animals rescues the PPS phenotype for all time intervals examined (Fig. 6J). We conclude that imp13 is primarily required in muscles to regulate presynaptic release and that the increased presynaptic release in imp13 mutants is a result of a synaptic homeostatic response to muscular defects.
Loss of function of imp13 causes a presynaptic increase in intracellular Ca2+
At the Drosophila NMJ, the pore forming a1-subunit of the presynaptic CaV2.1 calcium channel is necessary for homeostasis (Frank et al., 2006). In addition, an elevated probability of release is frequently associated with high levels of intracellular Ca2+ at synaptic terminals. To assess whether intracellular Ca2+ ([Ca2+]i) is increased at rest in imp13 mutants compared with control animals, we measured the ratio of fluorescence that is emitted after forward filling the terminals with fura-2 dextran at two different excitation wavelengths, 340 and 380 nm. Ca2+-free dye absorbs optimally at 380 nm, whereas Ca2+-bound dye is excited primarily at 340 nm. The increased F340/F380 ratio in the mutant animals (imp13164) is statistically significant when compared with controls (Fig. 7), indicating that there are subtle but significant increased levels of intracellular Ca2+ levels within the boutons of the mutants.
Figure 7.

Levels of intracellular Ca2+ are increased within neuronal terminals. A–F, Analysis of [Ca2+]i within synaptic boutons in control imp13Ex2 (A, D), mutant imp13164 (B, E), and rescued (C, F) animals. A–C, Representative readouts of excitation of fura-2 dextran at 340 nm (F340 nm, calcium bound); D–F, representative images of readouts of excitation of fura-2 dextran at 380 nm (F380 nm, calcium free). G, Quantification of F340 nm/F380 nm in control and mutant animals. The numbers on the bars indicate the number of animals/the total number of synaptic areas (each including a minimum of 3 boutons) included in our quantification. *p < 0.05.
Discussion
In a forward genetic screen designed to identify novel genes implicated in neurotransmission, we isolated mutations in CG7212, which encodes the Drosophila homolog of imp13, a member of the importin β protein family (Mingot et al., 2001; Artero et al., 2003; Quan et al., 2008). The importin β family (otherwise named karyopherin β) are an evolutionarily conserved set of proteins that mediate the majority of nucleocytoplasmic shuttling of a wide variety of cargo molecules within the cell, through both a classical and nonclassical pathway (Harel and Forbes, 2004; Mosammaparast and Pemberton, 2004; Conti et al., 2006; Cook et al., 2007). The classical nuclear localization pathway is involved in both developmental and functional processes in neurons, such as neuronal connectivity (Ting et al., 2007), relay of a neuronal injury signal from the distal axons to the nucleus (Hanz et al., 2003; Yudin et al., 2008), and in long-term synaptic plasticity (Thompson et al., 2004).
Imp13 is an atypical importin, which has been shown to interact with a plethora of different substrates, including transcription factors, posttranslational modifiers, and RNA binding proteins (Mingot et al., 2001; Ploski et al., 2004; Kahle et al., 2005; Tao et al., 2006; Yamaguchi et al., 2006; Shoubridge et al., 2007; Liang et al., 2008). However, the reported interactions are based on biochemical or cell culture studies, yet the in vivo role of imp13 in eukaryotes remains to be established. In the present study, we identified loss-of-function alleles of imp13 and discovered that its loss affects synapse function. In fact, despite the abundance of the documented molecular interactions of Imp13, we find that imp13 mutations confer surprisingly specific phenotypes in the visual system of the adult fly and at the larval NMJ.
According to a recent phylogenetic analysis, the importin β protein family in Drosophila consists of a total of 16 members (Quan et al., 2008). Imp13 and TRN-SR (the fly homolog of TNPO3) appear to have evolved from the same ancestral gene, similar to the yeast MTR10, through gene duplication and subsequent diversification (Quan et al., 2008). The common evolutionary roots of Imp13 and TRN-SR may underlie a certain degree of functional redundancy or compensation.
