

Abstract
Angiotensin converting enzyme 2 (ACE2) is a lately discovered enzyme catalyzing Angiotensin II into Angiotensin (1-7). Angiotensin II has been reported to impair endothelial progenitor cell (EPC) function and is detrimental to stroke. Here, we studied the role of ACE2 in regulating EPC function in vitro and in vivo. EPCs were cultured from human renin and angiotensinogen transgenic (R+A+) mice and their controls (R-A-). In in vitro experiments, EPCs were transduced with lentivirus-ACE2 (Lenti-ACE2) or Lenti-GFP. The effects of ACE2 over-expression on EPC function and endothelial oxide synthase (eNOS)/NADPH oxidase (Nox) expression were determined. ACE2, eNOS and Nox inhibitors were used for pathway validation. In in vivo studies, the therapeutic efficacy of EPCs over-expressing ACE2 was determined at day 7 after ischemic stroke induced by middle cerebral artery occlusion. We found that 1) Lenti-ACE2 transduction resulted in a four-fold increase of ACE2 expression in EPCs. This was accompanied with an increase in eNOS expression and nitric oxide production, and a decrease in Nox2, 4 expression and reactive oxygen species production. 2) ACE2 over-expression improved the abilities of EPC migration and tube formation which were impaired in R+A+ mice. These effects were inhibited by ACE2 or eNOS inhibitor and further enhanced by Nox inhibitor. 3) Transfusion of Lenti-ACE2 primed EPCs reduced cerebral infarct volume and neurologic deficits, increased cerebral microvascular density and angiogenesis. Our data demonstrate that ACE2 improves EPC function via regulating eNOS and Nox pathways and enhances the efficacy of EPC-based therapy for ischemic stroke.
Keywords: ACE2, EPCs, ROS, NO, ischemic stroke
Introduction
Angiotensin converting enzyme 2 (ACE2) is a newly discovered enzyme of the renin-angiotensin system (RAS), which catalyses the conversion of angiotensin I (Ang I) into Ang (1-9) and Ang II into Ang (1-7) (1). Accumulating evidence demonstrate that ACE2 provides vascular protective effects by counteracting the deleterious effects of Ang II and possesses great potential for developing new avenues to treat vascular diseases (2;3). Endothelial cell (EC) dysfunction/injury is well known as the common pathological basis for vascular diseases. It has been reported that ACE2 alleviates the development of early atherosclerotic lesions by improving EC function (4). The antiatherosclerotic effect of ACE2 involves in down-regulation of the Ang II-activated reactive oxygen species (ROS). Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) derived ROS plays a key role in Ang II-induced endothelial damage (5). Oxidative stress induced EC dysfunction is accompanied by the reduction of nitric oxide (NO) bioavailability and plays an important role in cerebrovascular disease (6).
Endothelial progenitor cells (EPCs) which are defined as bone marrow (BM) derived immature cells with the ability to differentiate into mature ECs are suggested to play important roles in vascular homeostasis and angiogenesis (7). Reduced number and impaired function of circulating EPCs are associated with poor cardiovascular outcomes in patients with coronary artery disease (8). It is recently reported that Ang II through type 1 angiotensin (AT1) receptor activation and oxidative stress impairs EPC function in vitro and in vivo (9). Ang II induces EPC senescence via activation of NADPH oxidase (10). Our previous study shows that the BM derived EPCs are reduced and dysfunctional in human renin and angiotensinogen transgenic (R+A+) mice, and that blockade of Ang II/AT1signaling with losartan is able to improve these defeats (11). Endothelial nitric oxide synthase (eNOS) down-regulation and decreased NO generation can also induce impairment of EPC function and senescence (12). Although the role of ACE2 in counteracting the effects of Ang II has been well detailed, the role of ACE2 in EPC function and the specific mechanism have not been explored.
In this study, we determined the role of ACE2 in regulating EPC function. We analyzed the eNOS/NO and Nox/ROS pathways in order to explore the underlying mechanisms. To further investigate the possible implication of ACE2 over-expression in EPCs, we evaluated the therapeutic efficiency of EPCs over-expressing ACE2 on experimental ischemic stroke.
