

Abstract
In this report we present a new strategy for targeting chemotherapeutics to tumors, based on targeting extracellular DNA. A gemcitabine prodrug was synthesized, termed H-gemcitabine, which is composed of Hoechst conjugated to gemcitabine. H-gemcitabine has low toxicity because it is membrane impermeable, however it still has high tumor efficacy because of its ability to target gemcitabine to E-DNA in tumors. We demonstrate here that H-gemcitabine has a wider therapeutic window than free gemcitabine.
Gemcitabine is an anticancer drug with tremendous clinical potential, but has had limited efficacy due to its high toxicity and inactivation in serum, due to deamination of its N-4 amine.1-3 Numerous gemcitabine prodrugs have been developed, which are capable of protecting against deamination, however, the clinical efficacy of these prodrugs has been limited because of their high toxicity.4-6
In this report we present for the first time a gemcitabine prodrug, termed H-gemcitabine, which has a wider therapeutic window than gemcitabine. H-gemcitabine (1) is composed of the DNA binding agent Hoechst conjugated to gemcitabine (Figure 1). H-gemcitabine is designed to reduce the systemic toxicity of gemcitabine by targeting it to the necrotic core of tumors, and also protect against deamination of its N-4 amine via protection with an amide bond. H-gemcitabine targets tumors via its Hoechst moiety, which binds extracellular DNA (E-DNA) present in the necrotic core of tumors.7 We show here that H-gemcitabine is significantly more effective than free gemcitabine at treating xenograft human colon tumors in nude mice, and also has a maximum tolerated dose equivalent to gemcitabine. We anticipate numerous applications of H-gemcitabine, given its unique combination of high efficacy and low toxicity.
Figure 1.

Chemical structure of H-gemcitabine (1). H-gemcitabine is a gemcitabine prodrug designed to target extracellular DNA in tumors.
The synthesis of H-gemcitabine (1) is described in Scheme 1 and followed a convergent route in which the two fragments 4 and 7 were synthesized and coupled to obtain 1. Our initial synthetic strategies focused on modifying commercially available Hoechst directly at its phenolic hydroxyl. However we were unable to alkylate Hoechst under a variety of experimental conditions and therefore had to base our synthetic scheme around the previously synthesized Hoechst derivative 2.7 The compound 4 is a thiol functionalized Hoechst and was obtained in two steps from 2 in a 50% yield. The compound 7 is a gemcitabine derivative with a thiopyridyl group and was prepared in three steps from commercially available gemcitabine (5). In detail, TBTU mediated acylation of 5 with 3-(tritylthio)propionic acid 3 followed by deprotection of the trityl group generated the thiol 6 (48.5% yield, for both steps). Activation of the thiol 6 with 2,2'-dithiodipyridine provided 7 in 41% yield. Finally, H-gemcitabine (1) was obtained via a disulfide exchange reaction between 4 and 7 in 30% yield.
Scheme 1. Synthesis of H-gemcitabine (1).

We measured the binding affinity of H-gemcitabine to DNA (a 21 bp oligonucleotide), to determine if attachment of gemcitabine lowers the binding affinity of Hoechst to DNA. Figure S1 (Supporting Information) demonstrates that H-gemcitabine has a DNA dissociation constant (Kd) of 11 nM, which is similar in magnitude to free Hoechst,8-10 and indicates that the attachment of gemcitabine does not interfere with the ability of Hoechst to bind DNA. Importantly, H-gemcitabine's affinity for DNA is similar in magnitude to numerous other ligand receptor pairs used for targeting drugs to tumor, such as antibodies.11,12
H-gemcitabine is designed to circulate through tissue, accumulate in tumors, and then release free gemcitabine. H-gemcitabine has two cleavable linkages, a disulfide and an aromatic amide and the hydrolysis rate of H-gemcitabine's aromatic amide is a key factor that will influence the efficacy in vivo. To obtain an understanding of the amide bond hydrolysis in H-gemcitabine, we performed a semiquantitative study of the degradation kinetics of H-gemcitabine, in phosphate buffer (pH 7.4) at 37 °C, via high-performance liquid chromatography (HPLC). Figure 2 demonstrates that in phosphate buffer the aromatic amide of H-gemcitabine undergoes hydrolytic cleavage to release free gemcitabine and has a half life of approximately 11.4 h, thus suggesting that H-gemcitabine will have sufficient time to circulate through the body and accumulate within the tumors.
