

Abstract
Thiol redox state (TRS) refers to the balance between reduced thiols and their corresponding disulfides and is mainly reflected by the ratio of reduced and oxidized glutathione (GSH/GSSG). A decrease in GSH/GSSG, which reflects a state of thiol oxidative stress, as well as thiol modifications such as S-glutathionylation have been shown to have important implications in a variety of cardiovascular diseases. Therefore, research models for inducing thiol oxidative stress are important tools for studying the pathophysiology of these disease states as well as examining the impact of pharmacological interventions on thiol pathways. The purpose of this study is to evaluate the use of a dithiocarbamate derivative, 2-acetylamino-3-[4-(2-acetylamino-2-carboxyethylsulfanylthiocarbonylamino)phenylthiocarbamoylsulfanyl] propionic acid (2-AAPA) as a pharmacological model of thiol oxidative stress by examining the extent of thiol modifications induced in H9c2 rat cardiomyocytes and its impact on cellular functions. The extent of thiol oxidative stress produced by 2-AAPA was also compared to other models of oxidative stress including hydrogen peroxide (H2O2), diamide, buthionine sulfoximine (BSO) and N,N′-bis(2-chloroethyl)-N-nitroso-urea (BCNU). Results indicated that 2-AAPA effectively inhibited glutathione reductase (GR) and thioredoxin reductase (TrxR) activities and decreased the GSH/GSSG ratio by causing a significant accumulation of GSSG. 2-AAPA also increased the formation of protein disulfides as well as S-glutathionylation. The alteration in TRS lead to loss of mitochondrial membrane potential, release of cytochrome c and an increase in reactive oxygen species (ROS) production. Compared to other models, 2-AAPA is more potent in creating a state of thiol oxidative stress with lower cytotoxicity, higher specificity and more pharmacological relevance, and could be utilized as a research tool to study TRS-related normal and abnormal biochemical processes in cardiovascular diseases.
Keywords: oxidative stress, thiol redox state, glutathione, glutathione reductase, enzyme inhibitor, cardiomyocytes
Introduction
Oxidative stress reflects an imbalance between the production of free radicals and ROS and the cell’s antioxidant defense mechanisms. An increase in free radicals and ROS can cause damage to the cell and disruption of cellular functions even though they are also involved in normal cell processes [1]. Oxidative stress is believed to play an important role in the pathogenesis of a variety of diseases including cardiovascular disorders such as ischemia-reperfusion injury [2], atherosclerosis [3], heart failure [4], doxorubicin cardiotoxicity [5], and diabetic cardiomyopathy [6].
Thiols are important in the cell’s protection against oxidative stress as thiol groups can be oxidized to disulfides upon exposure to free radicals and reactive oxygen species [7], and disulfides can be reversibly reduced by the related enzymes including GR, glutaredoxin, and the thioredoxin (Trx)/TrxR system [4, 8]. The balance between reduced thiols and their corresponding disulfides is referred to as TRS [9, 10]. In the cell, total thiols are contributed by all cysteine residues found in proteins and small non-protein thiols such as GSH. GSH is a tripeptide with a central cysteine amino acid. It is the most abundant thiol in cells with concentrations ranging from 0.5 to 10 mM [11]. The cell normally maintains a high ratio of GSH to its oxidized forms GSSG (~100:1) as the principal redox buffer against oxidative stress, and this ratio is often used in the literature as a parameter of TRS [11–13]. A decrease in the GSH/GSSG ratio reflects a state of thiol oxidative stress.
TRS is closely associated with intracellular redox homeostasis as well as a crucial mediator of various essential metabolic, redox signaling, and transcription processes in cells [9, 14–16]. Alterations in TRS and related enzymes have been shown to have important implications in the protection of the heart against oxidative stress processes. During myocardial ischemia and reperfusion, a decrease in myocardial GSH as well as GSH/GSSG ratio within the ischemic tissues has been observed, and the extent of the myocardial injury is inversely dependent on the myocardial GSH content [17]. Treatment with γ-glutamylcysteine ethyl ester can increase intracellular reduced GSH concentrations and has been shown to result in a dose-dependent reduction in infarct size in a canine model of occlusion-reperfusion [18]. The beneficial effects of carvedilol in congestive heart failure were also shown to significantly correlate with a change in GSH/GSSG [19]. In a rabbit diabetes model, the activities of glutathione peroxidase and glutathione reductase were both shown to increase in heart tissue. The level of total thiols in the cardiomyocytes also increased in this model [20]. This has implications for the role of thiols in protection against diabetic cardiomyopathy. Thiols modification has also been shown to be involved in ischemia-reperfusion injury. S-glutathionylation is the formation of mixed disulfides between proteins thiols and GSH in response to oxidative stress and has important implications for protein structure and function. Following ischemia-reperfusion injury, S-glutathionylation of actin has been observed and is believed to contribute to declines in contractility observed during ischemia-reperfusion injury [21]. In addition, S-glutathionylation of an active site cysteine moiety in creatine kinase during ischemia-reperfusion injury has been implicated in the development of cardiac injury [22].
Because of the important role of alterations in TRS in the development of cardiovascular disorders, models for studying intracellular TRS in cardiomyocytes are important for understanding the molecular changes that occur during the oxidative stress state as well as examining potential pharmacological interventions and the impact of medications on thiol pathways. Several models have been developed to study oxidative stress in cardiomyocytes. Induction of ischemia-reperfusion injury in vivo via coronary artery ligation [23] or Langendorff perfusion [24] are models for inducing an oxidative stress state. However, these models require special expertise and facilities for conducting the surgical procedure and require the use of animals. Pharmacological models would be useful tools for inducing thiol oxidative stress in vitro and in vivo. The most frequently used compounds to induce oxidative stress in cardiomyocytes are H2O2 [25–27] and tertiary butyl hydroperoxide [28–30]. However, they are not ideal for increasing thiol oxidative stress because of the nonspecific oxidative nature of these agents. Diamide is also used to oxidize thiols to disulfides to create thiol oxidative stress [31]. The drawback of this reagent is that it can also react with other functional groups such as carboxylic acids [32] and alcohols [33] leading to unwanted effects.
Creation of thiol oxidative stress can also be achieved by using enzyme inhibitors of the glutathione pathway. Intracellular GSH level is maintained by two pathways: the de novo synthesis of glutathione from the constituent amino acids and the reduction of GSSG back to GSH by GR [13]. Depletion of GSH in cardiomyocytes can be achieved by BSO, an inhibitor of GSH synthesis [34, 35]. Inhibition of GR is a more attractive model for alteration of TRS as GR inhibition creates a state of thiol oxidative stress by an increase in GSSG, which is expected to have a more significant impact on thiol redox potential than GSH depletion considering the high ratio of GSH/GSSG. BCNU is the most commonly used irreversible GR inhibitor in the literature [34–36]. However, BCNU is an alkylating agent which can complicate the use of BCNU as a research tool to study TRS [36, 37].
