

Abstract
Neuroinflammation affects the pathobiology of Alzheimer’s disease (AD). Notably, beta-amyloid (Aβ) deposition induces microglial activation and the subsequent production of pro-inflammatory neurotoxic factors. In maintaining brain homeostasis, microglia plasticity also enables phenotypic transition between toxic and trophic activation states. One important control for such cell activation is through the CC-chemokine ligand 2 (CCL2) and its receptor, the CC-chemokine receptor 2 (CCR2). Both affect microglia and peripheral macrophage immune responses and for the latter, cell ingress across the blood brain barrier. However, how CCL2-CCR2 signaling contributes to AD pathogenesis is not well understood. To this end, we report that CCL2 deficiency influences behavioral abnormalities and disease progression in Aβ precursor protein/presenilin-1 double-transgenic mice. Here, increased cortical and hippocampal Aβ deposition is coincident with the formulation of Aβ oligomers. Deficits in peripheral Aβ clearance and in scavenger, neuroprogenitor and microglial cell functions are linked to deficient Aβ uptake. All can serve to accelerate memory dysfunction. Taken together, these data support a role of CCL2 in innate immune functions relevant to AD pathogenesis.
Keywords: Chemokine, Microglia, Phagocytosis, Memory function, Neurogenesis
1. Introduction
Alzheimer’s disease (AD) is both progressive and debilitating with limited treatment options. Its cause, while incompletely understood, is linked to misfolded protein accumulation and subsequent neurodegeneration (Muchowski, 2002; Selkoe, 2004). In recent years the interplay between immunity and neurodegeneration has gained increased attention in unraveling disease mechanisms and for therapeutic development. Center stage in neuroinflammation is the microglia, a principal immune competent cell of the nervous system responsible for a range of functions affecting brain homeostasis and disease (Ransohoff and Perry, 2009). Microglial neurotoxicity is linked to a broad range of neurodegenerative disorders that include, but not limited to, AD, Parkinson’s disease, multiple sclerosis, stroke, spinal cord and traumatic brain injuries, viral encephalitis and amyotrophic lateral sclerosis. However, the precise role played by microglia in the pathobiology of AD has only recently been addressed (Liu and Hong, 2003). In both animal models and human disease, activated microglia and astrocytes accumulate in AD-associated senile plaques composed of densely packed fibrils of beta-amyloid (Aβ) peptides processed from the β-amyloid precursor protein (APP) (Frautschy et al., 1998; Rogers et al., 1996; Selkoe, 2002). A high level of microglial plasticity enables it to rapidly change phenotype and function (Schwartz et al., 2006). On the one hand, microglia serve to scavenge and eliminate misfolded proteins thereby protecting neurons while also eliciting inflammatory responses. This occurs by the production of pro-inflammatory cytokines, chemokines and other neurotoxic factors. The microglial phenotype that emerges depends on immune responses to misfolded protein aggregates and the brain microenvironment(El Khoury et al., 1998; Lucas et al., 2006; Paresce et al., 1996; Skaper, 2007). All can affect cell migration and differentiation.
The CC-chemokine ligand 2 (CCL2), also known as monocyte chemotactic protein-1, is a β-chemokine responsible for monocyte-macrophage tissue recruitment during infectious and inflammatory events affecting disease processes (Shi and Pamer, 2011). Notably, CCL2 is produced by Aβ-stimulated microglia and astrocytes and a critical part of the neuroinflammatory response (El Khoury et al., 2003; Smits et al., 2002). Its ligand binding receptor is the CC-chemokine receptor 2 (CCR2) (Hickman and El Khoury, 2010; Serbina et al., 2008), which is constitutively expressed on the cell surface (Boddeke et al., 1999; Mack et al., 2001) and regulates cell infiltration into tissues including the brain (Babcock et al., 2003; D’Mello et al., 2009; Izikson et al., 2000). While CCL2-overexpressing AD mice (APP/CCL2 double-transgenic (Tg) mice) show accelerated β-amyloidosis, microgliosis and cognitive impairments (Kiyota et al., 2009a; Yamamoto et al., 2005), CCR2 deficiency also effects disease tempo and progression (El Khoury et al., 2007; Naert and Rivest). Both increased CCL2 signaling and CCR2 deficiency in AD mouse models worsen pathology. While unexpected, an understanding of how this occurs remains pivotal. We reasoned that as CCR2, the receptor for CCL2, CCL2-CCR2 signaling might be a key defining set of microglial activities. To this end, we investigated AD-associated processes linked to CCL2 deficiency. Our data now demonstrate that CCL2-CCR2 signaling affects a range of microglia and neuroprogenitor cell functions that not only influence disease pathobiology, but also may be harnessed towards novel therapeutic developments.
2. Methods
2.1. Transgenic mice
APP mice (Tg2576 strain) expressing the Swedish mutation of human APP695 were obtained from Drs. G. Carlson and K. Hsiao-Ashe through Mayo Medical Venture (Hsiao et al., 1996). Presenilin (PS1) mice overexpressing mutant PS1 (M146L line 6.2) were provided by Dr. K. Duff through University of South Florida (Duff et al., 1996). Both mice were maintained in a B6/129 hybrid background (Kiyota et al., 2009b). CCL2KO mice [B6.129S4-Ccl2tm1Rol/J] were purchased from the Jackson Laboratory, Bar Harbor, ME, USA. Male APP or PS1 mice were crossed with female CCL2KO mice to generate APP/CCL2+/− or PS1/CCL2+/−, followed by back-cross with CCL2KO mice to generate APP/CCL2−/−(KO) or PS1/CCL2KO mice. APP/CCL2KO were crossed with PS1/CCL2KO mice to generate APP/PS1/CCL2KO mice. Non-Tg and APP/PS1 mice were developed in parallel (Kiyota et al., 2009b). All animal studies adhered to the guidelines established by the Institutional Animal Care and Use Committee at University of Nebraska Medical Center.