Figure 2.

Time dependent release kinetics of free gemcitabine (5). Gemcitabine is released from H-gemcitabine (1) with a half life of 11.4 h (mean ± STD, n=3).
A key requirement for H-gemcitabine is that it should be membrane impermeable, to prevent it from binding intracellular DNA. We evaluated the membrane permeability of H-gemcitabine in HT29 cells (a human colon cancer cell line) and in methanol permeabilized HT29 cells. Figure 3a demonstrates that H-gemcitabine has minimal permeability to live cells. For example, live cells incubated with H-gemcitabine generate low levels of intracellular fluorescence (left panel in Figure 3a), indicating that H-gemcitabine has very low cell permeability, presumably because of the hydrophilicity of gemcitabine and the large size of H-gemcitabine. In contrast, methanol permeabilized cells incubated with H-gemcitabine generated high levels of intracellular fluorescence (right panel in Figure 3a). This confirms that the low level of fluorescence observed in live cells incubated with H-gemcitabine was due to its low membrane permeability and not due to a reduction in its DNA-binding ability.
Figure 3.
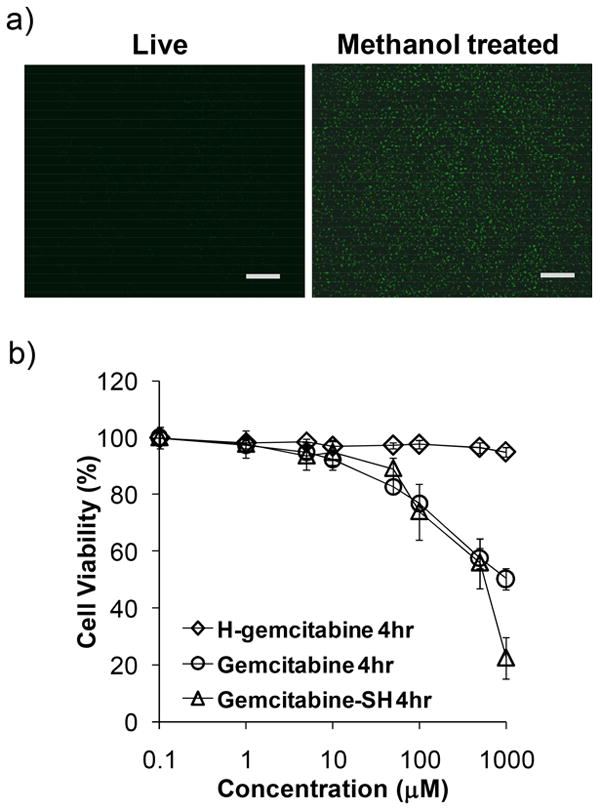
H-gemcitabine is membrane impermeable and has minimal toxicity. a) Cell permeability of H-gemcitabine. Live or methanol fixed monolayer cells (HT29) were treated with H-gemcitabine. Live cells treated with H-gemcitabine have low cellular fluorescence (the scale bar indicates 100 μm). b) Cell toxicity of H-gemcitabine. HT29 monolayer cells were treated with either H-gemcitabine, free gemcitabine or gemcitabine-SH for 4 h. H-gemcitabine has minimal toxicity at a 1 mM concentration (diamonds) (mean ± STD, n=9).
