

Abstract
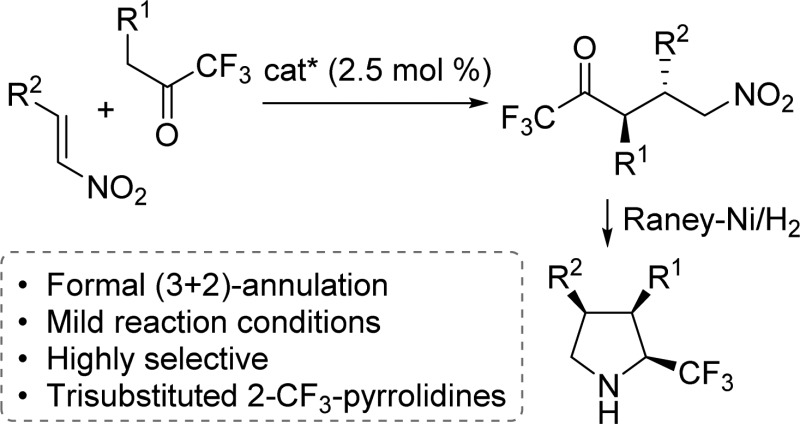
The stereoselective synthesis of trisubstituted 2-trifluoromethyl pyrrolidines by asymmetric Michael addition/hydrogenative cyclization is described. The direct organocatalytic addition of 1,1,1-trifluoromethylketones to nitroolefins proceeds under mild reaction conditions and low catalyst loadings to provide Michael adducts in high yield with excellent diastereo- and enantioselectivity. Catalytic hydrogenation of the Michael adducts stereoselectively generates 2-trifluoromethylated pyrrolidines bearing three contiguous stereocenters. A stereospecific route to epimeric 2-trifluoromethyl pyrrolidines from a common intermediate is described.
This letter describes a simple reaction platform for the enantio- and diastereoselective preparation of 2-trifluoromethyl pyrrolidines. Pyrrolidines are highly desirable building blocks due to their prevalence in the pharmaceutical and agrochemical industries.1 Given the unique physicochemical properties engendered by the trifluoromethyl (CF3) group,2 considerable effort has been directed toward new methodologies for the preparation of CF3-containing compounds.3 A productive merger of these functionalities has led to interest in the development of methodologies for the construction of N-containing organofluorine compounds.4 Consequently, methods toward the generation of optically active 2- and 3-trifluoromethylated pyrrolidines have garnered increasing attention in the literature. A common strategy for the synthesis of 3-trifluoromethylated proline derivatives utilizes the asymmetric 1,3-dipolar cycloaddition of azomethine ylides and CF3-substituted alkenes.5 A majority of the methods for the preparation of optically active 2-trifluoromethylated pyrrolidines rely on the use of chiral starting materials or auxiliaries.6 A more attractive method to generate these 2-trifluoromethylated pyrrolidines would utilize de novo pyrrolidine syntheses to rapidly generate molecular complexity from simple starting materials via asymmetric catalysis.7 Here, we report a formal (3 + 2)-annulation strategy for the preparation of trisubstituted 2-trifluoromethyl pyrrolidines via organocatalytic asymmetric Michael addition of 1,1,1-trifluoromethylketones to nitroolefins followed by diastereoselective reductive cyclization.
A number of highly functionalized pyrrolidines have previously been accessed via asymmetric Michael addition/reductive cyclization protocols employing aldehydes,8 aryl ketones,9 α-keto ester/amides,10 and β-keto esters11 as two-carbon donor synthons with nitroolefins for the de novo synthesis of substituted pyrrolidines. We postulated that an analogous (3 + 2)-annulation approach could be utilized in the preparation of trisubstituted 2-trifluoromethyl pyrrolidines 4 (Scheme 1a). The realization of this approach would require the invention of a heretofore unknown catalytic, enantioselective Michael addition of 1,1,1-trifluoromethylketones 1 to nitroolefins 2 that would provide the requisite γ-nitro carbonyl intermediate 3. An attractive feature of the projected experimental plan was the high level of atom efficiency associated with the catalytic addition/hydrogenative cyclization sequence (Scheme 1b).
Scheme 1. Strategy for the Synthesis of Highly Substituted 2-Trifluoromethylated Pyrrolidines.