2-AAPA (Figure 1), a dithiocarbamate derivative, is a novel irreversible inhibitor of GR and glutaredoxin developed in this laboratory [38, 39]. Previously, 2-AAPA has been shown to be capable of producing disturbances in cellular thiol status in the CV-1 (monkey kidney) cell line without the production of free radicals or alteration of the expression of other cellular antioxidant systems [40]. In addition, 2-AAPA is 10 times more potent than BCNU as a yeast GR inhibitor [38]. These features make 2-AAPA a good candidate for creating a model for alterations in TRS in cardiomyocytes. Therefore, the objective of this study was to evaluate the potential for 2-AAPA to be utilized as a research tool to induce thiol oxidative stress in cardiomyocytes.
Figure 1.
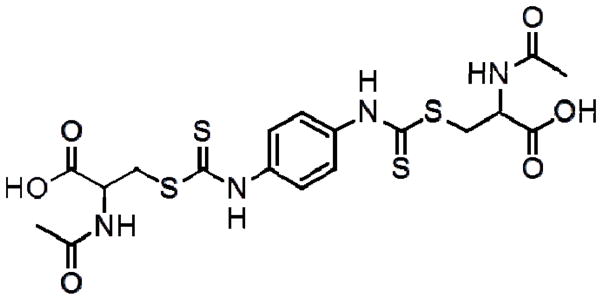
Chemical structure of 2-acetylamino-3-[4-(2-acetylamino-2-carboxyethylsulfanylthiocarbonylamino)phenylthiocarbamoylsulfanyl]propionic acid (2-AAPA)
Materials and Methods
Materials
GSH, GSSG, 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), sodium borohydride (NaBH4), BSO, BCNU, H2O2, diamide, p-aminobenzoic acid, sulfosalicylic acid, dimethyl sulfoxide (DMSO), ethylenediaminetetraacetic acid (EDTA), recombinant human thioredoxin, insulin and Bradford reagent were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI, USA). Dulbecco’s Modified Eagle’s medium (DMEM) was purchased from the American Type Culture Collection (Bethesda, MD, USA). Fetal bovine serum was obtained from Atlanta Biologicals (Lawrenceville, GA, USA). Penicillin/streptomycin, trypsin and phosphate-buffered saline were purchased from Mediatech (Herndon, VA, USA). BCA protein assay kit was bought from Thermo Scientific (Rockford, IL, USA). 2-AAPA was synthesized in this laboratory by a previously reported method [38].
Stock solutions
Solutions of DTNB, p-aminobenzoic acid, and NaBH4 were made in 0.15 M NaH2PO4 (pH 7.5). Solutions of GSH and GSSG were prepared in 0.1% formic acid aqueous solution. 2-AAPA (2 mM) was dissolved in DMEM containing 10% FBS and 1% penicillin/streptomycin and prepared fresh each time before using. BSO (20 mM) was made in ultrapure water (Milli-Q purification system; Millipore, Bedford, MA, USA) with pH adjusted to 10. Diamide (20 mM) and H2O2 (20 mM) were prepared directly in ultrapure water. BCNU (20 mM) was dissolved in ethanol. Solutions of 2-AAPA, diamide and BSO were filtered through a Medical Millex-GP filter (0.22 μm, sterilized; Millipore). All stock solutions were stored at −80°C before use except NaBH4, which was prepared fresh and stored over ice for 1-day assay.
Cell Culture
H9c2 cell line (embryonic rat myocardium) was obtained from ATCC (Bethesda, MD, USA) and maintained in DMEM with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin at 37 °C with 5% CO2. In this study, only cells with passage number below 15 were utilized.
Cell Viability
Cell viability was measured by a colorimetric assay in a 96-well plate with MTT (3-(4,5)-dimethylthiazol-2,5-diphenyltetrazolium bromide) [41]. Briefly, H9c2 cells were plated in 96 well plates at a density of 1,000 cells/well. After a 24 h attachment, the cells were treated with increasing concentrations of 2-AAPA dissolved in growth medium (150 μL/well) ranging from 1nM to 1mM for another 6 days. At the end of cell growth, MTT assay was performed by replacing the medium with an MTT solution (0.5 mg/ml, 50 μl/well) and incubated in the dark at 37°C for 4 h. The formed purple formazan product was dissolved by DMSO (150 μl/well, 1 h). The absorbance of each well was quantified with a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA) using a test wavelength of 570 nm and a reference wavelength of 650 nm. Cell viability of H9c2 cells treated with H2O2, diamide, BSO and BCNU were determined under the same conditions as descripted above.
Enzyme Inhibition in H9c2 Cells
5 million H9c2 cells were placed in a 175-cm2 flask in DMEM and incubated for 24 h for attachment. At the end of 24 h, the cells were treated with 2-AAPA (25, 50, 75 and 100 μM) in a 5% CO2 incubator at 37 °C for different duration ranging from 20 min to 12 h. The medium was collected, and the cells were rinsed with phosphate-buffered saline and detached by trypsinization. The medium and the cell suspension were combined and centrifuged at 1,000 g for 5 min. The pellet was washed with 5 ml of cold phosphate-buffered saline with 1mM EDTA, suspended in hypotonic phosphate buffer (1 ml, 1 mM) containing 1 mM EDTA, and homogenized over ice with an Omni 5000 homogenizer. The homogenate was centrifuged at 120,000 × g for 30 min at 4 °C. The supernatant was collected and used to determine GR and TrxR activities. To determine GR enzyme activity, 350 μl of the supernatant was added to a 0.1 M sodium phosphate buffer containing NADPH (0.2 mM). The enzymatic reaction was initiated by addition of GSSG (1 mM). To determine TrxR enzyme activity, 350 μl of the supernatant was added to a 0.1 M sodium phosphate buffer pH 7.4, containing NADPH (0.24 mM) and 0.45 units/mL Trx. The enzymatic reaction was initiated by addition of insulin (1.07 mg/mL). Both GR and TrxR activities were measured by the initial rates of disappearance of NADPH determined spectrophotometrically at λ=340 nm. Results were standardized to protein content determined by the method of Bradford [42].
Intracellular GSH and GSSG
H9c2 cells were treated with 2-AAPA in a 175-cm2 flask and collected as described above. The cell pellet was washed with cold phosphate-buffered saline (0.5 mL× 2) and resuspended in 0.5 ml of 3% (w/v) sulfosalicylic acid. The cell suspension was sonicated over ice using a Qsonica Q500 sonicator with a cup horn probe for 5 min and centrifuged at 20,817 × g for another 5 min at 4 °C. The supernatants of cell lysates were diluted with 0.1% (w/v) formic acid for analysis of GSH and GSSG by following a validated LC/MS/MS method developed in this laboratory. The analysis was carried out on an Agilent 1100 series HPLC system coupled with a Finnigan LCQ-DECA mass spectrometer, which is equipped with an ESI ion source. Chromatographic separation of GSH and GSSG was achieved on an Agilent Eclipse XDB-C18 column (1.0×150mm, 3.5μm) with mobile phase composed of 0.1% formic acid and acetonitrile at an isocratic ratio of 94/6 (v/v). The electrospray ion source was operated in positive ionization mode and selected reaction monitoring (SRM) was performed to detect GSH and GSSG at the mass transition of m/z 308→179 and m/z 613→484, respectively.