H-gemcitabine is a targeted anticancer prodrug and needs to have minimal toxicity in its intact form, however after targeting tumors the released free gemcitabine or gemcitabine-SH (6) needs to have high anticancer efficacy. We therefore measured the toxicities of H-gemcitabine, free gemcitabine, and gemcitabine-SH on HT29 cells after a 4 hour incubation, using the MTT assay.13 Figure 3b demonstrates that H-gemcitabine has minimal toxicity to HT29 cells after 4 hours of incubation at a concentration of 1 mM, whereas free gemcitabine and gemcitabine-SH caused over 50% toxicity under these conditions. Gemcitabine needs to be intracellular to kill cells, and H-gemcitabine's low toxicity is therefore probably due to its membrane impermeability. Finally, gemcitabine-SH had a similar level of cell toxicity as free gemcitabine, which is consistent with previous gemcitabine modifications at its amine terminus.14
Finally, we investigated if H-gemcitabine had greater efficacy in vivo than free gemcitabine, and if it could target tumors. Colon tumor cells were implanted subcutaneously into nude mice and the mice were treated with four doses once every three days, with either H-gemcitabine (25 mg/kg of gemcitabine equivalents, 95 μmol/kg), or free gemcitabine (100 mg/kg, 380 μmol/kg). These doses of H-gemcitabine and gemcitabine were chosen because they represent the maximum possible doses that could be given via an i.v. injection, due to solubility limitations in the case of H-gemcitabine or toxicity issues in the case of gemcitabine. The tumor size, lifespan, and the accumulation of H-gemcitabine in the tumors was then measured.
Figure 4a demonstrates that H-gemcitabine at a dose of 25 mg/kg of gemcitabine equivalents is significantly more effective than free gemcitabine given at a 100 mg/kg. For example, the tumor doubling time for gemcitabine treated tumors was only 6 days, whereas for H-gemcitabine it was 15 days. In addition, H-gemcitabine treated mice had a significantly higher survival rate than gemcitabine treated mice, having a survival rate of 80% after 30 days, as opposed to only 13% for free gemcitabine (Figure 4b). In addition we measured the accumulation of H-gemcitabine in the tumors via analysis of Hoechst fluorescence, and observed accumulation of 1.7±0.1% of the injected dose, per gram tissue. Importantly, tumor studies done with free gemcitabine demonstrate that gemcitabine does not have significant accumulation within tumors after 24 hours,15,16 suggesting that H-gemcitabine has the potential to target tumors in vivo and increase the efficacy of gemcitabine.
Figure 4.

H-gemcitabine has greater efficacy than gemcitabine in treating tumors in vivo. a) Tumor bearing nude mice were injected with either H-gemcitabine (25 mg/kg of gemcitabine equivalents, 95 μmol/kg) or free gemcitabine (100 mg/kg, 380 μmol/kg) on days 0, 3, 6, and 9. Tumors treated with H-gemcitabine have minimal growth, whereas tumors treated with gemcitabine increase in size (mean ± STD, n=5, *p≤0.05 vs gemcitabine). b) Tumor bearing mice have a higher survival rate at 30 days after treatment with H-gemcitabine (80%) than with free gemcitabine (13%). The Survival curve was generated by determining the percentage of mice surviving with respect to the start of the study. Log-rank test was performed to determine statistical significance between the survival curves (*p < 0.05, n = 10 for H-gemcitabine and n = 9 for gemcitabine).
Finally, we investigated the toxicity of H-gemcitabine and gemcitabine, to determine if H-gemictabine's therapeutic window will be wider than free gemcitabine's. Figure 5 demonstrates that neither H-gemcitabine nor free gemcitabine, had toxicity at the dose used in the tumor study presented in Figure 5. To assess H-gemcitabine's toxicity we performed a chronic toxicity study, in which H-gemcitabine and free gemcitabine were given via a daily i.p. injection. Figure S2 (Supporting Information) demonstrates that H-gemcitabine and gemcitabine have similar levels of toxicity, demonstrating that the therapeutic window of H-gemcitabine is wider than free gemcitabine.
Figure 5.

H-gemcitabine has similar toxicity to gemcitabine. Tumor bearing nude mice were injected with either H-gemcitabine (25 mg/kg of gemcitabine equivalents) or free gemcitabine (100 mg/kg) on days 0, 3, 6, and 9 via the jugular vein.
In summary, we demonstrate here that efficacy of gemcitabine can be dramatically improved by conjugating it to the DNA binding agent Hoechst. We anticipate numerous applications of H-gemcitabine and the general strategy of targeting E-DNA, given the widespread presence of E-DNA in tumors and its ability to be targeted with small molecules.