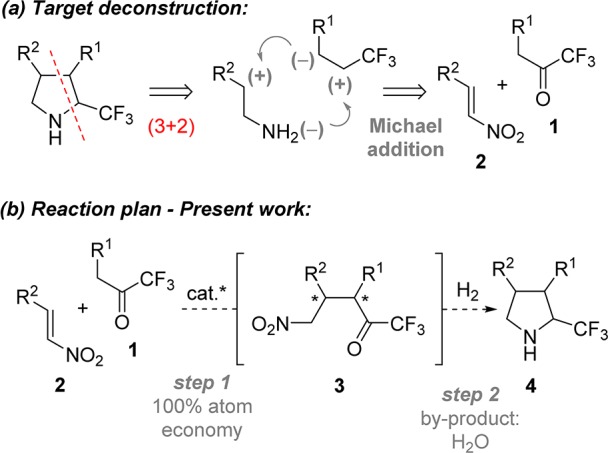
Despite significant interest in the synthetic utility of 1,1,1-trifluoromethylketones as electrophiles, their application as nucleophiles has gone relatively unexplored.12 Yan et al. recently reported the formal (4 + 2)-cycloaddition of 4,4,4-trifluoroacetoacetates to β,γ-unsaturated-α-keto esters;13 however, the direct catalytic asymmetric α-functionalization of simple 1,1,1-trifluoromethylketones is to the best of our knowledge unknown. While the electron-withdrawing nature of the CF3 group engenders enhanced C–H acidity relative to normal ketones, by corollary it also stabilizes the resultant enolate, rendering trifluoromethylketone enolates poorly nucleophilic.12 We anticipated that application of a bifunctional catalyst bearing thiourea and tertiary amine moieties would provide a pseudointramolecular pathway to overcome this inherent barrier to reactivity.14 We sought to harness the Brønsted acid/base ambifunctionality of these catalyst systems to develop the Michael addition of 1,1,1-trifluoromethylketones to nitroolefins.15
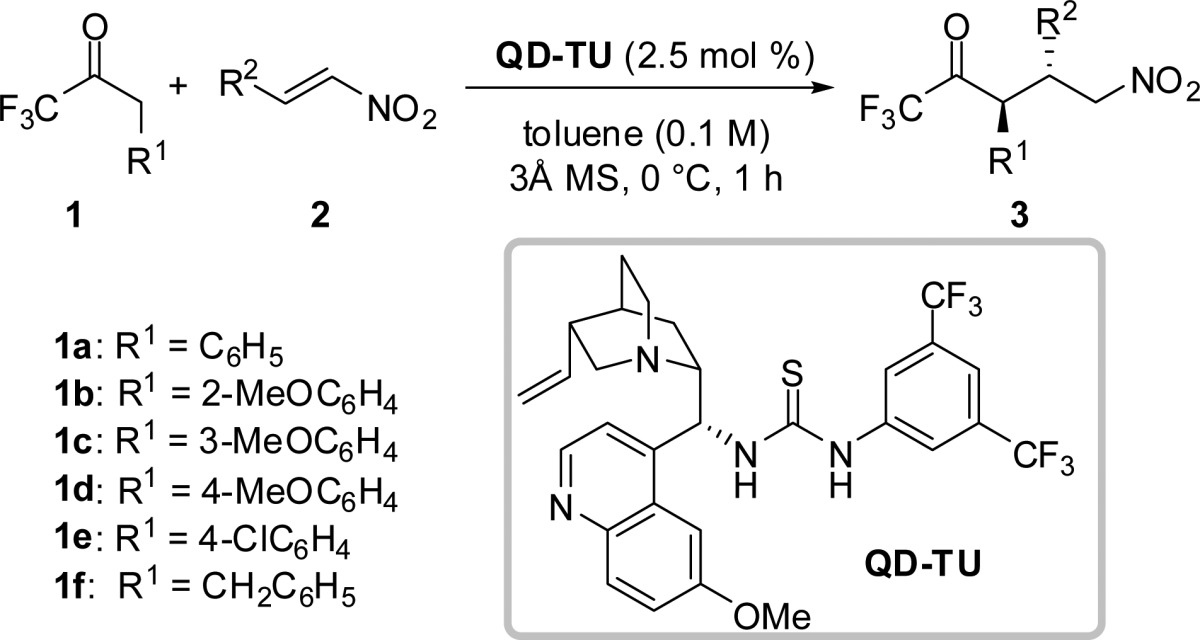
We began our studies by evaluating the feasibility of the Michael addition of 1,1,1-trifluoromethylketone 1a to nitroolefin 2a. Our experiments led to the use of bifunctional catalyst QD-TU,16 affording the desired γ-nitro carbonyl 3aa in quantitative yield with >20:1 dr and 95:5 er (Table 1, entry 1; see the Supporting Information (SI) for full optimization studies). A variety of reaction partners were next examined in the asymmetric Michael addition of 1,1,1-trifluoromethylketones 1 to nitroolefins 2 (Table 1). Electron-releasing and -withdrawing ortho-substituted aromatic nitroolefins were well tolerated, providing Michael adducts 3ab–3ae as single diastereomers with excellent levels of enantioselectivity (entries 2–5). Less sterically encumbering substitution at the meta-position of the nitroolefin resulted in a slight drop in diastereo- and enantiocontrol providing 3af in 98% yield with 19:1 dr and 93.5:6.5 er (entry 6). Examination of electron-releasing and -withdrawing para-substituents on the aromatic nitroolefin revealed