2.2. Bromodeoxyuridine (BrdU) administration and tissue preparation
BrdU was intraperitoneally injected (50 mg/kg of body weight) twice daily every 12 h for 2.5 days to label proliferating cells (Butovsky et al., 2006). Three weeks after the first BrdU injection, mice were euthanized with isoflurane, blood samples were collected, mice were perfused transcardially with 25 ml of ice-cold PBS, and the brains were rapidly removed. The left hemisphere was immediately frozen in dry ice for biochemical testing. The right hemisphere was immersed in freshly depolymerized 4% paraformaldehyde for 48 hours at 4 °C, and protected by successive 24-hour immersions in 15 and 30% sucrose in 1x PBS. Fixed, cryopreserved brains were sectioned coronally using a Cryostat (Leica, Bannockburn, IL, USA) with serial sections collected and stored at −80 °C for immunohistochemical tests.
2.3. Immunohistochemistry
Immunohistochemistry was performed as described previously (Kiyota et al., 2011) using specific antibodies (Abs) to identify pan-Aβ (rabbit polyclonal, 1:100, Zymed, San Francisco, CA, USA), Iba1 (rabbit polyclonal, 1:1000, Wako, Richmond, VA, USA), and doublecortin (Dcx, goat polyclonal, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunodetection was visualized using biotin-conjugated anti-rabbit or anti-goat IgG was used as a secondary Ab, followed by a tertiary incubation with Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA, USA). 1% thioflavin S in 50% EtOH was used for counterstaining of compact plaque (Sigma, St. Louis, MO, USA). For immunofluorescence, sections were co-incubated with FITC-conjugated anti-BrdU (mouse monoclonal, 6 μg/ml, Roche Diagnostics, Indianapolis, IN, USA), biotin-conjugated anti-NeuN (mouse monoclonal, 1:500, Millipore, Billerica, MA, USA), followed by incubation with Streptavidin-Alexa Fluor®568 (1:1000, Invitrogen, Carlsbad, CA, USA). For quantification analysis, the areas of Aβ loads and thioflavin S-positive (TS+) plaques were analyzed by Nuance EX multispectral imaging system (Cambridge Research & Instruments, Woburn, MA, USA) at 300 μm intervals in ten 30 μm coronal sections from each mouse. Five brains of mice per group were analyzed. The numbers of Iba1-positive microglia were quantified using an image analysis software (ImageJ, NIH, Bethesda, MD, USA). The numbers of Dcx+ and BrdU+/NeuN+ cells per dentate gyrus were calculated by stereology based on the Cavalieri principle (West, 1999). Ten 30 μm coronal sections at 300 μm intervals from each mouse were used for the calculation. Five brains of mice per group were analyzed (Kiyota et al., 2011).
2.4. Immunoprecipitation, immunoblots and ELISA tests
Protein extraction of extracellular, intracellular and membrane-enriched fractions and immunoblot tests were performed as described (Lesne et al., 2006). For Aβ immunoblotting, 250 μg of protein/brain sample were incubated with unconjugated pan-Aβ monoclonal Ab (6E10, 2 μg/ml, Covance, Emeryville, CA, USA) in TNE buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 2 mM EDTA, 1 mM PMSF, 1x protease inhibitor cocktail (Sigma, St. Louis, MO, USA)) at 4 ºC for 1h, followed by incubation with 40 μl of Protein A/G Plus agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 ºC for overnight. Precipitants were collected by centrifugation at 3000 rpm, 4 ºC for 5 min, reconstituted with sample buffer, incubated at 95 ºC for 5 min, and then analyzed by electrophoresis on 16% SDS-polyacrylamide Tris-Tricine gels (Schagger, 2006) and electroblotted to 0.2-μm pore size polyvinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories, Richmond, CA, USA, USA). Membranes were blocked in 3% BSA/TBST (Tris-Buffered Saline-Tween 20), and incubated with biotinylated 6E10 monoclonal Ab (1:1000, Covance, Emeryville, CA, USA), followed by incubation with HRP-conjugated streptavidin (Thermo Scientific, Rockford, IL, USA). For the other immunoblotting, membrane-enriched protein samples were run on Tris-Glycine polyacrylamide gel and electroblotted to 0.45-μm pore size PVDF membranes (Immobilon-P, Millipore, Billerica, MA, USA), followed by incubation with CD36, CD47 rabbit polyclonal (1:300, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or β-actin monoclonal Ab (Sigma, St. Louis, MO, USA). HRP-conjugated anti-rabbit or mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was used as a secondary Ab. Immunoreactive bands were detected with SuperSignal West Pico Chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA) and quantitatively analyzed by normalizing band intensities relative to controls on scanned films by ImageJ software (NIH, Bethesda, MD, USA). Protein extraction and Aβ42 ELISA was performed as described (Kiyota et al., 2011).