Methods and Materials
Culture and characterization of early outgrowth EPCs
BM derived EPCs were generated from R+A+ mice (R+A+ EPCs) and their controls (R-A-) (R-A- EPCs) and characterized as we previously reported (13). After 7 days of culture, cells double-positive for Di-LDL and Bs-Lectin staining were considered as early outgrowth EPCs (14). In addition, EPCs were stained with PE-conjugated CD31 (10 μl, BD Bioscience, San Jose, CA, USA), PE-conjugated CD34 (10 μl, AbD Serotec), FITC-conjugated VE-Cadherin (1 μl, eBioscience), PE-Cy7-conjugated vascular endothelial growth factor receptor-2 (VEGFR-2, 5 μl, BD Bioscience), PE-conjugated von Willebrand Factor (vWF, 10 μl, BD Bioscience), FITC-conjugated CD133 (5 μl, BD Bioscience), FITC-conjugated CD45 (1 μl, eBioscience) or FITC-conjugated CD146 (1 μl, eBioscience) antibodies. The phenotype of EPCs was analyzed by flow cytometry (Accuri C6 flow cytometer) as we described before (13).
EPCs transduction with lentivirus-ACE2 and pathway blocking studies
The lentivirus containing murine ACE2 cDNA (Lenti-ACE2) and lentivirus containing green fluorescence protein (Lenti-GFP) were produced as previously described (15). EPCs were transduced with Lenti-ACE2 (Lenti-ACE2-EPCs) or Lenti-GFP (Lenti-GFP-EPCs) as previously reported (16). In brief, after cultivation for 7 days, EPCs were cultured in six-well plates (1×105/well) and incubated with serum-free EPC culture medium containing the lentivirus (at 5×106 infection-forming units) for 4 hrs, after which the serum-free medium was replaced with 10%(v/v) FBS medium for 24 hrs. Transduction efficiency (the percentage of GPF expressing cells) was quantified by direct counting using an optical grid.
For blocking experiments, after transduction with Lenti-GFP or Lenti-ACE2, EPCs were incubated with ACE2 inhibitor (DX600, 1 μmol/L), eNOS inhibitor (L-NAME, 1 mmol/L) or Nox inhibitor (apocynin, 10 μmol/L) for 24 hrs. Then, the EPCs were harvested for the analyses of function and gene expression.
EPC proliferate assay
The EPC proliferation was performed according to the manufacturer's protocol (Cell proliferation enzyme-linked immunosorbent assay, bromodeoxyuridine [BrdU; colorimetric], Roche) as previous reported (17). Briefly, EPCs were seeded on 96-well plates (1×104 cells/well). The cells were grown in complete culture medium which was changed after 12 hrs with complete media supplemented with 10 μM BrdU. BrdU incorporation was measured after 24 hrs using a microtiterplate reader (Packard) at 450 nm with a reference wavelength of 690 nm. BrdU uptake was calculated as the percentage of the untransduced EPCs.
Real-time RT PCR
The levels of ACE2 in EPCs transduced with Lenti-ACE2 were determined using real-time RT-PCR method (18). EPC total mRNAs were isolated using RNeasy Mini kit and reverse-transcripted with the high capacity cDNA archive kit (Qiagen). The real-time PCR was run using SYBR Green reagents (Qiagen). The primer sequences for ACE2 were 5′- AAGCTAGCATAGCCAGGTCCTCCTGGCTCCTTC-3′ and 5′- AAGTCGACCTAAAAGGAAGTCTGAGCATCATCACTG-3′. β-actin was chosen as the housekeeping gene for normalizing the data of gene expression. The mRNA level of ACE2 in EPCs from R-A- mice transduced with Lenti-GFP was defined as 100%.
EPC function assays
The migration and tube formation abilities of EPCs were evaluated by using Boyden chamber (Chemicon) and tube formation assay kit (Chemicon) methods as we previously described (13).
Measurement of ROS generation
Intracellular ROS generation in EPCs was determined by dihydroethidium (DHE) staining as previously described (19). Cells were incubated with DHE (2 μmol/L) in dark for 30 min. After washing with phosphate buffer saline (PBS), cells were then lysed with 50 μl lysis buffer on ice. The lysates were transferred into black 96-well plates for fluorescence measurement using a spectrofluorometer.
Determination of NO production
The membrane-permeable indicator diaminofluorescein (DAF-FM) diacetate (Invitrogen, Grand Island, NY, USA) was used to assess NO production released by EPCs (20). Briefly, the EPCs were loaded with 2 μmol/L DAF-FM diacetate in serum free EBM-2 (37°C for 30 min), washed twice with PBS and incubated with DAF-FM diacetate-free in EBM-2 (20 min) for deesterification of the indicator. DAF-FM fluorescence was measured using a spectrofluorometer.