significant electronic effects with respect to the diastereo- and enantioselectivity of the reaction (entries 7–11). Whereas electron-withdrawing groups (Br, NO2, CN) provided Michael adducts 3ag–3ai with high levels of selectivity, electron-releasing groups (Me and OMe) provided 3aj and 3ak, respectively, in diminished diastereo- and enantioselectivity. This electronic effect was also observed in the reaction of electron-rich heteroaromatic 2-thienyl- and 3-N-Ts-indoyl-substituted nitroolefins 2l and 2m, which afforded their respective Michael adducts 3al and 3am in diminished enantioselectivity (entries 12 and 13). Despite noticeably reduced reactivity, alkenyl and aliphatic nitroolefins were also found to be competent reaction partners providing 3an and 3ao in moderate conversion, but with high diastereo- and enantiocontrol (entries 14 and 15).
Table 1. Scope of Michael Addition of 1,1,1-Trifluoromethylketones 1 to Nitroolefins 2a.
| entry | 1 | 2 | 3 | yield (%)b | drc | erd |
|---|---|---|---|---|---|---|
| 1 | 1a | C6H5 | 3aa | 98 | >20:1 | 95:5 |
| 2 | 1a | 2-BrC6H4 | 3ab | 95 | >20:1 | 96.5:3.5 |
| 3 | 1a | 2-NO2C6H4 | 3ac | 94 | >20:1 | 97.5:2.5 |
| 4 | 1a | 2-CF3C6H4 | 3ad | 96 | >20:1 | 96.5:3.5e |
| 5 | 1a | 2-OMeC6H4 | 3ae | 98 | >20:1 | 94:6 |
| 6 | 1a | 3-ClC6H4 | 3af | 98 | 19:1 | 93.5:6.5 |
| 7 | 1a | 4-BrC6H4 | 3ag | 99 | >20:1 | 95.5:4.5 |
| 8 | 1a | 4-NO2C6H4 | 3ah | 99 | 7:1 | 95.5:4.5 |
| 9 | 1a | 4-CNC6H4 | 3ai | 96 | >20:1 | 96:4 |
| 10 | 1a | 4-MeC6H4 | 3aj | 96 | 8:1 | 93:7 |
| 11 | 1a | 4-OMeC6H4 | 3ak | 91 | 6:1 | 86.5:13.5 |
| 12 | 1a | 2-thienyl | 3al | 97 | 16:1 | 91.5:8.5 |
| 13f | 1a | 3-N-Ts-indoyl | 3am | 92 | >20:1 | 87.5:12.5 |
| 14 | 1a | CH=CHC6H5 | 3an | 62 (65)g | >20:1 | 87:13 |
| 15 | 1a | cyclohexyl | 3ao | 42 (43)g | >20:1 | 94:6 |
| 16 | 1b | 4-BrC6H4 | 3bg | 97 | >20:1 | 96:4 |
| 17 | 1c | 4-BrC6H4 | 3cg | 96 | >20:1 | 88:12 |
| 18 | 1d | 4-BrC6H4 | 3dg | 97 | >20:1 | 87:13 |
| 19 | 1e | 4-BrC6H4 | 3eg | 95 | >20:1 | 93:7 |
| 20h | 1f | C6H5 | 3fa | 73 (82)g | >20:1 | 97:3 |
Reactions were performed with 1 (0.21 mmol) and 2 (0.20 mmol) and proceeded to full conversion as adjudged by TLC.