2.5. Microglia cultivation and Aβ phagocytosis assays
Mouse primary microglia was cultured as described previously (Kiyota et al., 2009a). Five x 104 microglial cells per well (in 96-well plate for immunofluorescence and microplate reading) were seeded for 1 day in Dulbecco’s modified eagle medium (DMEM) supplemented with heat-inactivated 10% fetal bovine serum (FBS), 50 μg/ml penicillin/streptomycin (all from Invitrogen, Carlsbad, CA, USA) and macrophage colony stimulating factor (MCSF, a generous gift from Pfizer Pharmaceuticals, Cambridge, MA). Aβ1–42 (Aβ42) peptide was commercially purchased (Invitrogen, Carlsbad, CA, USA). The Aβ42 was dissolved in cold hexafluoro-2-propanol (HFIP), and incubated at room temperature for at least 1 hour to establish monomerization. The HFIP was removed by evaporation, and the Aβ42 peptide was stored as a film at −20 °C. For oligomer, Aβ42 was dissolved in anhydrous DMSO at 5 mM, diluted into 100 μM in PBS, and then incubated for 24 h at 4 °C. For monomer, Aβ42 was dissolved in anhydrous DMSO diluted in DMEM, and then immediately used for treatment. Microglia were incubated with 10 μM Aβ for 1 h, washed with PBS three times, and then fixed with freshly depolymerized 4% paraformaldehyde for 15 min for immunofluorescence. Standard immunofluorescence was performed using pan-Aβ (rabbit polyclonal, 1:100) and Alexa Fluor®488-conjugated anti-mouse IgG (1:1000), followed by counterstaining with Hoechst 33342 (all from Invitrogen, Carlsbad, CA, USA). Fluorescent intensities were measured by SpectraMAX M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at excitation and emission wavelengths (Ex/Em) of 488/519 nm for Alexa Fluor®488 and 350/461 nm for Hoechst 33342.
2.6. Radial arm water maze test
The radial arm water maze (RAWM) task was run as described with minor modifications (Arendash et al., 2001; Kiyota et al., 2009a). Animals were introduced into the perimeter of a circular water-filled tank 110 cm in diameter and 91 cm in height (San Diego Instruments, San Diego, CA) with triangular inserts placed in the tank to produce six swim paths radiating from a central area. Spatial cues for mouse orientation were present on the tank walls. At the end of one arm, a 10 cm circular plexiglass platform was submerged 1 cm deep and as such hidden from the mice. The platform was located in the same arm for four consecutive acquisition trials (T1 through T4), and one 30-min delayed retention trial (T5), but in a different arm on different days. For T1–T4, the mouse started the task from a different randomly chosen arm, excluding the arm with the platform. After four trials, the mouse was returned to its cage for 30 min, and then administered the retention trial (T5) starting from the same arm as in T4. Each trial lasted 1 minute and an error was scored each time when the mouse, excluding tail, entered the wrong arm, entered the arm with the platform but did not climb on it, or did not make a choice for 20 sec. The trial ended when the mouse climbed onto and remained on the hidden platform for 10 seconds. The mouse was given 20 seconds to rest on the platform between trials. The time taken by the mouse to reach the platform was recorded as its latency. If the mouse did not reach the platform, 60 sec was recorded as its latency and the mouse was gently guided to the submerged platform. The errors over 9-day test were divided into three blocks, and the errors in each block consisting of 3-day test were averaged for statistical analysis.
2.7. Statistics
All data were normally distributed and presented as mean values ± standard errors of the mean (S.E.M.). In the case of single mean comparison, data were analyzed by Student’s t-test. In case of multiple mean comparisons, the data were analyzed by one-way ANOVA and Newman-Keuls post-hoc or two-way repeated measures ANOVA, followed by Bonferroni multiple comparison tests using statistics software (Prism 4.0, Graphpad Software, San Diego, CA). A value of p < 0.05 was regarded as a significant difference.
3. Results
3.1. Accelerated Aβ deposits and oligomers in CCL2-deficient APP/PS1 mice
Aβ deposits develop between 9 to 12 months of age in single Tg2576 strain expressing familial AD mutants of APP (Hsiao et al., 1996). APP/PS1 double-Tg mice, which also overexpress mutant PS1, show accelerated Aβ deposition as early as 5 to 6 months with robust microglial responses (Arendash et al., 2001; Holcomb et al., 1998; Morgan et al., 2000). Notably, β-amyloidosis occurs through oligomer formation that evolves into diffuse and eventually TS+-compact plaques. To examine how CCL2 deficiency affects β-amyloidosis, APP/PS1/CCL2KO mice were used to assess Aβ loads in the cortex and hippocampus (Fig. 1A–D). Total Aβ load, composed of diffuse and compact plaques, was determined by anti-Aβ staining. Aβ load was increased in brains of APP/PS1/CCL2KO mice (154 % of APP/PS1 group in the cortex, p = 0.0304, 175 % of APP/PS1 group in the hippocampus, p = 0.0386, Fig. 1A–B, E). TS+ compact plaques were also increased, but only the increases in TS+ compact plaques were significant in the cortex for the APP/PS1/CCL2KO as compared to APP/PS1 mice (234 % of APP/PS1 group in the cortex, p = 0.0478, 188 % of APP/PS1 group in the hippocampus, p = 0.1880, Fig. 1C–D, F). To quantify Aβ42 levels, brain tissue homogenates were separated into SDS-soluble fractions containing stable oligomers and SDS-insoluble fractions. The latter included fibrillar Aβ. Although insoluble Aβ42 increased (APP/PS1: 8.121 ± 0.319 μg/μg protein versus APP/PS1/CCL2KO: 8.908 ± 0.726 μg/μg protein; p = 0.3701, Fig. 1H), only soluble Aβ42 increases were significant for the APP/PS1/CCL2KO as compared to APP/PS1 mice (APP/PS1: 10.13 ± 1.592 ng/μg protein versus APP/PS1/CCL2KO: 17.44 ± 2.703 ng/μg protein; p = 0.0420, Fig. 1G). Plasma Aβ42 levels were decreased in APP/PS1/CCL2KO animals compared to APP/PS1 mice (APP/PS1: 1.302 ± 0.083 ng/ml plasma vs APP/PS1/CCL2KO: 0.928 ± 0.124 ng/ml plasma; p = 0.0270, Fig. 1I).