The numerous reported Imp13 interactors may not represent the full range of Imp13 substrates. Still, they emphasize a common characteristic among importin β family members, namely their functional diversity, which is conferred by their structural flexibility (Mosammaparast and Pemberton, 2004; Conti et al., 2006). Distinct sets of protein domains have been identified to interact with various importins (Mosammaparast and Pemberton, 2004, their Fig. 1). Hence, different functions may be served by each importin and is therefore possible that distinct phenotypes will be associated with loss of function of different importins. For example, hypomorphic mutations of dcas (the fly homolog of CSE1L) phenocopy gain of function of Notch signaling during cell fate specification of the external mechanosensory organs on the thorax (Tekotte et al., 2002). In addition, in the developing eye of Drosophila, overexpression of dominant-negative forms of human importin β disrupt axonal guidance and positioning of the photoreceptors, which are further deteriorated by removal of one copy of Ketel (the fly homolog of importin β) (Kumar et al., 2001). We do not observe these defects in the visual system of imp13 mutants. It is therefore likely that the specificity of the phenotypes in imp13 mutants is caused by the spatiotemporal requirements of imp13 function and more importantly, the nature of its substrate.
In the visual system of Drosophila, imp13 is required presynaptically, in the photoreceptors, to control the phototransduction cascade and transmission of the light stimuli, without affecting earlier aspects of development such as cell fate determination or axonal pathfinding. At the ultrastructural level, R1–R6 photoreceptors contain normal numbers of synapses, mitochondria, and capitate projections, but the morphology of the capitate projections is aberrant. Because capitate projections are sites of endocytosis and their morphology is affected in some endocytic and exocytic mutants (Fabian-Fine et al., 2003; Ohyama et al., 2007), imp13 may regulate the distribution and/or function of endocytic or exocytic proteins as well as proteins that mediate the phototransduction cascade.
To investigate whether imp13 does indeed regulate synaptic transmission, we focused on the Drosophila NMJ. The loss of imp13 does not result in major changes of the development of the NMJ. However, imp13 controls synapse function by regulating presynaptic neurotransmitter release and quantal content. Importantly, imp13 is required postsynaptically (in the muscle), suggesting that imp13 functions in the context of synaptic homeostasis. i.e., adjustment of the neuronal output.
To analyze how imp13 is involved in synaptic homeostasis, we first examined the composition of the postsynaptic glutamate receptors, because they are important for the appropriate control of neurotransmitter release (DiAntonio et al., 1999). DGluRIIA is one of the nonessential subunits of the postsynaptic glutamate receptors that play a pivotal role in synaptic homeostasis. In GluRIIA null mutants, the amplitude of mEJPs is severely reduced, but the evoked response of the muscles is similar to control animals, suggesting that more synaptic vesicles are released to compensate for the muscle defect (Petersen et al., 1997). However, during loss of function of only GluRIIB nonessential subunits, there is compensation through synaptic homeostasis without any effect on the amplitude of mEJPs (DiAntonio et al., 1999). In imp13 mutants, we find that the amplitude and frequency of the mEJPs are similar to control animals. As expected, the levels and localization of GluRIIA are not altered. In addition, GluRIIB nonessential subunits are also correctly expressed and localized. Hence, the composition of the postsynaptic receptors is not the cause of the homeostatic response in imp13 mutants.
An additional mechanism at the NMJ that is responsible for a homeostatic neuronal response is dependent on chronic hyperpolarization of the muscle. However, neither the resting potential nor the passive properties of membrane of the muscles are significantly different in mutant and control animals, indicating that chronic hyperpolarization is not the cause of synaptic homeostasis in imp13 mutants.
Another interesting mechanism of triggering synaptic homeostasis is the postsynaptic inhibition of CaMKII (Haghighi et al., 2003). Under these conditions, the amplitude of mEJP is not affected, but the evoked response is higher than normal, leading to an increase in quantal content. It remains to be determined whether CaMKII plays a role in imp13-mediated homeostasis.
Disruption of dystrophin (dys), a member of the Dystrophin–glycoprotein complex, also sparks synaptic homeostasis at the Drosophila NMJ by affecting the amplitude of EJPs but not the amplitude of mEJPs (van der Plas et al., 2006). Interestingly, in dys mutants, the composition of GluRIIA and GluRIIB glutamate receptor subunits is not altered, the number of active zones is unaffected, the size of the releasable pool is not changed, but the probability of release is increased (van der Plas et al., 2006). These aspects of the phenotype are similar to our observations in imp13 mutants. We therefore attempted to assess the levels of Dystrophin in imp13 mutants, albeit unsuccessfully.