Isolated yield. The diastereomers were not separable, and this represents the combined yield.
The diastereomeric ratio was determined by 19F NMR spectroscopic analysis of the crude product.
The enantiomeric ratio was determined by HPLC or SFC analysis on a chiral stationary phase.
The enantiomeric ratio was determined following reduction of 3ad with NaBH4 (see the SI).
The reaction was performed at 0 °C for 3 h.
Number in parentheses is conversion of nitroolefin as determined by 1H NMR spectroscopic analysis of the crude product.
The reaction was performed employing QD-TU (10 mol %) at 0 °C for 12 h.
We next turned our attention to examining electronic and structural variations of the 1,1,1-trifluoromethylketone donor 1 by placing methoxy groups at the ortho-, meta-, and para-positions of the aromatic ring. Although sterically demanding ortho-substituted 1b required longer reaction times (3 h) to achieve full conversion, 3bg was obtained in 97% yield with >20:1 dr and 96:4 er (entry 16). Less sterically encumbered electron-releasing nucleophiles 1c and 1d proceeded efficiently under the standard reaction conditions to provide 3cg and 3dg, respectively, in excellent yield and diastereoselectivity, but with reduced enantioselectivity (entries 17 and 18). This electronic effect was confirmed by utilizing electron-withdrawing para-substituted 1e as the nucleophile, which provided 3eg in 95% yield with 17:1 dr and 93:7 er (entry 19). It is worth noting that subjecting 3eg, which possesses enhanced C–H acidity, to silica gel chromatography resulted in erosion of diastereoselectivity from 17:1 to 2:1 dr presumably due to facile epimerization via enol formation. Lastly, we employed aliphatic 1,1,1-trifluoromethylketone 1f in the Michael addition to nitroolefin 2a to provide 3ea in 73% yield with >20:1 dr and 97:3 er, although the reaction required a higher catalyst loading and longer reaction times to achieve acceptable conversion (entry 20). The majority of adducts are crystalline solids. A single recrystallization of 3ai provided a useful upgrade in the enantiomeric composition (to 99.5:0.5 er).
Having developed a highly diastereo- and enantioselective Michael addition to access γ-nitro trifluoromethyl ketones 3 (Table 1), we sought to exploit this functional array toward the synthesis of 3,4-diaryl-2-trifluoromethyl pyrrolidines 4 (Scheme 2). Subjecting the crude products obtained via Michael addition to Raney-Ni hydrogenation conditions resulted in the clean formation of the desired pyrrolidines with excellent levels of diastereocontrol (>20:1 in all cases) bearing an all cis-relationship as determined by NOESY analysis. Attempts employing a one-pot protocol were promising; however, the molecular sieves from step 1 were found to be detrimental to the hydrogenation step, resulting in only moderate conversions. In addition to bis(phenyl) pyrrolidine 4a, ortho- and para-substituents were tolerated at both the 3- and 4-positions of the pyrrolidine providing products 4b–4e in high yield and enantioselectivity. No erosion in diastereo- or enantiomeric composition was observed during the hydrogenation, indicating that neither epimerization nor retro-Michael pathways are operative during the reaction.
Scheme 2. Synthesis of Enantioenriched 2-Trifluoromethyl Pyrrolidines.

Reactions were performed as described in Table 1. The yield is for both diastereomers. The diastereomeric ratio was determined by 19F NMR spectroscopic analysis of the crude product. The enantiomeric ratio was determined by HPLC analysis on a chiral stationary phase. b Number in parentheses is dr following column chromatography. c The Michael addition was performed at −10 °C for 1 h.