Protein Thiols, Protein Disulfides and S-glutathionylation
The lysate pellets were washed thoroughly with 3% (w/v) sulfosalicylic acid (0.5 ml × 3) to ensure that no residual non-protein thiols were left before being resuspended in 0.5 ml of 3% (w/v) sulfosalicylic acid. The quantification of protein thiols, protein disulfides and S-glutathionylation followed a previously reported procedure developed in this laboratory [43]. Briefly, protein thiols were determined by reacting the thiols in suspension with DTNB and measuring the subsequent formation of 2-nitro-5-thiobenzoic acid (TNB) using UV-HPLC at a detection wavelength of 326 nm. Total thiol content was determined in the same way as protein thiols except that a reaction to reduce disulfides was performed using NaBH4 before the reaction with DTNB. S-glutathionylation was measured by releasing the GSH from protein upon NaBH4 reduction and detecting the conjugate of GSH with 2-nitro-5-thiobenzoic acid (GS-TNB) formed in the reaction with DTNB. The reactions were performed individually for each sample and manual injections were employed to shoot the samples into HPLC. While protein thiols and S-glutathionylation were expressed directly as equivalent of GSH, protein disulfide was quantified indirectly by subtracting the protein thiols from total thiol content and expressed as equivalent of GSSG.
ROS Production
ROS production was assessed using cell-permeable probe carboxy-H2DCFDA (Invitrogen, Life Technologies). Upon entry into the cytoplasm, this probe is cleaved by cellular esterase and oxidized by ROS to yield fluorescence. Briefly, H9c2 cells were plated in 96 well plates at a density of 10,000 cells/well. After 24 h attachment, the cells were pretreated with carboxy-H2DCFDA for 30 min followed by treatment with 2-AAPA at concentration of 25, 50, 75 and 100 μM. Fluorescence intensity was measured at 492 nm (excitation) and 527 nm (emission) on a SpectraMax M2 fluorescence microplate reader (Molecular Devices, Sunnyvale, California) at different time points (20 min, 1 h, 2 h and 4 h).
Mitochondrial Membrane Potential
H9c2 cells were plated in 96 well plates at a density of 10,000 cells/well. After 24 h attachment, the cells were treated with 2-AAPA 100 μM for 20 min and 2 h. Mitochondrial membrane potential was determined by incubating the cells with a fluorescent dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1, 0.5 μg/mL) for 15 min. JC-1 is a cationic dye which exhibits potential-dependent accumulation in mitochondria indicated by a fluorescence emission shift from green (monomer) to red (aggregates). Fluorescence intensity was measured on a SpectraMax M2 fluorescence microplate reader (Molecular Devices, Sunnyvale, California). For red fluorescence, an excitation wavelength of 550 nm and an emission wavelength of 600 nm were used while an excitation wavelength of 485 nm and an emission wavelength of 610 nm were used to measure green fluorescence. The ratio of fluorescence intensity of red to green in cells was used as a marker of mitochondrial membrane potential.
Cytochrome C Release
15 million H9c2 cells were treated with 2-AAPA 100 μM for 20 min and 2 h. The cells were harvested and intact mitochondria were isolated from cytosolic component by differential centrifugation using a mitochondria isolation kit (Thermo Scientific, Rockford, IL). The cytosolic fractions were analyzed using Western blotting to detect the release of cytochrome c from mitochondria. Briefly, equal amounts of protein (30 μg) from each sample were resolved by SDS-PAGE (15%) and transferred onto a 0.22 μm nitrocellulose membrane. The membrane was blocked using 5% (w/v) dried milk powder in Tris-buffered saline (TBS) [10 mM Tris-HCl (pH 7.5) and 0.15 M NaCl] with 0.1 % Tween 20 at room temperature and incubated with primary antibodies raised against antigens of rat origin for 12 h in TBS with 0.1 % Tween 20 [rabbit polyclonal cytochrome c (Cell Signaling Technology, Danvers, MA) and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA)] at 4 °C. Secondary antibody [Horseradish peroxidase (HRP)-conjugated antibody (Santa Cruz Biotechnology, Santa Cruz, CA)] was then added to the membrane for 1 h at room temperature. Thereafter, the membrane was washed, exposed to HRP substrate (Amersham, UK), and visualized using UVP Bioimaging system. The band intensity was quantified by Lab Works™ image acquisition and analysis software (UVP, Inc., Upland, CA).
Extent of Thiol Oxidative Stress in Other Models
The thiol oxidative stress produced by 2-AAPA was compared to other commonly used pharmacological models of oxidative stress: H2O2, diamide, BSO and BCNU under the same conditions. A concentration of 100 μM was selected for all compounds based on literature reports. While the incubation time of H2O2, diamide and BCNU with H9c2 cells was 2 h, the incubation time of BSO with H9c2 cells was 24 h to allow enough time for GSH depletion. The thiol content including GSH, GSSG, protein thiols, protein disulfides and S-glutathionylation was evaluated as described above and compared to the data obtained from 2-AAPA.
Statistical Analysis
Data are expressed as means ± standard deviation (SD) from three independent experiments and were analyzed using one-way ANOVA and Tukey’s post hoc test. A p value < 0.05 was considered to be statistically significant.
Results
IC50 of 2-AAPA, H2O2, diamide, BSO and BCNU in H9c2 cells
The IC50, the concentration of 2-AAPA that decreases the cell viability by 50%, was determined to be 68 ± 7 μM in H9c2 cells. IC50s of H2O2, diamide, BSO and BCNU in H9c2 cells were determined to be 277 ± 68 μM, 47 ± 11 μM, 16 ± 5 μM and 55± 6 μM, respectively.
2-AAPA inhibited GR and TrxR activities in H9c2 cells
The inhibitory effects of 2-AAPA on GR were determined in H9c2 cells over a 12 h period. Results from GR activity assays are shown in Figure 2. The average GR activity in the control was determined to be 0.033 units/mg protein. When the cells was incubated with 2-AAPA, GR activity was inhibited and the maximum inhibition was observed within the first 2 h with remaining activity determined to be 49%, 33%, 20% and 13% at concentrations of 25, 50, 75, and 100 μM, respectively. The GR activity started to return after 2 h and almost reached to the level of control at the end of 12 h for all concentrations. 2-AAPA also inhibited TrxR activity in H9c2 cells. The remaining activity of TrxR in H9c2 cells treated with 100 μM 2-AAPA for 20 min was determined to be 34%.
Figure 2.