Fig. 1.
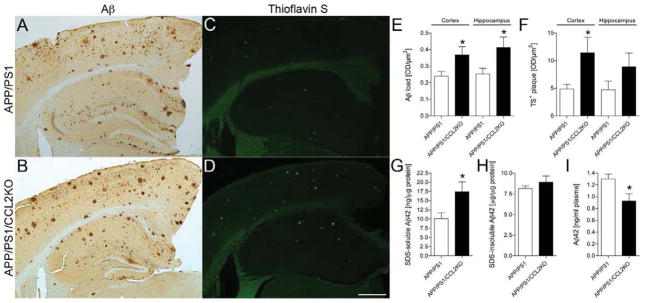
CCL2 deficiency accelerates β-amyloidosis in the brain. (A–D) Representative images of Aβ3 and thioflavin S (TS) staining in the cortex and hippocampus of APP+PS1 and APP/PS1/CCL2KO mice. Scale bar: 500μm. (E–F) Quantification of total Aβ load and TS+ plaque in the cortex and hippocampal regions (n = 5 per group, 10 sections per brain). (G–I) The level of SDS-soluble Aβ42, SDS-insoluble Aβ42 in the brain, and Aβ42 in plasma were measured by human Aβ42 specific ELISA (n = 5). Bars represent mean ± S.E.M. * denotes p < 0.05 as determined by Student’s t-test.
We next quantitated Aβ oligomers by immunoblotting extracellular-enriched mouse brain homogenates (Fig. 2) (Lesne et al., 2006). Although monomers (1-mer) were unchanged between APP/PS1 and APP/PS1/CCL2KO (p = 0.1665, Fig. 2B), trimer (3-mer), dodecamer (12-mer) and 75kDa Aβ (Aβ75) were increased in APP/PS1/CCL2KO mice (148, 191, and 156% of APP/PS1 mice; p = 0.0010, 0.0309 and 0.0082, respectively, Fig. 2B). Aβ oligomeric dimers, tetramers, and hexamers were not observed. These results suggest that forms of soluble Aβ oligomeric species are modulated through CCL2 and as such could contribute to the total brain Aβ load.
Fig. 2.

Aβ oligomer formation in the APP/PS1 or APP/PS1/CCL2KO mouse brain. (A) Extracellular-enriched fractions of the APP/PS1 and APP/PS1/CCL2KO mouse brain tissues were subjected to immunoprecipitation with 6E10 antibody, followed by immunoblotting using biotinylated 6E10 antibody showing specific Aβ oligomers. Arrowheads indicate respective migration positions of monomer and oligomers. The arrow indicates sAPPα fragments processed from full-length APP. A short-term exposed images (5 minutes, top two panels) are shown to provide single bands of sAPPα and 75kDa Aβ (Aβ75), in addition to overnight exposed image (bottom). NTg: non-Tg mouse brain as a negative control. (B) Band luminescent intensities for 1, 3, 12-mers and Aβ75 were quantified by Image J software. Bars represent mean ± S.E.M. (n = 10 per group). *, ** or *** denotes p < 0.05, 0.01 or 0.001 versus APP/PS1, as determined by Student’s t-test.
3.2. Microglia accumulate in areas of Aβ plaques in CCL2-deficient APP/PS1 mice
Aβ peptides and deposition induce reactive microgliosis and subsequent neuroinflammation linked to neuronal injury (Tan et al., 2002). In contrast, microglia also affect Aβ clearance (Bard et al., 2000). To determine if CCL2 deficiency affects microglial accumulation in the APP/PS1/CCL2KO brain, Iba1-immunoreactive cells were quantified (Fig. 3A–C). Numbers of Iba1+ microglia, in both the cortex and hippocampus, were reduced in APP/PS1/CCL2KO mice (APP/PS1: 306.6 ± 13.12/mm2 versus APP/PS1/CCL2KO: 180.4 ± 18.62/mm2 in the cortex; p = 0.0005, APP/PS1: 248.3 ± 14.59/mm2 versus APP/PS1/CCL2KO: 174.1 ± 5.02/mm2 in the hippocampus; p = 0.0013, Fig. 3D). However, the number of Iba1-immunoreactive microglia surrounding TS+ Aβ compact plaques in APP/PS1/CCL2KO mice was increased as compared to APP/PS1 mice (APP/PS1: 2.000 ± 0.377/plaque versus APP/PS1/CCL2KO: 3.350 ± 1.412/plaque; p = 0.0094, Fig. 3E).
Fig. 3.