Experimental Section
General chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and were used as received, unless otherwise specified. Gemcitabine (2'-Deoxy-2',2'-difluoro-D-cytidine) was purchased from Carbosynth Ltd. (Berkshire, UK). Dimethyl formamide, ethanol, dichloromethane and methanol were purified using standard methods. Flash chromatography was carried out using silica gel (230-400 mesh). NMR spectra were recorded on a Varian spectrometer (400 MHz for 1H NMR and 100 MHz for 13C) with chloroform (7.26) or methanol (3.30) as internal reference. High-resolution mass spectra (HRMS) were obtained on a Karatos MS9 and are reported as m/z (relative intensity). Accurate masses are reported for the molecular ion (M+) or a suitable fragment ion.
Synthesis of H-gemcitabine (1)
An oven dried 25 mL flask with a magnetic stir bar was charged with Hoechst thiol 4 (0.08 g, 0.1 mmol) and gemcitabine dithiopyridine 7 (0.12 g, 0.2 mmol) under an argon atmosphere. To this mixture anhydrous DMF (1.4 mL) was added followed by triethylamine (19 μL, 0.1 mmol) and stirred at rt overnight. The solvent was removed under reduced pressure and the resulting residue was purified by silica gel flash chromatography using a gradient of 10-15% methanol in dichloromethane containing 0.1% triethylamine. The compound 1 was obtained as a pale yellow solid (0.04 g, 0.04 mmol) in 30% yield. 1H NMR (400 MHz, CD3OD+DMSO): δ 8.33 (d, J = 7.2 Hz, 2H), 8.14 (d, J = 8.8 Hz, 2H), 8.03 – 7.09 (m, 1H), 7.77 – 7.71 (m, 1H), 7.56 – 7.53 (m, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.19 – 7.17 (m, 3H), 7.08 (dd, J = 9.2, 2.0 Hz, 1H), 6.25 (t, J = 7.2 Hz, 1H), 4.42 (s, 1H), 4.31 - 4.25 (m, 2H), 3.81 - 3.73 (m, 3H), 3.68 – 3.65 (m, 2H), 3.56 (t, J = 5.6 Hz, 2H), 3.38 (t, J = 5.6 Hz, 2H), 3.20 (s, 3H), 3.12 (t, J = 5.2 Hz, 4H), 2.82 (t, J = 6.8 Hz, 4H), 2.76 – 2.74 (m, 2H), 2.55 (t, J = 5.2 Hz, 4H), 2.46 (t, J = 6.8 Hz, 2H), 2.25 (s, 3H); 13C NMR (100 MHz, DMSO): δ 172.6, 170.5, 163.2, 160.5, 154.6, 153.2, 148.1, 145.3, 128.7, 125.9, 124.8, 123.4, 122.9, 118.6, 115.3, 114.0, 111.6, 96.4, 84.5, 84.2, 81.4, 70.3, 70.0, 69.5, 69.3, 69.0, 68.7, 68.5, 67.8, 59.2, 55.3, 50.3, 49.0, 46.2, 46.0, 39.0, 36.7, 35.3, 34.3, 33.1, 33.0, 21.6; HRMS (ESI) m/z: [M+H]+ calcd for C46H55N10O9F2S2, 993.3557; found, 993.3596.
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institute of Health, Department of Health and Human Services, under Contract No. HHSN268201000043C, NSF-BES-0546962 Career Award (N.M.) and NIH RO1 HL096796-01 (N.M.).