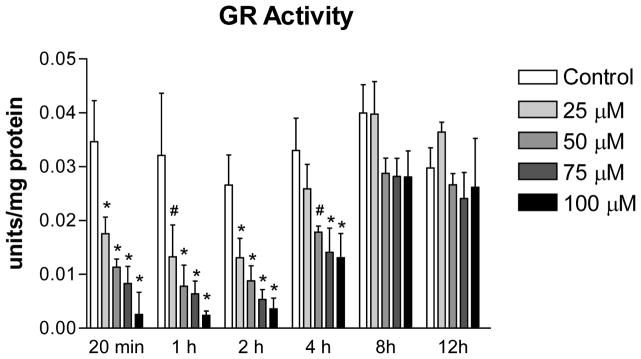
GR activity in the control and 2-AAPA treated H9c2 cells. The data are presented as units/mg protein and expressed as the means ± SD of three independent experiments. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 and * p < 0.01 compared with control group.
GR inhibition by 2-AAPA decreased GSH/GSSG
To determine the extent of thiol oxidative stress induced by GR inhibition, intracellular GSH and GSSG in H9c2 cells were quantified. The average values of GSH and GSSG in control samples over a 4 h period were determined to be 32.9 ± 2.3 and 0.20 ± 0.02 nmol/mg protein (n=24), respectively. As shown in Figure 3, a 2 to 20 fold increase in GSSG was observed in 2-AAPA treated cells depending on the concentrations and incubation time studied. For intracellular GSH, slight depletion by 2-AAPA (~28%) was seen only at the first 20 minutes with a concentration of 100 μM, and induction was found with lower concentrations of 2-AAPA (25, 50 and 75 μM) after 1 h incubation. The intracellular GSH/GSSG ratios were calculated and presented in Figure 4. In control H9c2 cells, the GSH/GSSG ratios were determined to be 165:1. In 2-AAPA treated H9c2 cells, the GSH/GSSG ratios were significantly decreased to be in a range of 11:1–108:1 depending on concentrations and incubation time studied.
Figure 3.

Intracellular GSH (A), GSSG (B) and GSH/GSSG ratio (C) in the control and 2-AAPA treated H9c2 cells. The data are presented as nmol/mg protein and expressed as the means ± SD of three independent experiments. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 and * p < 0.01 compared with control group.
Figure 4.

Protein thiols (A) and protein disulfides (B) in the control and 2-AAPA treated H9c2 cells. The data are presented as nmol/mg protein and expressed as the means ± SD of three independent experiments. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 and * p < 0.01 compared with control group.
2-AAPA increased protein disulfides and S-glutathionylation
The extent of thiol oxidative stress in 2-AAPA treated H9c2 cells was also evaluated by determining the content of protein thiols, protein disulfides and S-glutathionylation. As shown in Figure 4, protein thiols content was found to decrease in 100 μM 2-AAPA treated H9c2 cells (~20% reduction) throughout the 4 h period. For cells incubated with lower concentrations of 2-AAPA, the content of protein thiols was also found to decrease but was only statistically significant at 20 min and 4 h with the concentration of 75 μM. By contrast, the content of protein disulfides was significantly increased in a concentration-dependent manner. In addition, by detecting the GSH released from protein-glutathione mixed disulfides upon reduction with NaBH4, a significant amount of protein S-glutathionylation was found in 2-AAPA treated cells (except the concentration of 25 μM) while S-glutathionylation was not detectable in control samples (Table 1). The formation of protein S-glutathionylation was found to be highest at the first 1 h and started to decrease afterward. No GSH release was detected in 2-AAPA treated H9c2 cells after 2 h incubation.
Table 1.
Protein S-glutathionylation in the control and 2-AAPA treated H9c2 cells. Protein S-glutathionylation was determined by quantification of GSH released from proteins after NaBH4 reduction as described under “Materials and Methods”. The data are presented as nmol/mg protein and expressed as the means ± SD of three independent experiments.
| Time | GSH released from proteins (nmol/mg protein)
|
||||
|---|---|---|---|---|---|
| Control | 25 μM | 50 μM | 75 μM | 100 μM | |
| 20 min | ND* | ND* | 0.8 ± 0.2 | 2.7 ± 0.3 | 4.4 ± 0.6 |
| 1 h | ND* | ND* | 1.5 ± 0.4 | 3.0 ± 0.6 | 4.7 ± 1.2 |
| 2 h | ND* | ND* | ND* | 0.9 ± 0.1 | 1.5 ± 0.3 |
| 4 h | ND* | ND* | ND* | ND* | ND* |
ND, not detected.
2-AAPA increased intracellular ROS production in H9c2 cells
The time course profile of ROS production in 2-AAPA treated H9c2 cells is shown in Figure 5. Interestingly, no significant difference in ROS production was noticed between the control and the 2-AAPA treated cells within the first hour. Increased ROS was observed when the cells were incubated with 2-AAPA for longer times (Figure 5).
Figure 5.
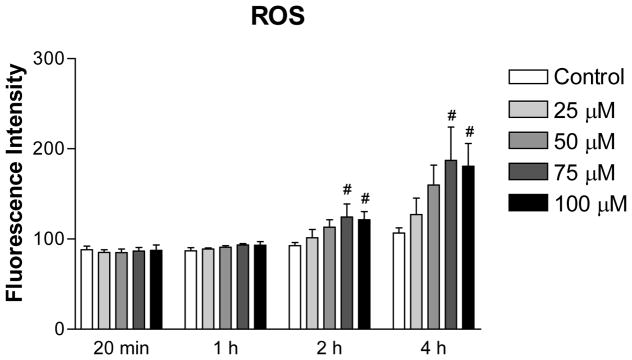
ROS production in the control and 2-AAPA treated H9c2 cells. The data are presented as fluorescence intensity and expressed as the means ± SD of three independent experiments. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 compared with control group.
2-AAPA induced mitochondrial membrane potential loss in H9c2 cells
Mitochondrial membrane potential of H9c2 cells in the presence of 2-AAPA was measured to evaluate the effect of an altered TRS on mitochondrial function. As shown in Figure 6, the ratio of red/green fluorescence intensity of JC-1 was decreased in 2-AAPA treated H9c2 cells at both 20 min and 2 h time points with concentrations of 75 μM and 100 μM, which indicated the loss of mitochondrial membrane potential in cells. Imaging of the stained cells under fluorescence microscopy also demonstrated the results by observations of decreased red fluorescence and increased green fluorescence in 2-AAPA treated cells (data not shown).
Figure 6.