We also report that there are postsynaptic abnormalities in the formation of SSR in imp13 mutants. However, SSR formation may not be the primary cause for the abnormalities in neurotransmitter release in imp13 mutants, because there is no apparent correlation between the development of the SSR and synaptic transmission. For example, mutations in dlg or overexpression of Par-1 lead to a significant reduction of SSR yet, contrary to the imp13 mutants, they exhibit a severe reduction in synaptic transmission (Zhang et al., 2007). In straightjacket mutants, in which SSR formation is also affected, synaptic transmission and quantal content is reduced, unlike in imp13 mutants (Ly et al., 2008). Finally, in dpix mutants, in which the SSR is almost entirely absent, the EJPs are only reduced by 10% (Parnas et al., 2001). Therefore, the SSR defects in imp13 mutants indicate a role for imp13 in the muscle during synapse development, but they may not contribute to the electrophysiological defects that we observe in imp13 mutants.
Presynaptically, synaptic homeostasis is accompanied by structural and functional alterations. For example, under conditions of postsynaptic inhibition of CaMKII or in GluRIIA null mutants, the number of active zones is increased to facilitate increased presynaptic release (Haghighi et al., 2003). However, presynaptic release does not only depend on the number of release sites but also on the size of the releasable pool and the probability of release of synaptic vesicles. Our data indicate that the number of active zones in imp13 mutant and control animals is similar and that there are no changes in the presynaptic ultrastructure with respect to the size, density, and subcellular distribution of synaptic vesicles near the active zones. This suggests that the probability of release is increased. The increase in probability of release can be most easily explained by the observed increase in [Ca2+]i levels in imp13 mutant presynaptic terminals. Thus, mechanisms that regulate intracellular Ca2+ levels at the nerve terminal are tightly linked with the phenomenon of synaptic homeostasis, as previously suggested by presynaptic loss-of-function mutations in the voltage-gated channel cacophony (Frank et al., 2006).
We propose that imp13 regulates muscle properties. When these properties are disturbed during chronic loss of imp13 function, mechanisms that mediate the communication between postsynaptic and presynaptic compartments trigger synaptic homeostasis. In that respect, imp13 might be indirectly linked with synaptic homeostasis.
Synaptic homeostasis plays an important role during development as well as in diseases such as myasthenia gravis by coupling neuronal activity to synaptic efficacy and membrane excitability (Davis and Bezprozvanny, 2001; Davis, 2006). Altered kinetics of neurotransmission have also been implicated in models of muscular dystrophy disorders (van der Plas et al., 2006; Bogdanik, 2008; Wairkar, 2008). Therefore, the identification of imp13 as a novel factor in synaptic homeostasis provides the opportunity to gain additional insight in the mechanistic basis of retrograde regulation of presynaptic release.
Footnotes
Confocal microscopy was supported by the Baylor College of Medicine Mental Retardation and Developmental Disabilities Research Center. N.G. was supported by a long-term European Molecular Biology Organization postdoctoral fellowship and the Howard Hughes Medical Institute. H.J.B. is a Howard Hughes Medical Institute investigator. We thank P. R. Hiesinger for the identification of the 3R23 complementation group, his expert advice on the visual system experiments, and comments on this manuscript. We thank R. Atkinson for help with confocal microscopy, Y. He and H. Pan for technical help and injections, and K. Schulze for help with this manuscript. We thank M. Jaiswal, C. V. Ly, T. Ohyama, S. Yamamoto, C.-K. Yao, and A.-C. Tien for helpful discussions. We thank C.-H. Lee, A. DiAntonio, J. Noordermeer, L. G. Fradkin, K. Broadie, and V. Budnik for sharing fly stocks and antibodies. We thank the Bloomington Stock Center at the University of Indiana, Bloomington, the Szeged Drosophila Stock Center in Szeged, Hungary, and the Developmental Studies Hybridoma Bank at the University of Iowa.