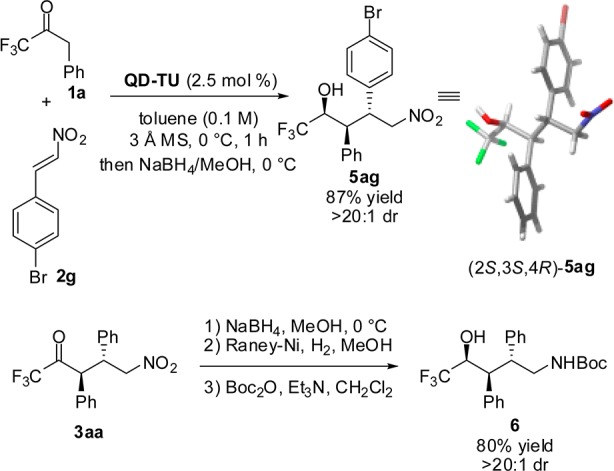
In addition to providing expedient access to new classes of enriched 2-trifluoromethyl pyrrolidines, the obtained Michael adducts 3 are also amenable to a number of other secondary transformations (Scheme 3). Employing a modified workup to the Michael addition of 1a to 2g by addition of NaBH4/MeOH mixture results in the highly diastereoselective reduction of the carbonyl to provide 5ag in 87% yield. An X-ray diffraction study of alcohol 5 was performed to assign the relative and absolute stereochemistries as (2S,3S,4R).17 By analogy, the Michael adducts were assigned as (3S,4R)-3 and the pyrrolidines as (2S,3S,4R)-4. Next, sequential carbonyl/nitro reduction was employed to provide the Boc-protected amino alcohol 6 bearing three contiguous stereocenters in 80% overall yield from 3aa as a single diastereomer.
Scheme 3. Secondary Transformations of Michael Adducts and Determination of Relative and Absolute Stereochemistries.
The high syn-selectivity observed in the NaBH4 reduction of Michael adducts 3aa and 3ag (Scheme 3) led us to pursue a unified strategy to access both C(2)-epimers of 2-trifluoromethylated pyrrolidine 4a. We envisioned stereospecific intramolecular SN2-displacement of an alcohol derivative by the terminal amine would provide 2-epi-4a (Scheme 4). Reduction of the alcohol with NaBH4, conversion of the free alcohol to its derived mesylate, and hydrogenation of the nitro group provided the unstable primary amine intermediate 7. The latter was immediately treated with DBU to induce cyclization to diastereomerically pure 2-epi-4a in 52% yield over the four steps.
Scheme 4. Access to Diastereomerically Pure Epimeric 2-Trifluoromethylated Pyrrolidines.

In conclusion, we have developed a formal (3 + 2)-annulation strategy for the highly selective synthesis of trisubstituted 2-trifluoromethyl pyrrolidines via asymmetric Michael addition/reductive cyclization. A direct catalytic Michael addition of 1,1,1-trifluoromethylketones to nitroolefins was developed, providing access to 3,4-disubstituted-5-nitro-1,1,1-trifluoromethylketones with excellent levels of diastereo- and enantioselectivity. The obtained Michael adducts were then utilized in a diastereoselective reductive cyclization to afford functionalized 3,4-disubstituted-2-trifluoromethyl pyrrolidines bearing three contiguous stereocenters. We also demonstrated that C(2)-epimeric 2-trifluoromethyl pyrroldines can be accessed from a common intermediate with excellent diastereocontrol utilizing an intramolecular SN2-displacement.
Acknowledgments
The project described was supported by Award No. R01 GM103855 from the National Institute of General Medical Sciences. X-ray crystallography was performed by Dr. Peter S. White (UNC Chapel Hill).