Abbreviations
- TBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- DIEA
N,N-diisopropylethylamine
- Trt
triphenylmethyl
- TFA
trifluoroacetic acid
- TES
triethylsilane
Footnotes
Supporting Information. Detailed synthetic procedures and spectroscopic data of all synthesized compounds, cell culture, in vivo tumor and toxicology experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beumer JH, Eiseman JL, Parise RA, Joseph E, Covey JM, Egorin MJ. Modulation of gemcitabine (2',2'-difluoro-2'-deoxycytidine) pharmacokinetics, metabolism, and bioavailability in mice by 3,4,5,6-tetrahydrouridine. Clin Cancer Res. 2008;14:3529–35. doi: 10.1158/1078-0432.CCR-07-4885. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert JA, Salavaggione OE, Ji Y, Pelleymounter LL, Eckloff BW, Wieben ED, Ames MM, Weinshilboum RM. Gemcitabine pharmacogenomics: cytidine deaminase and deoxycytidylate deaminase gene resequencing and functional genomics. Clin Cancer Res. 2006;12:1794–803. doi: 10.1158/1078-0432.CCR-05-1969. [DOI] [PubMed] [Google Scholar]
- 3.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Cellular elimination of 2',2'-difluorodeoxycytidine 5'-triphosphate: a mechanism of self-potentiation. Cancer Res. 1992;52:533–539. [PubMed] [Google Scholar]
- 4.Bergman AM, Adema AD, Balzarini J, Bruheim S, Fichtner I, Noordhuis P, Fodstad O, Myhren F, Sandvold ML, Hendriks HR, Peters GJ. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs. 2011;29:456–466. doi: 10.1007/s10637-009-9377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy LH, Couvreur P. Novel approaches to deliver gemcitabine to cancers. Curr pharm Des. 2008;14:1124–1137. doi: 10.2174/138161208784246216. [DOI] [PubMed] [Google Scholar]
- 6.Pasut G, Canal F, Dalla Via L, Arpicco S, Veronese FM, Schiavon O. Antitumoral activity of PEG-gemcitabine prodrugs targeted by folic acid. J Control Release. 2008;127:239–248. doi: 10.1016/j.jconrel.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Dasari M, Lee S, Sy J, Kim D, Brown M, Davis M, Murthy N. Hoechst-IR: an imaging agent that detects necrotic tissue in vivo by binding extracellular DNA. Org Lett. 2010;12:3300–3303. doi: 10.1021/ol100923d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho J, Rando RR. Specific binding of Hoechst 33258 to site 1 thymidylate synthase mRNA. Nucleic acids Res. 2000;28:2158–2163. doi: 10.1093/nar/28.10.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkinson JA, Barber J, Douglas KT, Rosamond J, Sharples D. Minor-groove recognition of the self-complementary duplex d(CGCGAATTCGCG)2 by Hoechst 33258: a high-field NMR study. Biochemistry. 1990;29:10181–10190. doi: 10.1021/bi00496a005. [DOI] [PubMed] [Google Scholar]
- 10.Loontiens FG, Regenfuss P, Zechel A, Dumortier L, Clegg RM. Binding characteristics of Hoechst 33258 with calf thymus DNA, poly[d(A-T)], and d(CCGGAATTCCGG): multiple stoichiometries and determination of tight binding with a wide spectrum of site affinities. Biochemistry. 1990;29:9029–39. doi: 10.1021/bi00490a021. [DOI] [PubMed] [Google Scholar]
- 11.Goldmacher VS, Kovtun YV. Antibody–drug conjugates: using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther Delivery. 2011;2:397–416. doi: 10.4155/tde.10.98. [DOI] [PubMed] [Google Scholar]
- 12.Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the Dock-and-Lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med. 2008;49:158–163. doi: 10.2967/jnumed.107.046185. [DOI] [PubMed] [Google Scholar]
- 13.Freimoser FM, Jakob CA, Aebi M, Tuor U. The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities. Appl Environ Microbiol. 1999;65:3727–3729. doi: 10.1128/aem.65.8.3727-3729.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Immordino ML, Brusa P, Rocco F, Arpicco S, Ceruti M, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Controll Release. 2004;100:331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Bergman AM, Eijk PP, Ruiz van Haperen VW, Smid K, Veerman G, Hubeek I, Ijssel P, Ylstra B, Peters GJ. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005;65:9510–9516. doi: 10.1158/0008-5472.CAN-05-0989. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Li M, Rinehart JJ, Zhang R. Pretreatment of dexamethasone increases antitumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: in vivo activity, pharmacokinetics, and clinical inplications for cancer chemotherapy. Clin Cancer Res. 2004;10:1633–1644. doi: 10.1158/1078-0432.ccr-0829-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.