The total number of microglial cells decreases in the brain but increases around Aβ plaques in CCL2-deficient APP/PS1 mice. (A–B) Representative images of Iba1 staining in the cortex and hippocampus of APP+PS1 and APP/PS1/CCL2KO mice. Scale bar: 500μm. (C) High magnification of Iba1 staining in the APP+PS1 and APP/PS1/CCL2KO mouse brains. (D) Quantification of the average number of Iba1-positive cells per area (mm2) in the APP+PS1 and APP/PS1/CCL2KO mouse brains (n = 5 per group, 10 sections per brain). (E) Representative images of Iba1 staining around TS+ Aβ plaques. The number of Iba1-positive cells surrounding the plaques was quantified (n = 5 per group, 10 sections per brain, 3 plaques/ section). Bars represent mean ± S.E.M. * or ** denotes p < 0.05 or 0.01 as determined by Student’s t-test.
3.3. Aβ phagoyctosis is decreased in CCL2-deficient microglia
Why Aβ oligomers and deposits increase in the brain may be linked to deficits in microglial function. As the cells serve as scavengers (El Khoury et al., 1998; Lucas et al., 2006; Paresce et al., 1996), we investigated whether CCL2 deficiency could affect phagocytic functions. CD36 and CD47 expression was explored by immunoblotting with membrane-enriched brain fractions (El Khoury et al., 2003; Koenigsknecht and Landreth, 2004) (Fig. 4A). While CD36 expression was unchanged in APP/PS1 and APP/PS1/CCL2KO mice (p = 0.0659, Fig. 4B), CD47 expression was reduced in APP/PS1/CCL2KO animals (70.1 % of the APP/PS1 group; p = 0.0113, Fig. 4B). Aβ microglial phagocytosis from non-Tg and CCL2KO mice were tested (Fig. 5A–O). Immunofluorescent levels were measured after treatment of microglia with monomeric or oligomeric Aβ42 for 1 h normalized by nuclear staining (Kiyota et al., 2009a). Interestingly, immunofluorescent intensity, against both monomeric and oligomeric Aβ42, was reduced in CCL2KO, compared to non-Tg microglia (monomeric: 74 % of non-Tg microglia, p < 0.01, oligomeric: 80 % of non-Tg microglia, p < 0.001, Fig. 5P–Q).
Fig. 4.
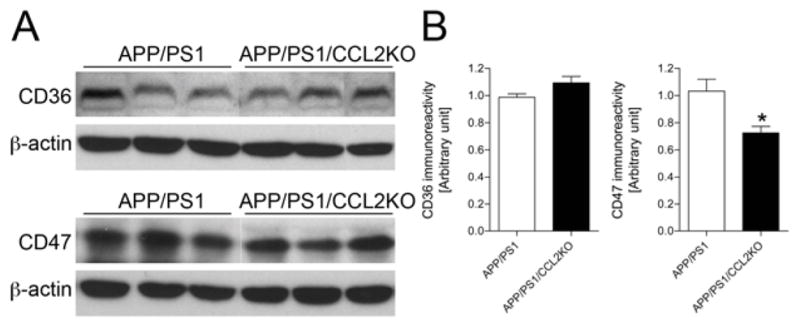
Reduced expression of CD47, but not CD36. (A) Immunoblotting of CD36 and CD47 in membrane-enriched fraction of the mouse brain in APP/PS1 and APP/PS1/CCL2KO mice. (B) Quantification CD36 and CD47 expression. Bars represent mean ± S.E.M. * denotes p < 0.05 as determined by Student’s t-test (n = 6 per group).
Fig. 5.

Impaired phagocytosis of Aβ in CCL2-deficient primary microglia. (A–O) Primary mouse microglia were incubated with monomeric or oligomeric Aβ42 for 1 h, followed by immunofluorescence with anti-Aβ Ab (green) and Hoechst33342 for nuclear staining (blue). Merged captured images were shown. Scale bar, 50 μm. (P–Q) Fluorometric quantification of Aβ fluorescent signal (Ex/Em = 488 nm/519 nm) normalized by Hoechst33342 signal (Ex/Em = 350 nm/461 nm). Bars represent mean ± S.E.M. ** or *** denotes p < 0.01 or 0.001 as determined by one-way ANOVA and Newman–Keuls post-test, respectively (n = 4 per group).
3.4. CCL2 deficiency effects cognitive function
Enhanced β-amyloidosis in AD is disease signature. In particular, oligomeric forms of Aβ accumulate and effect Aβ-induced neurophysiological (Shankar et al., 2008; Walsh et al., 2002), hippocampal (Lambert et al., 1998), and memory functions (Cleary et al., 2005; Lesne et al., 2006). APP/PS1 mice show impaired hippocampal function, memory acquisition and retention. This occurs by 6–7 months of age as seen by RAWM tests (Diamond et al., 1999; Arendash et al., 2001; Jensen et al., 2005; Kiyota et al.,). To assess if CCL2 deficiency affects memory, we employed a RAWM task to assess memory acquisition and retention in non-Tg, CCL2KO, APP/PS1, and APP/PS1/CCL2KO mice. Three 3-day blocks for trial 1 (T1; randomized initial trial), T4 (final acquisition trial), and T5 (delayed retention trial) were used to evaluate the memory function at 6–7 months of age (Fig. 6) (Jensen et al., 2005). Although all animal groups showed reduced error numbers by T4 through three blocks, the average number in non-Tg or CCL2KO was lower than that in APP/PS1 and APP/PS1/CCL2KO mice. By T5, APP/PS1/CCL2KO mice showed the highest number of errors with significant differences compared to the other groups. APP/PS1 group also showed higher numbers than did non-Tg or CCL2KO animals, but lower than APP/PS1/CCL2KO in blocks 1 and 3. While limitations of the set are acknowledged through the KO mice alone, the data do support the idea that impaired memory acquisition and retention are operative in APP/PS1 and APP/PS1/CCL2KO mice and as such CCL2 deficiency can accelerate such impairments in the setting of AD-like disease.