References
- Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- Artero R, Furlong EE, Beckett K, Scott MP, Baylies M. Notch and Ras signaling pathway effector genes expressed in fusion competent and founder cells during Drosophila myogenesis. Development. 2003;130:6257–6272. doi: 10.1242/dev.00843. [DOI] [PubMed] [Google Scholar]
- Babcock MC, Stowers RS, Leither J, Goodman CS, Pallanck LJ. A genetic screen for synaptic transmission mutants mapping to the right arm of chromosome 3 in Drosophila. Genetics. 2003;165:171–183. doi: 10.1093/genetics/165.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazigou E, Apitz H, Johansson J, Lorén CE, Hirst EM, Chen PL, Palmer RH, Salecker I. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell. 2007;128:961–975. doi: 10.1016/j.cell.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Bogdanik L, Framery B, Frolich A, Franco B, Mornet D, Bockaert J, Sigrist SJ, Grau Y, Parmentier ML. Muscle dystroglycan organizes the postsynapse and regulates presynaptic neurotransmitter release at the Drosophila neuromuscular junction. PLoS ONE. 2008;3:e2084. doi: 10.1371/journal.pone.0002084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 1996;17:627–640. doi: 10.1016/s0896-6273(00)80196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Campanella JJ, Bitincka L, Smalley J. MatGAT: an application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics. 2003;4:29. doi: 10.1186/1471-2105-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casso D, Ramírez-Weber F, Kornberg TB. GFP-tagged balancer chromosomes for Drosophila melanogaster. Mech Dev. 2000;91:451–454. doi: 10.1016/s0925-4773(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Chou TB, Noll E, Perrimon N. Autosomal P[ovoD1] dominant female-sterile insertions in Drosophila and their use in generating germ-line chimeras. Development. 1993;119:1359–1369. doi: 10.1242/dev.119.4.1359. [DOI] [PubMed] [Google Scholar]
- Conti E, Müller CW, Stewart M. Karyopherin flexibility in nucleocytoplasmic transport. Curr Opin Struct Biol. 2006;16:237–244. doi: 10.1016/j.sbi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Cook A, Bono F, Jinek M, Conti E. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem. 2007;76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- Coombe PE, Heisenberg M. The structural brain mutant vacuolar medulla of Drosophila melanogaster with specific behavioral defects and cell degeneration in the adult. J Neurogenet. 1986;3:135–158. doi: 10.3109/01677068609106845. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, Bezprozvanny I. Maintaining the stability of neural function: a homeostatic hypothesis. Annu Rev Physiol. 2001;63:847–869. doi: 10.1146/annurev.physiol.63.1.847. [DOI] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci. 1999;19:3023–3032. doi: 10.1523/JNEUROSCI.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Verstreken P, Hiesinger PR, Horne JA, Kostyleva R, Zhou Y, Bellen HJ, Meinertzhagen IA. Endophilin promotes a late step in endocytosis at glial invaginations in Drosophila photoreceptor terminals. J Neurosci. 2003;23:10732–10744. doi: 10.1523/JNEUROSCI.23-33-10732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita SC, Zipursky SL, Benzer S, Ferrús A, Shotwell SL. Monoclonal antibodies against the Drosophila nervous system. Proc Natl Acad Sci U S A. 1982;79:7929–7933. doi: 10.1073/pnas.79.24.7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futai K, Kim MJ, Hashikawa T, Scheiffele P, Sheng M, Hayashi Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95-neuroligin. Nat Neurosci. 2007;10:186–195. doi: 10.1038/nn1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- Harel A, Forbes DJ. Importin beta: conducting a much larger cellular symphony. Mol Cell. 2004;16:319–330. doi: 10.1016/j.molcel.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol. 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle J, Baake M, Doenecke D, Albig W. Subunits of the heterotrimeric transcription factor NF-Y are imported into the nucleus by distinct pathways involving importin beta and importin 13. Mol Cell Biol. 2005;25:5339–5354. doi: 10.1128/MCB.25.13.5339-5354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Koh TW, Bellen HJ. Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci. 2003;26:413–422. doi: 10.1016/S0166-2236(03)00195-4. [DOI] [PubMed] [Google Scholar]