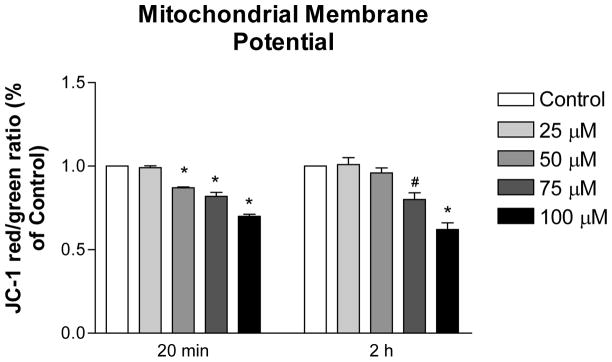
Mitochondrial membrane potential in the control and 2-AAPA treated H9c2 cells. The data are presented as the ratio of JC-1 red/green fluorescence from three independent experiments and normalized with control group. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 and * p < 0.01 compared with control group.
2-AAPA induced cytochrome c release from mitochondria
The mitochondrial integrity in 2-AAPA treated H9c2 cells was evaluated by determining the cytochrome c in cytosol. Western blotting results are shown in Figure 7. Release of cytochrome c to cytosol from mitochondria was detected as early as 20 minutes in 100 μM 2-AAPA treated H9c2 cells.
Figure 7.
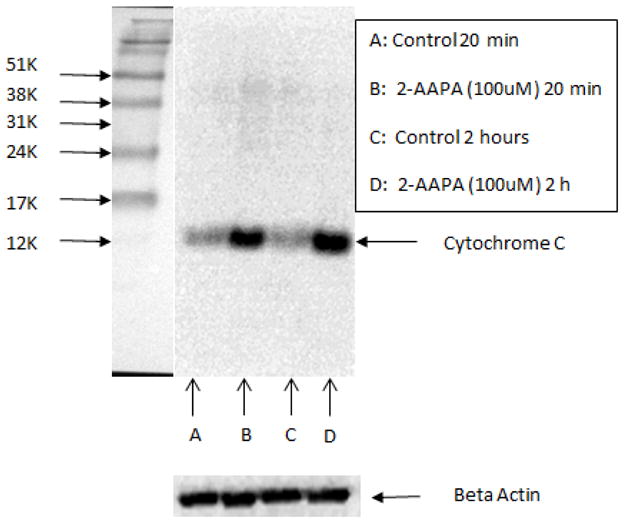
Cytochrome c release in the control (A: 20 min; C: 2 h) and 100 μM 2-AAPA treated H9c2 cells (B: 20 min; D: 2h) determined by Western blot analysis.
H2O2, diamide, BSO and BCNU induced less extent of thiol oxidative stress in H9c2 cells
The extent of thiol oxidative stress produced by 2-AAPA was compared to existing pharmacological models of thiol oxidative stress including H2O2, diamide, BSO and BCNU at a concentration of 100 μM. Intracellular GSH, GSSG, protein thiols, protein disulfides and S-glutathionylation were measured and summarized in Figure 8. Depletion of both GSH and GSSG was seen in BSO and BCNU treated H9c2 cells. Slight depletion of GSH (33%) was also found in H2O2 treated cells but no significant difference was observed in GSSG compared to control. For diamide, no significant difference in either GSH or GSSG was found between control and treatment at concentration of 100 μM, and depletion of GSH and an increase of GSSG were seen only when the concentration of diamide was increased to 250 μM (data not shown). GSH/GSSG ratios were decreased in BCNU (81:1) and H2O2 (91:1) treated cells. No significant difference was found when comparing the GSH/GSSG ratio among diamide, BSO and control. For protein thiols, slight depletion was observed in H2O2 (20%) and BCNU (8%) treated H9c2 cells and no significant difference was found in diamide and BSO treated cells compared to control. Increased protein disulfides were only seen in in H2O2 and BCNU treated cells.
Figure 8.


Intracellular GSH (A), GSSG (B), GSH/GSSG (C), protein thiols (D), protein disulfides (E) in the control and H2O2, diamide, BSO and BCNU treated H9c2 cells. The data are presented as nmol/mg protein and expressed as the means ± SD of three independent experiments. One-way ANOVA and Tukey’s post hoc test were performed to analyze data at each time point. # p < 0.05 and * p < 0.01 compared with control group.
Discussion
GR inhibition could serve as a valuable research tool in creating thiol oxidative stress to study TRS-related normal and abnormal biochemical processes in cardiovascular diseases. For various reasons, BCNU has been the most commonly used GR inhibitor in the literature. However, the DNA-alkylating property complicates the use of BCNU as a GR inhibitor. Therefore, a GR inhibitor that can selectively modify intracellular TRS is more desirable. In this study, we used a dithiocarbamate derivative, 2-AAPA, to create a model of thiol oxidative stress in cardiomyocytes. The compound was initially designed and developed as a GR inhibitor, although later it was also found to be an inhibitor of glutaredoxin. Overall this compound is still a selective TRS modulator as it showed only minimal inhibition of two glutathione-related enzymes, glutathione S-transferase and glutathione peroxidase, and had no inhibition of the enzymes involved in GSH synthesis or other antioxidant enzymes including catalase and superoxide dismutase [38].
We assessed the impact of 2-AAPA on thiol oxidative stress parameters and cellular functions in H9c2 rat cardiomyocytes. This cell line was chosen because it has been used in numerous literature reports studying the effects of oxidative stress in cardiovascular diseases [5, 25–27, 29, 30, 35]. In addition, this cell line is relatively easy to maintain. We selected four concentrations of 2-AAPA including 25, 50, 75 and 100 μM to be evaluated in the study. The selection was based on the dose-response curve from cell viability assays, and concentrations of 50 μM and below produced over 80% viability.
As demonstrated by GR activity assays, 2-AAPA produced GR inhibition in H9c2 cells. The maximal GR inhibition was observed within the first two hours, and the GR activity started to return afterward and reached to the normal level of control at the end of 12 h (Figure 2). 2-AAPA is known to inhibit GR through thiocarbamoylation of thiols at the active site. Although the primary mode of 2-AAPA inhibition is irreversible, the labile thiocarbamate bonds formed between 2-AAPA and enzyme might have undergone hydrolysis to release the enzyme during incubation in cells [38]. Another possible explanation for the return of GR activity in H9c2 cells is the synthesis of new GR proteins as indicated in an earlier reported literature [44]. This paper reported a 50% return of GR activity in a murine leukemia cell line within 12 h after completely inhibition by BCNU, which is also an irreversible GR inhibitor. The return of activity observed in this paper was demonstrated to be dependent on the synthesis of new proteins [44].