Fig. 6.

CCL2 deficiency accelerates memory impairment in APP+PS1 mice. Non-Tg, CCL2KO, APP/PS1 and APP/PS1/CCL2 mice were tested by the RAWM task at 6–7 months of age. Non-Tg serves as a positive control for the spatial learning task. The compiled average errors for day 1–3, 4–6 and 7–9 are shown. Bars represent mean ± S.E.M. (n = 12 per group). *, ** or *** denotes p < 0.01 or 0.001 as determined by two-way ANOVA and Bonferroni tests.
3.5. Impaired neurogenesis in CCL2-deficient APP/PS1 mice
A number of studies imply correlations between cognitive function and adult neurogenesis in the hippocampal dentate gyrus (Bruel-Jungerman et al., 2007; Deng et al.; Kiyota et al., 2011; Lledo et al., 2006). Since CCL2-deficient APP/PS1 mice show accelerated impairment in memory retention, we reasoned this could be linked to deficits in neurogenesis. To this end, we examined how CCL2 deficiency might affect neurogenesis and differentiation in the subgranular zone (SGZ) of the dentate gyrus. We examined expression of doublecortin (Dcx), a marker for newly generated premature neurons in the SGZ (Fig. 7A–D) (Rao and Shetty, 2004). The numbers of Dcx-positive (Dcx+) cells in the dentate gyrus of APP/PS1 mice was significantly reduced (1003 ± 113.5 versus non-Tg: 1677 ± 255.4; p < 0.01, Fig. 7I). In addition, APP/PS1/CCL2KO mice show reduced numbers of Dcx+ cells compared to APP/PS1 (456.5 ± 77.82 in APP/PS1/CCL2KO; p < 0.001 versus non-Tg and p < 0.05 vs APP/PS1) (Fig. 7I), suggesting impaired neuronal differentiation in APP/PS1/CCL2KO mice. To evaluate cell proliferative responses in the SGZ, BrdU was injected intraperitoneally three weeks prior to euthanasia to track neuronal maturation (NeuN+/BrdU+ cells) (Fig. 7E–H). The total BrdU+/NeuN+ cell counts in the SGZ were decreased in APP/PS1 mice (534.0 ± 50.52 versus non-Tg: 757.5 ± 57.23; p < 0.05) (Fig. 7J), and were further decreased in APP/PS1/CCL2KO as compared to APP/PS1 mice (362.4 ± 18.10 in APP/PS1/CCL2KO; p < 0.001 versus non-Tg and p < 0.01 versus APP/PS1) (Fig. 7J). These data suggest that CCL2 deficiency affects neuronal differentiation and neuronal-linked stem cell proliferation, and also provides a link between alterations in neurogenesis and memory impairment.
Fig. 7.

CCL2 deficiency accelerates impaired hippocampal neurogenesis in APP/PS1 mice. (A–D) Representative images of Dcx staining in the subgranular zone (SGZ) of the dentate gyrus (DG) in non-Tg, CCL2KO, APP/PS1 or APP/PS1/CCL2KO mice. Scale bar: 200μm. (E–H) Mice received i.p. injection with BrdU 3 weeks prior to the euthanasia at 7 months of age. Representative images of immunofluorescence for BrdU (green) and NeuN (red) in SGZ of non-Tg, CCL2KO, APP/PS1 or APP/PS1/CCL2KO mice were shown. Scale bar: 100μm. (I–J) Quantification of Dcx+ or BrdU+Neu+ cells per DG. Bars represent mean ± S.E.M. (n = 5 per group, 10 sections per brain). *, ** or *** denotes p < 0.05, 0.01 or 0.001 versus non-Tg, and # or ## denotes P < 0.05 or 0.01 between APP/PS1 and APP/PS1/CCL2KO groups, as determined by one-way ANOVA and Newman–Keuls post-test.
DISCUSSION
We demonstrate that CCL2 deficiency results in increased Aβ that includes both oligomers and deposits in the brain. This is heralded by the elimination of CCL2-CCR2 signaling affecting microglial chemotaxis as shown by reduced numbers of microglial cells in the brains of CCL2KO mice. The results are consistent with prior reports demonstrating that CCR2 deficiency leads to impaired microglial accumulation and accelerated AD pathobiology (El Khoury et al., 2007; Naert and Rivest, 2011). Although CCL2 is a major chemokine involved in microglial chemotaxis, it is upregulated in AD brain tissue (Ishizuka et al., 1997). In addition, CCL2 levels in cerebrospinal fluid and sera are linked to neurodegeneration as they show a significant negative correlation with cognition in mildly impaired AD patients. This suggests that elevated CCL2 is an early event in AD pathogenesis (Galimberti et al., 2006a; Galimberti et al., 2006b). However, microglia accumulated in brain regions surrounding TS+ Aβ deposits in CCL2KO mice. This may be caused by the other ligands including CCL7 that affect microglial nervous system chemotaxis (Chen et al., 2001).