- Koh TW, Korolchuk VI, Wairkar YP, Jiao W, Evergren E, Pan H, Zhou Y, Venken KJ, Shupliakov O, Robinson IM, O'Kane CJ, Bellen HJ. Eps15 and Dap160 control synaptic vesicle membrane retrieval and synapse development. J Cell Biol. 2007;178:309–322. doi: 10.1083/jcb.200701030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar JP, Wilkie GS, Tekotte H, Moses K, Davis I. Perturbing nuclear transport in Drosophila eye imaginal discs causes specific cell adhesion and axon guidance defects. Dev Biol. 2001;240:315–325. doi: 10.1006/dbio.2001.0468. [DOI] [PubMed] [Google Scholar]
- Lee CH, Herman T, Clandinin TR, Lee R, Zipursky SL. N-cadherin regulates target specificity in the Drosophila visual system. Neuron. 2001;30:437–450. doi: 10.1016/s0896-6273(01)00291-4. [DOI] [PubMed] [Google Scholar]
- Liang J, Ke G, You W, Peng Z, Lan J, Kalesse M, Tartakoff AM, Kaplan F, Tao T. Interaction between importin 13 and myopodin suggests a nuclear import pathway for myopodin. Mol Cell Biochem. 2008;307:93–100. doi: 10.1007/s11010-007-9588-1. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Bellen HJ, Perin MS. Expression of synaptotagmin in Drosophila reveals transport and localization of synaptic vesicles to the synapse. Development. 1993;118:1077–1088. doi: 10.1242/dev.118.4.1077. [DOI] [PubMed] [Google Scholar]
- Ly CV, Yao CK, Verstreken P, Ohyama T, Bellen HJ. straightjacket is required for the synaptic stabilization of cacophony, a voltage-gated calcium channel alpha1 subunit. J Cell Biol. 2008;181:157–170. doi: 10.1083/jcb.200712152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod GT, Hegström-Wojtowicz M, Charlton MP, Atwood HL. Fast calcium signals in Drosophila motor neuron terminals. J Neurophysiol. 2002;88:2659–2663. doi: 10.1152/jn.00515.2002. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Suster ML, Charlton MP, Atwood HL. Single neuron activity in the Drosophila larval CNS detected with calcium indicators. J Neurosci Methods. 2003;127:167–178. doi: 10.1016/s0165-0270(03)00127-4. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SQ, Hiesinger PR, Beronja S, Zhai RG, Schulze KL, Verstreken P, Cao Y, Zhou Y, Tepass U, Crair MC, Bellen HJ. Mutations in Drosophila sec15 reveal a function in neuronal targeting for a subset of exocyst components. Neuron. 2005;46:219–232. doi: 10.1016/j.neuron.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Mingot JM, Kostka S, Kraft R, Hartmann E, Görlich D. Importin 13: a novel mediator of nuclear import and export. EMBO J. 2001;20:3685–3694. doi: 10.1093/emboj/20.14.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosammaparast N, Pemberton LF. Karyopherins: from nuclear-transport mediators to nuclear-function regulators. Trends Cell Biol. 2004;14:547–556. doi: 10.1016/j.tcb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Verstreken P, Ly CV, Rosenmund T, Rajan A, Tien AC, Haueter C, Schulze KL, Bellen HJ. Huntingtin-interacting protein 14, a palmitoyl transferase required for exocytosis and targeting of CSP to synaptic vesicles. J Cell Biol. 2007;179:1481–1496. doi: 10.1083/jcb.200710061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak WL, Grossfield J, White NV. Nonphototactic mutants in a study of vision of Drosophila. Nature. 1969;222:351–354. doi: 10.1038/222351a0. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Ploski JE, Shamsher MK, Radu A. Paired-type homeodomain transcription factors are imported into the nucleus by karyopherin 13. Mol Cell Biol. 2004;24:4824–4834. doi: 10.1128/MCB.24.11.4824-4834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Y, Ji ZL, Wang X, Tartakoff AM, Tao T. Evolutionary and transcriptional analysis of karyopherin beta superfamily proteins. Mol Cell Proteomics. 2008;7:1254–1269. doi: 10.1074/mcp.M700511-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossano AJ, Macleod GT. Loading Drosophila nerve terminals with calcium indicators. J Vis Exp. 2007;2007:250. doi: 10.3791/250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder E, Blows F, Ashburner M, Bautista-Llacer R, Coulson D, Drummond J, Webster J, Gubb D, Gunton N, Johnson G, O'Kane CJ, Huen D, Sharma P, Asztalos Z, Baisch H, Schulze J, Kube M, Kittlaus K, Reuter G, Maroy P, et al. The DrosDel collection: a set of P-element insertions for generating custom chromosomal aberrations in Drosophila melanogaster. Genetics. 