By inhibiting GR, 2-AAPA blocked the reduction of GSSG to GSH resulting in significant accumulation of GSSG in H9c2 cells as expected (Figure 3). However, there is a discrepancy between the GR activity and the GSSG accumulation as no drop in GSSG was observed as the GR activity returned. This discrepancy is likely due to the limited capacity of GR in cardiomyocytes as the heart tissue has been reported to have a low GR activity compared to other major organs [45]. In this study, the basal GR activity in control H9c2 cells was determined to be 0.033 units/mg protein which is much lower than in a monkey kidney cell line (0.3 units/mg protein) [40]. Therefore, even though there is return of GR activity observed in H9c2 cells, the limited GR capacity might not be enough to effectively decrease the accumulated GSSG. Another possible explanation for the discrepancy is the limitation of GSSG efflux system either by the saturation of accumulated GSSG or even inhibition by 2-AAPA. In a separate study in this laboratory, we have found that verapamil, which has been reported to inhibit GSSG efflux [46], increased GSSG accumulation (~3 fold) in BCNU treated H9c2 cells (Xie J and Seefeldt T, unpublished data), and interestingly BCNU alone as a GR inhibitor was shown to deplete GSSG instead of causing GSSG accumulation as indicated in Figure 8. This indirectly indicated that GSSG efflux might be inhibited in 2-AAPA treated H9c2 cells leading to the persistent accumulation of GSSG despite returned GR activity. However, there is also other alternative possibility regarding GSSG transport system for explanation of the discrepancy. A recent published study revealed that GSSG that is not immediately reduced in the cytosol is rapidly transported into the vacuole in yeast cells by the ABC-C transporter Ycf1 [47]. Although further evidences are needed, it is possible that mammalian cells under oxidative stress can also transport cytosolic GSSG into a subcellular compartment such as endoplasmic reticulum that is not accessible for reduction by the newly synthesized cytosolic GR. If so, the discrepancy might be due to the inability of the method employed to distinguish the cytosolic GSSG from subcellular GSSG as the GSSG levels were measured after lysis of the complete cells.
For intracellular GSH, we initially expected the GSH to be depleted to some degree by 2-AAPA. However, the depletion was observed only in the treatment of 100 μM 2-AAPA at the first 20 minutes. Intracellular GSH was unexpectedly increased when the cells were treated with lower concentrations of 2-AAPA for longer times (Figure 3). The elevation of GSH is likely due to an increase in γ-glutamylcysteine synthetase (γ-GCS) activity, which currently is being investigated in a separate study. γ-GCS is an enzyme responsible for catalyzing the rate limiting reaction in GSH de novo synthesis. Even though the GSH level was elevated substantially (23–154%), the more profound accumulation in GSSG (up to 20 fold) significantly decreased the GSH/GSSG ratio in 2-AAPA treated H9c2 cells (165:1 in control compared to 11:1–108:1 in 2-AAPA treated, Figure 3). Because the cells were completely lysed by 3% sulfosalicylic acid, the measured GSH/GSSG ratios reflect the overall redox state in H9c2 cells. In literatures, the reported GSH/GSSG ratios are highly varied due to the different methods employed in measurement. Therefore, it is difficult to compare the GSH/GSSG ratios measured in this paper to those reported in other literatures.
Nevertheless, the significantly decreased GSH/GSSG ratios in 2-AAPA treated H9c2 cells indicated a state of thiol oxidative stress created by 2-AAPA. Besides decreasing the GSH/GSSG ratio, 2-AAPA also increased the protein disulfide bond formation as well as S-glutathionylation in H9c2 cells (Figure 4). The increase in protein disulfides is consistent with the finding of inhibited TrxR activity in this study. TrxR is the enzyme catalyzing the reduction of Trx and hence is the central component in the Trx system. Together with Trx and NADPH, this system is responsible for protein disulfide reduction in cells and is important in regulating TRS. The Trx/TrxR system has also been shown to have important role in the development of cardiovascular diseases [4]. The ability to inhibit TrxR is therefore a strong asset to 2-AAPA in creating the thiol oxidative stress model in H9c2 cells.
The altered TRS induced by 2-AAPA readily impacts the normal cellular functions of cardiomyocytes as demonstrated by the decreased mitochondrial membrane potential and the release of cytochrome c from mitochondria observed at the first 20 min. The disruption in mitochondrial function is likely due to the opening of the permeability transition pore, which is sensitive to a change in TRS and has been implicated in ischemia-reperfusion injury in heart [48, 49]. Release of cytochrome c subsequently initiated the apoptosis processes in H9c2 cells as detected in Annexin-V staining assays (data not shown). Interestingly, while cytochrome c release has been detected as early as in 20 min, no increase in ROS was detected in 2-AAPA treated H9c2 cells at the first hour. A significant increase in ROS was detected only when the cells were treated for a longer time which is not unexpected as a disruption in mitochondrial electron transport chain will cause leakage of free radicals.
Compared to other pharmacological models of oxidative stress including H2O2, diamide, BSO and BCNU, 2-AAPA has several advantages in creating a state of thiol oxidative stress in H9c2 cells. First, 2-AAPA has relatively lower cytotoxicity in H9c2 cells. The IC50 of 2-AAPA in H9c2 cells was determined to be 68 ± 7 μM, which is much higher than that of BSO (IC50 = 16 ± 5 μM). In addition, while BSO requires a long incubation time (12–24h) to deplete the intracellular GSH, 2-AAPA alters TRS almost instantly as a decreased GSH/GSSG ratio was detected as early as 20 min.
Second, 2-AAPA is more potent in increasing intracellular GSSG and decreasing GSH/GSSG. As indicated earlier, a decrease in GSH/GSSG has been shown to strongly correlate with oxidative stress induced cardiac injuries [17–19]. Although lower GSH/GSSG ratios have also been observed in H2O2 and BCNU treated cells (not in BSO and diamide treated cells, Figure 8), the decreases were due to a more significant depletion of GSH and not because of an actual increase in GSSG. In fact, other than 2-AAPA, no significant increase in GSSG has been found with any compounds selected in this study, and even in BSO and BCNU treated cells, intracellular GSSG was significantly depleted as well as GSH (Figure 8). By contrast, even the lowest concentration of 2-AAPA (25 μM) studied can produce a 3-fold increase in GSSG in 2 h. Such an increase in GSSG might have more physiological relevance with cardiovascular diseases, because myocardial GSSG has been shown to significantly increase in the left ventricle (up to 4 fold) during heart failure in a coronary artery ligation model of myocardial infarction in rats [50].
Third, 2-AAPA is able to induce protein S-glutathionylation in H9c2 cells which was not detected with other compounds selected in this study. Protein S-glutathionylation is involved in redox regulation of various cellular protein functions and is known to maintain sulfhydryl homeostasis by protecting protein cysteine residues from irreversible oxidation under oxidative stress conditions [8, 51]. It also has been shown to have important implications in the development of cardiac injury such as ischemia-reperfusion injury as indicated earlier. Deglutathionylation of proteins are mainly catalyzed by the enzyme glutaredoxin, and 2-AAPA has been identified as an irreversible inhibitor of human glutaredoxin-1 recently [39]. The ability of 2-AAPA in inducing protein S-glutathionylation makes it a good tool for studying the important role of redox signaling in the development of cardiovascular diseases.
Last but not least, the effect of 2-AAPA on intracellular TRS and cellular functions of H9c2 cells is concentration and time dependent. Thus, 2-AAPA might be capable to meet the needs of different cardiovascular research by selection of different concentrations and incubation times. For example, 2-AAPA does not induce ROS production in H9c2 cells within the first hour. This can differentiate 2-AAPA from H2O2, which produces a general state of oxidative stress with nonspecific oxidative nature, and makes 2-AAPA a more specific model of thiol oxidative stress for cardiovascular diseases.