Microglia profoundly influences Aβ clearance and degradation (Lee and Landreth, 2010). Microglia produce proteolytic enzymes such as insulin-degrading enzyme (IDE) and matrix metalloproteinases (MMPs) for Aβ degradation (Mentlein et al., 1998; Miners et al., 2011) and expressing receptors promoting Aβ phagocytosis and clearance (El Khoury et al., 1996; Koenigsknecht and Landreth, 2004; Weldon et al., 1998). Although expression levels of IDE, MMP9 and neprilysin in APP/PS1/CCL2KO were equivalent in APP/PS1 mice (data not shown), significant impairments in phagocytosis were demonstrated for both monomeric and oligomeric Aβ in CCL2-deficient microglia. This linked to reduced expression of CD36 and CD47, scavenger molecules on the MP surface (El Khoury et al., 2003; Koenigsknecht and Landreth, 2004). Taken together, these data suggest that accelerated β-amyloidosis, as shown by Aβ load and soluble Aβ42 levels in CCL2-deficient APP/PS1 mice, is due to impairments of Aβ phagocytosis and clearance. In contrast, CCL2-overexpressing mice showed accelerated AD-pathogenesis including β-amyloidosis. This may be due to elevated apolipoprotein E relevant to Aβ assembly and microglial activation where Aβ aggregates are released (Kiyota et al., 2009a; Yamamoto et al., 2005). Therefore, both CCL2- overexpressing and deficient AD mice may show similar finding by very divergent mechanisms.
Increased Aβ burden is significantly associated with impaired cognition in AD mouse models (Morgan, 2003). We demonstrate that CCL2-deficient APP/PS1 mice show elevated Aβ and impaired memory. However, memory impairments were in retention, but not acquisition. These results, in part, could be linked to the age of the animals studied (Kiyota et al., 2010; Kiyota et al., 2011).
Recent studies have shown significant correlations between memory function and adult neurogenesis in the dentate gyrus of hippocampus (Coras et al., 2010). The enhanced number of mature neurons is especially critical, since the earlier study showed enhanced neuronal division but impaired maturation in AD brain (Jin et al., 2004). In the mouse model of AD, the neurogenesis was impaired in APP/PS1 mice after counting the reduced number of Dcx+ and BrdU+/NeuN+ cells as shown previously (Kiyota et al., 2011; Kiyota et al., 2012), but the even fewer number of the cells was found in APP/PS1/CCL2KO mice, demonstrating that the CCL2-deficiency accelerates impairment of the neurogenesis that is closely associated with improvement in spatial learning. Although the effect of Aβ species in different aggregated forms on neurogenesis has been a matter of debate (Waldau and Shetty, 2008), we have shown that increased Aβ oligomer species are significantly correlated with impaired neurogenesis. In accord with these results, the impaired neuronal maturation is affected by enhanced β-amyloidosis in CCL2-deficit APP/PS1 mice. This was shown to be correlated with both BrdU+/NeuN+ and Dcx+ cell counts. In addition, single Tg mice expressing PS1 M146 mutant does not affect cell proliferation in standard condition (Choi et al., 2008), and we did not observe any impaired neuronal proliferation and differentiation in CCL2KO mice, suggesting that worsen neurogenesis in APP/PS1/CCL2KO mice is caused by a synergistic effect of PS1 mutant in a CCL2KO background for impaired neurogenesis.
All together, we now demonstrate that CCL2-deficiency results in accelerated β-amyloidosis with increased oligomer species, decreased microglial cell immunostaining, impaired Aβ phagocytic ability, and memory impairment accompanied with reduced neurogenesis in the dentate gyrus of hippocampus. Taken together, these studies support an idea that CCL2 is a key molecule to maintain normal brain function in an animal model of AD, and offers new insights into the disease processes during progressive neurodegeneration.
Acknowledgments
The research was supported by the National Institutes of Health [1P01 DA028555, 2R01 NS034239, 2R37 NS36126, P01 NS31492, P20RR 15635, P30 MH062261, P01MH64570 and P01 NS43985 to H.E.G.], the Carol Swarts Emerging Neuroscience Fund, Start up funds from the Department of Pharmacology and Experimental Neuroscience, the Frani and Louis Blumking Foundation and a research grant from Baxter Healthcare. T.K. is the recipient of the Vada Kinman Oldfield Alzheimer’s Research Prize at the University of Nebraska Medical Center. The authors thank Drs. K. Hsiao-Ashe for providing Tg2576 mice, K. Duff for providing M146L PS1 mice, D. Morgan for RAWM test training and consultation.
Footnotes
Conflict of interest
The authors declare that there is no actual or potential Conflict of interest.
No author has any actual and potential conflict of interest.
The University of Nebraska Medical Center (UNMC) does not have any contracts related to this work.
No author or the UNMC has any financial interest in this work.
The data contained in this manuscript have not been previously published and is not under consideration elsewhere.