2004;167:797–813. doi: 10.1534/genetics.104.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H. Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science. 1991;254:112–114. doi: 10.1126/science.1681587. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. I. Fasciclin II controls synaptic stabilization and growth. Neuron. 1996;17:641–654. doi: 10.1016/s0896-6273(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Shoubridge C, Cloosterman D, Parkinson-Lawerence E, Brooks D, Gécz J. Molecular pathology of expanded polyalanine tract mutations in the Aristaless-related homeobox gene. Genomics. 2007;90:59–71. doi: 10.1016/j.ygeno.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Stevens CF. A comment on Martin's relation. Biophys J. 1976;16:891–895. doi: 10.1016/S0006-3495(76)85739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol [A] 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- Tahayato A, Sonneville R, Pichaud F, Wernet MF, Papatsenko D, Beaufils P, Cook T, Desplan C. Otd/Crx, a dual regulator for the specification of ommatidia subtypes in the Drosophila retina. Dev Cell. 2003;5:391–402. doi: 10.1016/s1534-5807(03)00239-9. [DOI] [PubMed] [Google Scholar]
- Tao T, Lan J, Lukacs GL, Haché RJ, Kaplan F. Importin 13 regulates nuclear import of the glucocorticoid receptor in airway epithelial cells. Am J Respir Cell Mol Biol. 2006;35:668–680. doi: 10.1165/rcmb.2006-0073OC. [DOI] [PubMed] [Google Scholar]
- Tekotte H, Berdnik D, Török T, Buszczak M, Jones LM, Cooley L, Knoblich JA, Davis I. Dcas is required for importin-alpha3 nuclear export and mechano-sensory organ cell fate specification in Drosophila. Dev Biol. 2002;244:396–406. doi: 10.1006/dbio.2002.0612. [DOI] [PubMed] [Google Scholar]
- Thompson KR, Otis KO, Chen DY, Zhao Y, O'Dell TJ, Martin KC. Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron. 2004;44:997–1009. doi: 10.1016/j.neuron.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Ting CY, Herman T, Yonekura S, Gao S, Wang J, Serpe M, O'Connor MB, Zipursky SL, Lee CH. Tiling of r7 axons in the Drosophila visual system is mediated both by transduction of an activin signal to the nucleus and by mutual repulsion. Neuron. 2007;56:793–806. doi: 10.1016/j.neuron.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- van der Plas MC, Pilgram GS, Plomp JJ, de Jong A, Fradkin LG, Noordermeer JN. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci. 2006;26:333–344. doi: 10.1523/JNEUROSCI.4069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Dürrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wairkar YP, Fradkin LG, Noordermeer JN, DiAntonio A. Synaptic defects in a Drosophila model of congential muscular dystrophy. J Neurosci. 2008;28:3781–3789. doi: 10.1523/JNEUROSCI.0478-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi YL, Tanaka SS, Yasuda K, Matsui Y, Tam PP. Stage-specific Importin13 activity influences meiosis of germ cells in the mouse. Dev Biol. 2006;297:350–360. doi: 10.1016/j.ydbio.2006.04.465. [DOI] [PubMed] [Google Scholar]
- Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben-Yaakov K, Hieda M, Yoneda Y, Twiss JL, Fainzilber M. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron. 2008;59:241–252. doi: 10.1016/j.neuron.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, Greenbaum MP, Bellen HJ. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci U S A. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Sweezey NB, Gagnon S, Muskat B, Koehler D, Post M, Kaplan F. A novel karyopherin-beta homolog is developmentally and hormonally regulated in fetal lung. Am J Respir Cell Mol Biol. 2000;22:451–459. doi: 10.1165/ajrcmb.22.4.3929. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guo H, Kwan H, Wang JW, Kosek J, Lu B. PAR-1 kinase phosphorylates Dlg and regulates its postsynaptic targeting at the Drosophila neuromuscular junction. Neuron. 2007;53:201–215. doi: 10.1016/j.neuron.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]