Supporting Information Available
Experimental procedures, characterization of the products, and CIF data for CCDC 979177. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a O’Hagan D. Nat. Prod. Rep. 1997, 14, 637–651. [Google Scholar]; b O’Hagan D. Nat. Prod. Rep. 2000, 17, 435–446. [DOI] [PubMed] [Google Scholar]
- a Welch J. T. Tetrahedron 1987, 43, 3123–3197. [Google Scholar]; b Müller K.; Faeh C.; Diederich F. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]; c O’Hagan D. Chem. Soc. Rev. 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]; d Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; e Hagmann W. K. J. Med. Chem. 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- a Kiselyov A. S.; Strekowski L. Org. Prep. Proced. Int. 1996, 28, 289–318. [Google Scholar]; b Langlois B. R.; Billard T. Synthesis 2003, 185–194. [Google Scholar]; c Ma J.-A.; Cahard D. Chem. Rev. 2004, 104, 6119–6146. [DOI] [PubMed] [Google Scholar]; d Billard T.; Langlois B. R. Eur. J. Org. Chem. 2007, 891–897. [Google Scholar]; e Shibata N.; Mizuta S.; Kawai H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. [Google Scholar]; f Zheng Y.; Ma J.-A. Adv. Synth. Catal. 2010, 352, 2745–2750. [Google Scholar]; g Nie J.; Guo H.-C.; Cahard D.; Ma J.-A. Chem. Rev. 2011, 111, 455–529. [DOI] [PubMed] [Google Scholar]; h Furuya T.; Kamlet A. S.; Ritter T. Nature 2011, 473, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Studer A. Angew. Chem., Int. Ed. 2012, 51, 8950–8958. [DOI] [PubMed] [Google Scholar]
- Fustero S.; Sanz-Cervera J. F.; Aceña J. L.; Sánchez-Roselló M. Synlett 2009, 525–549. [Google Scholar]
- a Llamas T.; Arrayás R. G.; Carretero J. C. Synthesis 2007, 950–956. [Google Scholar]; b Li Q.-H.; Tong M.-C.; Li J.; Tao H.-Y.; Wang C.-J. Chem. Commun. 2011, 47, 11110–11112. [DOI] [PubMed] [Google Scholar]; c Li Q.; Ding C.-H.; Li X.-H.; Weissensteiner W.; Hou X.-L. Synthesis 2012, 265–271. [Google Scholar]; d Li Q.-H.; Xue Z.-Y.; Tao H.-Y.; Wang C.-J. Tetrahedron Lett. 2012, 53, 3650–3653. [Google Scholar]
- Examples drawing from the chiral pool:; a Fustero S.; Jiménez D.; Sánchez-Roselló M.; del Pozo C. J. Am. Chem. Soc. 2007, 129, 6700–6701. [DOI] [PubMed] [Google Scholar]; b Bezdudny A. V.; Alekseenko A. N.; Mykhailiuk P. K.; Manoilenko O. V.; Shishkin O. V.; Pustovit Y. M. Eur. J. Org. Chem. 2011, 1782–1785. [Google Scholar]; c Fritz S. P.; West T. H.; McGarrigle E. M.; Aggarwal V. K. Org. Lett. 2012, 14, 6370–6373. [DOI] [PubMed] [Google Scholar]; d Khangarot R. K.; Kaliappan K. P. Eur. J. Org. Chem. 2013, 2692–2698. [Google Scholar]; Examples employing chiral auxiliaries:; e Okano T.; Fumoto M.; Kusukawa T.; Fujita M. Org. Lett. 2002, 4, 1571–1573. [DOI] [PubMed] [Google Scholar]; f Chaume G.; van Severen M.-C.; Marinkovic S.; Brigaud T. Org. Lett. 2006, 8, 6123–6126. [DOI] [PubMed] [Google Scholar]; g Caupène C.; Chaume G.; Ricard L.; Brigaud T. Org. Lett. 2009, 11, 209–212. [DOI] [PubMed] [Google Scholar]; h Chaume G.; van Severen M.-C.; Ricard L.; Brigaud T. J. Fluorine Chem. 2008, 129, 1104–1109. [Google Scholar]; i Chaume G.; Lensen N.; Caupène C.; Brigaud T. Eur. J. Org. Chem. 2009, 5717–5724. [DOI] [PubMed] [Google Scholar]; j Jlalia I.; Lensen N.; Chaume G.; Dzhambazova E.; Astasidi L.; Hadjiolova R.; Bocheva A.; Brigaud T. Eur. J. Med. Chem. 2013, 62, 122–129. [DOI] [PubMed] [Google Scholar]; Example employing asymmetric catalysis:; k Huang G.; Yin Z.; Zhang X. Chem.—Eur. J. 2013, 19, 11992–11998. [DOI] [PubMed] [Google Scholar]