Animal procedures were approved by the Institutional Animal Care and Use Committee at the UNMC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891(1–2):42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci. 2003;23(21):7922–7930. doi: 10.1523/JNEUROSCI.23-21-07922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Boddeke EW, Meigel I, Frentzel S, Gourmala NG, Harrison JK, Buttini M, Spleiss O, Gebicke-Harter P. Cultured rat microglia express functional beta-chemokine receptors. J Neuroimmunol. 1999;98(2):176–184. doi: 10.1016/s0165-5728(99)00096-x. [DOI] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Rampon C, Laroche S. Adult hippocampal neurogenesis, synaptic plasticity and memory: facts and hypotheses. Rev Neurosci. 2007;18(2):93–114. doi: 10.1515/revneuro.2007.18.2.93. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103(31):11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol. 2001;167(8):4585–4592. doi: 10.4049/jimmunol.167.8.4585. [DOI] [PubMed] [Google Scholar]
- Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, Sisodia SS. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59(4):568–580. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Coras R, Siebzehnrubl FA, Pauli E, Huttner HB, Njunting M, Kobow K, Villmann C, Hahnen E, Neuhuber W, Weigel D, Buchfelder M, Stefan H, Beck H, Steindler DA, Blumcke I. Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain. 2010;133(11):3359–3372. doi: 10.1093/brain/awq215. [DOI] [PubMed] [Google Scholar]
- D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29(7):2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11(5):339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Heman KL, Rose GM. Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus. 1999;9(5):542–552. doi: 10.1002/(SICI)1098-1063(1999)9:5<542::AID-HIPO8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382(6593):716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 1998;19(1 Suppl):S81–84. doi: 10.1016/s0197-4580(98)00036-0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197(12):1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152(1):307–317. [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corra B, Scalabrini D, Clerici F, Mariani C, Bresolin N, Scarpini E. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer’s disease. Neurobiol Aging. 2006a;27(12):1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Bouwman F, Venturelli E, Guidi I, Blankenstein MA, Bresolin N, Scarpini E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2006b;63(4):538–543. doi: 10.1001/archneur.63.4.538. [DOI] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):168–173. doi: 10.2174/187152710791011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4(1):97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-Yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci. 1997;51(3):135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD. Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med. 2000;192(7):1075–1080. doi: 10.1084/jem.192.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human beta-amyloid (1–42) protects Alzheimer’s transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130(3):667–684. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci USA. 2004;101(36):13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Ingraham KL, Jacobsen MT, Xiong H, Ikezu T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc Natl Acad Sci USA. 2011;108(49):E1339–1348. doi: 10.1073/pnas.1102349108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther. 2012;19(7):724–733. doi: 10.1038/gt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010;24(8):3093–3102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Xiong H, Lambert MP, Klein WL, Gendelman HE, Ransohoff RM, Ikezu T. CCL2 accelerates microglia-mediated Abeta oligomer formation and progression of neurocognitive dysfunction. PLoS One. 2009a;4(7):e6197. doi: 10.1371/journal.pone.0006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Schroder B, Jacobsen MT, Swan RJ, Lambert MP, Klein WL, Gendelman HE, Ransohoff RM, Ikezu T. AAV1/2-mediated CNS Gene Delivery of Dominant-negative CCL2 Mutant Suppresses Gliosis, beta-amyloidosis, and Learning Impairment of APP/PS1 Mice. Mol Ther. 2009b;17(5):803–809. doi: 10.1038/mt.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2007;24(44):9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117(8):949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304(1):1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7(3):179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, Bruhl H, Frink M, Anders HJ, Vielhauer V, Pfirstinger J, Stangassinger M, Schlondorff D. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166(7):4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- Mentlein R, Ludwig R, Martensen I. Proteolytic degradation of Alzheimer’s disease amyloid beta-peptide by a metalloproteinase from microglia cells. J Neurochem. 1998;70(2):721–726. doi: 10.1046/j.1471-4159.1998.70020721.x. [DOI] [PubMed] [Google Scholar]
- Miners JS, Barua N, Kehoe PG, Gill S, Love S. Abeta-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70(11):944–959. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- Morgan D. Learning and memory deficits in APP transgenic mouse models of amyloid deposition. Neurochem Res. 2003;28(7):1029–1034. doi: 10.1023/a:1023255106106. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Abeta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ. Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron. 2002;35(1):9–12. doi: 10.1016/s0896-6273(02)00761-4. [DOI] [PubMed] [Google Scholar]
- Naert G, Rivest S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011;31(16):6208–6220. doi: 10.1523/JNEUROSCI.0299-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17(3):553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19(2):234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P. Inflammation and Alzheimer’s disease pathogenesis. Neurobiol Aging. 1996;17(5):681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- Schagger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1(1):16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29(2):68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann NY Acad Sci. 2007;1122:23–34. doi: 10.1196/annals.1403.002. [DOI] [PubMed] [Google Scholar]
- Smits HA, Rijsmus A, van Loon JH, Wat JW, Verhoef J, Boven LA, Nottet HS. Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol. 2002;127(1–2):160–168. doi: 10.1016/s0165-5728(02)00112-1. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Crawford F, Mori T, DelleDonne A, Crescentini R, Obregon D, Flavell RA, Mullan MJ. Role of CD40 ligand in amyloidosis in transgenic Alzheimer’s mice. Nat Neurosci. 2002;5(12):1288–1293. doi: 10.1038/nn968. [DOI] [PubMed] [Google Scholar]
- Waldau B, Shetty AK. Behavior of neural stem cells in the Alzheimer brain. Cell Mol Life Sci. 2008;65(15):2372–2384. doi: 10.1007/s00018-008-8053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18(6):2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22(2):51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Horiba M, Buescher JL, Huang D, Gendelman HE, Ransohoff RM, Ikezu T. Overexpression of monocyte chemotactic protein-1/CCL2 in beta-amyloid precursor protein transgenic mice show accelerated diffuse beta-amyloid deposition. Am J Pathol. 2005;166(5):1475–1485. doi: 10.1016/s0002-9440(10)62364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]