Conclusions
In conclusion, 2-AAPA is capable of producing a state of thiol oxidative stress in H9c2 rat cardiomyocytes by increasing the intracellular GSSG, protein disulfides and S-glutathionylation. The alteration in TRS elicits a disruption in normal mitochondrial and cellular functions. In comparison to other compounds, 2-AAPA is superior as a model of thiol oxidative stress with lower cytotoxicity, higher specificity and more pharmacological relevance. The use of 2-AAPA as a model of thiol oxidative stress in H9c2 rat cardiomyocytes has been extensively and successfully employed in this laboratory to screen and study compounds in protection of heart against abnormal TRS.
Acknowledgments
Funding for this project was provided by a grant from the National Institutes of Health-National Heart, Lung, and Blood Institute (R15 HL102777).
Abbreviations
- TRS
thiol redox state
- 2-AAPA
2-acetylamino-3-[4-(2-acetylamino-2-carboxyethylsulfanylthiocarbonylamino)phenylthiocarbamoylsulfanyl]propionic acid
- GSH
reduced form glutathione
- GSSG
oxidized form of glutathione or glutathione disulfide
- ROS
reactive oxygen species
- GR
glutathione reductase
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- BCNU
N,N′-bis(2-chloroethyl)-N-nitroso-urea
- BSO
buthionine sulfoximine
- MTT
3-(4,5)-dimethylthiazol-2,5-diphenyltetrazolium bromide
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- TNB
2-nitro-5-thiobenzoic acid
- GS-TNB
the conjugate of GSH with 2-nitro-5-thiobenzoic acid
- PBS
phosphate-buffered saline
- EDTA
ethylenediaminetetraacetic acid
- DMSO
dimethyl sulfoxide
- DMEM
Dulbecco’s Modified Eagle’s medium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. The international journal of biochemistry & cell biology. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari R, Alfieri O, Curello S, Ceconi C, Cargnoni A, Marzollo P, Pardini A, Caradonna E, Visioli O. Occurrence of oxidative stress during reperfusion of the human heart. Circulation. 1990;81:201–211. doi: 10.1161/01.cir.81.1.201. [DOI] [PubMed] [Google Scholar]
- 3.Go YM, Jones DP. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation. 2005;111:2973–2980. doi: 10.1161/CIRCULATIONAHA.104.515155. [DOI] [PubMed] [Google Scholar]
- 4.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. American journal of physiology. Heart and circulatory physiology. 2007;292:H1227–1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 5.Shi R, Huang CC, Aronstam RS, Ercal N, Martin A, Huang YW. N-acetylcysteine amide decreases oxidative stress but not cell death induced by doxorubicin in H9c2 cardiomyocytes. BMC pharmacology. 2009;9:7. doi: 10.1186/1471-2210-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovascular toxicology. 2001;1:181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 7.Pullela PK, Chiku T, Carvan MJ, 3rd, Sem DS. Fluorescence-based detection of thiols in vitro and in vivo using dithiol probes. Analytical biochemistry. 2006;352:265–273. doi: 10.1016/j.ab.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 8.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Current opinion in pharmacology. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Arrigo AP. Gene expression and the thiol redox state. Free radical biology & medicine. 1999;27:936–944. doi: 10.1016/s0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 10.Patsoukis N, Georgiou CD. Determination of the thiol redox state of organisms: new oxidative stress indicators. Analytical and bioanalytical chemistry. 2004;378:1783–1792. doi: 10.1007/s00216-004-2525-1. [DOI] [PubMed] [Google Scholar]
- 11.Zeevalk GD, Razmpour R, Bernard LP. Glutathione and Parkinson’s disease: is this the elephant in the room? Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2008;62:236–249. doi: 10.1016/j.biopha.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free radical biology & medicine. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 13.Dickinson DA, Forman HJ. Cellular glutathione and thiols metabolism. Biochemical pharmacology. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- 14.Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochemical pharmacology. 2006;71:551–564. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 15.Ghezzi P, Bonetto V, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxidants & redox signaling. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 16.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxidants & redox signaling. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh A, Lee KJ, Lee CY, Goldfarb RD, Tsan MF. Relation between myocardial glutathione content and extent of ischemia-reperfusion injury. Circulation. 1989;80:1795–1804. doi: 10.1161/01.cir.80.6.1795. [DOI] [PubMed] [Google Scholar]
- 18.Hoshida S, Kuzuya T, Yamashita N, Nishida M, Kitahara S, Hori M, Kamada T, Tada M. gamma-Glutamylcysteine ethyl ester for myocardial protection in dogs during ischemia and reperfusion. Journal of the American College of Cardiology. 1994;24:1391–1397. doi: 10.1016/0735-1097(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 19.Kawai K, Qin F, Shite J, Mao W, Fukuoka S, Liang CS. Importance of antioxidant and antiapoptotic effects of beta-receptor blockers in heart failure therapy. American journal of physiology. Heart and circulatory physiology. 2004;287:H1003–1012. doi: 10.1152/ajpheart.00797.2003. [DOI] [PubMed] [Google Scholar]
- 20.Gumieniczek A, Hopkala H, Wojtowicz Z, Nikolajuk J. Changes in antioxidant status of heart muscle tissue in experimental diabetes in rabbits. Acta biochimica Polonica. 2002;49:529–535. [PubMed] [Google Scholar]
- 21.Chen FC, Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. American journal of physiology. Cell physiology. 2006;290:C719–727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 22.Reddy S, Jones AD, Cross CE, Wong PS, Van Der Vliet A. Inactivation of creatine kinase by S-glutathionylation of the active-site cysteine residue. The Biochemical journal. 2000;347(Pt 3):821–827. [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Tang K, Xie B, Li S, Rozanski GJ. Regulation of Kv4 channel expression in failing rat heart by the thioredoxin system. American journal of physiology. Heart and circulatory physiology. 2008;295:H416–424. doi: 10.1152/ajpheart.91446.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inafuku H, Kuniyoshi Y, Yamashiro S, Arakaki K, Nagano T, Morishima Y, Kise Y. Determination of Oxidative Stress and Cardiac Dysfunction after Ischemia/Reperfusion Injury in Isolated Rat Hearts. Annals of thoracic and cardiovascular surgery : official journal of the Association of Thoracic and Cardiovascular Surgeons of Asia. 2012 doi: 10.5761/atcs.oa.12.01896. [DOI] [PubMed] [Google Scholar]