- a Pichon M.; Figadre B. Tetrahedron: Asymmetry 1996, 7, 927–964. [Google Scholar]; b Felpin F.-X.; Lebreton J. Eur. J. Org. Chem. 2003, 3693–3712. [Google Scholar]; c Pandey G.; Banarjee P.; Gadre S. R. Chem. Rev. 2006, 106, 4484–4517. [DOI] [PubMed] [Google Scholar]
- Representative examples:; a Betancort J. M.; Barbas C. F. III. Org. Lett. 2001, 3, 3737–3740. [DOI] [PubMed] [Google Scholar]; b García-García P.; Ladépêche A.; Halder R.; List B. Angew. Chem. 2008, 120, 4797–4799. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. 2008, 47, 4719 - 4721. [DOI] [PubMed]; c Kastl R.; Wennemers H. Angew. Chem. 2013, 125, 7369–7373. [DOI] [PubMed] [Google Scholar]; Angew. Chem., Int. Ed. 2013, 52, 7228 - 7232. [DOI] [PubMed]
- Representative examples:; a Trost B. M.; Hisaindee S. Org. Lett. 2006, 8, 6003–6005. [DOI] [PubMed] [Google Scholar]; b Jiang X.; Zhang Y.; Chan A. S. C.; Wang R. Org. Lett. 2009, 11, 153–156. [DOI] [PubMed] [Google Scholar]; c Guo S.; Xie Y.; Hu X.; Huang H. Org. Lett. 2011, 13, 5596–5599. [DOI] [PubMed] [Google Scholar]; d Ma H.; Liu K.; Zhang F.-G.; Zhu C.-L.; Nie J.; Ma J.-A. J. Org. Chem. 2010, 75, 1402–1409. [DOI] [PubMed] [Google Scholar]
- a Nakamura A.; Lectard S.; Hashizume D.; Hamashima Y.; Sodeoka M. J. Am. Chem. Soc. 2010, 132, 4036–4037. [DOI] [PubMed] [Google Scholar]; b Xu Y.; Matsunaga S.; Shibasaki M. Org. Lett. 2010, 12, 3246–3249. [DOI] [PubMed] [Google Scholar]
- Representative examples:; a Ji J.; Barnes D. M.; Zhang J.; King S. A.; Wittenberger S. J.; Morton H. E. J. Am. Chem. Soc. 1999, 121, 10215–10216. [Google Scholar]; b Barnes D. M.; Ji J.; Fickes M. G.; Fitzgerald M. A.; King S. A.; Morton H. E.; Plagge F. A.; Preskill M.; Wagaw S. H.; Wittenberger S. J.; Zhang J. J. Am. Chem. Soc. 2002, 124, 13097–13105. [DOI] [PubMed] [Google Scholar]
- Kelly C. B.; Mercadante M. A.; Leadbeater N. E. Chem. Commun. 2013, 49, 11133–11148. [DOI] [PubMed] [Google Scholar]
- Wang J.-j.; Hu Z.-p.; Lou C.-l.; Liu J.-l.; Li X.-m.; Yan M. Tetrahedron 2011, 67, 4578–4583. [Google Scholar]
- a Ma J.-A.; Cahard D. Angew. Chem. 2004, 116, 4666–4683. [Google Scholar]; Angew. Chem., Int. Ed. 2004, 43, 4566 - 4583. [DOI] [PubMed]; b Miyabe H.; Takemoto Y. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar]; c Connon S. J. Chem. Commun. 2008, 44, 2499–2510. [DOI] [PubMed] [Google Scholar]; d Siau W.-Y.; Wang J. Catal. Sci. Technol. 2011, 1, 1298–1310. [Google Scholar]
- a Berner O. M.; Tedeschi L.; Enders D. Eur. J. Org. Chem. 2002, 1877–1894. [Google Scholar]; b Tsogoeva S. B. Eur. J. Org. Chem. 2007, 1701–1716. [Google Scholar]; c Aitken L. S.; Arezki N. R.; Dell’Isola A.; Cobb A. J. A. Synthesis 2013, 45, 2627–2648. [Google Scholar]
- a Vakulya B.; Varga S.; Csámpai A.; Soós T. Org. Lett. 2005, 7, 1967–1969. [DOI] [PubMed] [Google Scholar]; b McCooey S. H.; Connon S. J. Angew. Chem. 2005, 117, 6525–6528. [Google Scholar]; Angew. Chem., Int. Ed. 2005, 44, 6367 - 6370. [DOI] [PubMed]
- CCDC 979177 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Centre via www.ccdc.cam.ac.uk/data_request/cif. The structure in Scheme 3-D were generated with CYLview: Legault, C. Y. CYLview, version 1.0b; Université de Sherbrooke: Sherbrooke, QC, 2009; http://www.cylview.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.