- 25.Park C, So HS, Shin CH, Baek SH, Moon BS, Shin SH, Lee HS, Lee DW, Park R. Quercetin protects the hydrogen peroxide-induced apoptosis via inhibition of mitochondrial dysfunction in H9c2 cardiomyoblast cells. Biochemical pharmacology. 2003;66:1287–1295. doi: 10.1016/s0006-2952(03)00478-7. [DOI] [PubMed] [Google Scholar]
- 26.Han H, Long H, Wang H, Wang J, Zhang Y, Wang Z. Progressive apoptotic cell death triggered by transient oxidative insult in H9c2 rat ventricular cells: a novel pattern of apoptosis and the mechanisms. American journal of physiology. Heart and circulatory physiology. 2004;286:H2169–2182. doi: 10.1152/ajpheart.00199.2003. [DOI] [PubMed] [Google Scholar]
- 27.Turner NA, Xia F, Azhar G, Zhang X, Liu L, Wei JY. Oxidative stress induces DNA fragmentation and caspase activation via the c-Jun NH2-terminal kinase pathway in H9c2 cardiac muscle cells. Journal of molecular and cellular cardiology. 1998;30:1789–1801. doi: 10.1006/jmcc.1998.0743. [DOI] [PubMed] [Google Scholar]
- 28.Lenz ML, Michael LH, Smith CV, Hughes H, Shappell SB, Taylor AA, Entman ML, Mitchell JR. Glutathione disulfide formation and lipid peroxidation during cardiac ischemia and reflow in the dog in vivo. Biochemical and biophysical research communications. 1989;164:722–727. doi: 10.1016/0006-291x(89)91519-2. [DOI] [PubMed] [Google Scholar]
- 29.Luo C, Li Y, Wang H, Feng Z, Li Y, Long J, Liu J. Mitochondrial accumulation under oxidative stress is due to defects in autophagy. Journal of cellular biochemistry. 2013;114:212–219. doi: 10.1002/jcb.24356. [DOI] [PubMed] [Google Scholar]
- 30.Park J, Park E, Ahn BH, Kim HJ, Park JH, Koo SY, Kwak HS, Park HS, Kim DW, Song M, Yim HJ, Seo DO, Kim SH. NecroX-7 prevents oxidative stress-induced cardiomyopathy by inhibition of NADPH oxidase activity in rats. Toxicology and applied pharmacology. 2012;263:1–6. doi: 10.1016/j.taap.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 31.Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochemical and biophysical research communications. 1969;37:593–596. doi: 10.1016/0006-291x(69)90850-x. [DOI] [PubMed] [Google Scholar]
- 32.Tsunoda T, Yamamiya Y, Kawamura Y, Ito S. Mitsunobu Acylation of Sterically Congested Secondary Alcohols by N,N,N′,N′-Tetramethylazodicarboxamide-Tributylphosphine Reagents. Tetrahedron Lett. 1995;36:2529–2530. [Google Scholar]
- 33.Tsunoda T, Uemoto K, Nagino C, Kawamura M, Kaku H, Ito S. A facile one-pot cyanation of primary, secondary alcohols. Application of some new Mitsunobu reagents. Tetrahedron Lett. 1999;40:7355–7358. [Google Scholar]
- 34.Li S, Li X, Rozanski GJ. Regulation of glutathione in cardiac myocytes. Journal of molecular and cellular cardiology. 2003;35:1145–1152. doi: 10.1016/s0022-2828(03)00230-x. [DOI] [PubMed] [Google Scholar]
- 35.Chiu PY, Ko KM. Schisandrin B-induced increase in cellular glutathione level and protection against oxidant injury are mediated by the enhancement of glutathione synthesis and regeneration in AML12 and H9c2 cells. BioFactors. 2006;26:221–230. doi: 10.1002/biof.5520260401. [DOI] [PubMed] [Google Scholar]
- 36.FitzGerald GB, Bauman C, Hussoin MS, Wick MM. 2,4-Dihydroxybenzylamine: a specific inhibitor of glutathione reductase. Biochemical pharmacology. 1991;41:185–190. doi: 10.1016/0006-2952(91)90475-k. [DOI] [PubMed] [Google Scholar]
- 37.Kann HE, Jr, Schott MA, Petkas A. Effects of structure and chemical activity on the ability of nitrosoureas to inhibit DNA repair. Cancer research. 1980;40:50–55. [PubMed] [Google Scholar]
- 38.Seefeldt T, Zhao Y, Chen W, Raza AS, Carlson L, Herman J, Stoebner A, Hanson S, Foll R, Guan X. Characterization of a novel dithiocarbamate glutathione reductase inhibitor and its use as a tool to modulate intracellular glutathione. The Journal of biological chemistry. 2009;284:2729–2737. doi: 10.1074/jbc.M802683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sadhu SS, Callegari E, Zhao Y, Guan X, Seefeldt T. Evaluation of a dithiocarbamate derivative as an inhibitor of human glutaredoxin-1. Journal of enzyme inhibition and medicinal chemistry. 2013;28:456–462. doi: 10.3109/14756366.2011.649267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Seefeldt T, Chen W, Wang X, Matthees D, Hu Y, Guan X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Archives of biochemistry and biophysics. 2009;485:56–62. doi: 10.1016/j.abb.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Archives of biochemistry and biophysics. 1993;303:474–482. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- 42.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 43.Chen W, Zhao Y, Seefeldt T, Guan X. Determination of thiols and disulfides via HPLC quantification of 5-thio-2-nitrobenzoic acid. Journal of pharmaceutical and biomedical analysis. 2008;48:1375–1380. doi: 10.1016/j.jpba.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen MB, Duvel DL. Characterization of the inhibition of glutathione reductase and the recovery of enzyme activity in exponentially growing murine leukemia (L1210) cells treated with 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemical pharmacology. 1988;37:3317–3320. doi: 10.1016/0006-2952(88)90645-4. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki M, Muraoka H, Kurata M, Agar NS. Glutathione reductase activity and flavin concentration in guinea-pig tissues. Experimental animals / Japanese Association for Laboratory Animal Science. 1999;48:199–202. doi: 10.1538/expanim.48.199. [DOI] [PubMed] [Google Scholar]
- 46.Ellison I, Richie JP., Jr Mechanisms of glutathione disulfide efflux from erythrocytes. Biochemical pharmacology. 2012;83:164–169. doi: 10.1016/j.bcp.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 47.Morgan B, Ezerina D, Amoako TN, Riemer J, Seedorf M, Dick TP. Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nature chemical biology. 2013;9:119–125. doi: 10.1038/nchembio.1142. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. The Journal of biological chemistry. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sloan RC, Moukdar F, Frasier CR, Patel HD, Bostian PA, Lust RM, Brown DA. Mitochondrial permeability transition in the diabetic heart: contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. Journal of molecular and cellular cardiology. 2012;52:1009–1018. doi: 10.1016/j.yjmcc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 50.Hill MF, Singal PK. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation. 1997;96:2414–2420. doi: 10.1161/01.cir.96.7.2414. [DOI] [PubMed] [Google Scholar]
- 51.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-glutathionylation in protein redox regulation. Free radical biology & medicine. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]