Animals and procedures
Animals
Male adult (8-10 weeks of age, weight ranges from 25 g to 32 g) R+A+ mice and their age-matched controls (R-A-) with C57BL/6J genetic background were used for all experiments. The strains were maintained in our laboratory (founders were from Dr. Curt D. Sigmund's lab at the University of Iowa) (21;22). Mice were maintained in a 22°C room with a 12-hr light/dark cycle and fed with standard chow and drinking water ad libitum. All experimental procedures were approved by the Wright State University Laboratory Animal Care and Use Committee and were in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health (NIH).
Middle cerebral artery occlusion (MCAO) surgery and EPC transfusion
Focal ischemic stroke was induced in animals by MCAO surgery under anesthesia by inhaling 2.5% isoflurane as we reported previously (13). Two hours after MCAO, mice were injected via the tail vein with Lenti-GFP-EPCs or Lenti-ACE2-EPCs (2x105 cells/100 μl in PBS) or the same volume of PBS as previously described (13;23). EPCs were donated from R-A- mice. Pain and discomfort was minimized by an initial injection of Buprenorphine (0.1mg/kg, s.c) followed with another two injection every 12 hrs.
BrdU labeling
To label the new generated cells, mice were injected with BrdU (i.p., 65 μg/g/day) immediately after EPC infusion for continuous 7 days (24).
Functional evaluation of neurological deficits
On day 7, the neurological deficit score of each mouse was evaluated by using the 5-point scale method as previously described (22).
Measurements of cerebral infarct volume and microvascular density
On day 7 after EPC transfusion, the brains were immediately collected and fixed in 4% paraformaldehyde (PFA) overnight and in 4% PFA plus 30% sucrose for 3 days. The brains were then cut into coronal sections (20 μm). As we previously described (13;22), cerebral ischemic damage and cerebral microvascular density (cMVD) in the peri-infarct area were revealed by Fluoro-Jade (0.001%, Histo-chem) and CD31 (1:50, Invitrogen) staining, respectively.
Immunofluorescence analysis
Brain coronal sections were incubated with BrdU (1:50, Abcam), CD31 (1:50, BD Biosciences) or GFP (1:50, Santa Cruz) antibody overnight at 4°C. Next, brain sections were reacted with Cy5 (blue, for BrdU), Cy3 (red, CD31) or FITC (green, for GFP) conjugated secondary antibodies (1:250, Invitrogen) for 30 min at room temperature in the dark. The positive cells in the peri-infarct area of each section were visualized using confocal microscopy (Leica TCS SP2). Angiogenesis was determined as BrdU+CD31+ cells according to previous reports (24;25). In vivo tracking of transfused EPCs were recognized as GFP+ cells. Cell counting was performed from photographs in 6 random microscopic fields (200x) by an investigator who was unaware of grouping. The average of five sections from rostral to caudal represented the data for sample.
Western blot analysis
Proteins from EPCs were isolated with lysis buffer (Roche Diagnostic). The antibodies used were anti-ACE2 (1:1000, Cell Signaling Technology), eNOS (1:1000, Abcam), Nox2 (1:1000, Abcam) or Nox4 (1:250, Abcam) at 4°C overnight. β-actin (1:4000, Sigma) was used to normalize protein loading.
Statistical analysis
All data, excepting neurologic deficit scores, are presented as mean±SE. The neurologic deficit scores were expressed as median (range). The neurological deficit scores among different groups were compared by the Kruskal–Wallis test. When the Kruskal–Wallis test showed a significant difference, the Mann–Whitney U-tests were applied. For the rest measurements, comparisons for two groups were performed by the student's t test. Multiple comparisons were analyzed by one- or two-way ANOVA. For all tests, a P-value <0.05 was considered significant.
Results
Characterization of EPCs
As we reported previously, BM derived EPCs were defined as the cells up-taking Di-LDL and binding with Bs-Lectin (13), as well as expressing specific surface markers. The EPCs showed positive to CD34, VEGFR2, CD133, and negative to CD31, VE-Cadherin, vWF, CD45 and CD146 (Figure 1). There were no difference regarding cell markers expressed on the EPCs from the R-A- and R+A+ mice.
Figure 1.

The phenotype of cultured EPCs. Representative flow plot showing the expression of CD34, VEGFR2, CD133, CD31, VE-Cadherin, vWF, CD45 and CD146 on the EPCs cultured from R-A- (A) and R+A+ mice (B).
The Effects of Lentivirus Transduction on EPC proliferation
The transduction efficiency was about 94±2% (Figure 2A). Lentiviral transduction decreased EPC proliferation rate by 15%. The proliferation rate of Lenti-ACE2-EPCs was increased when compared to Lenti-GFP-EPCs (by 35%) or untransduced EPCs (by 20%) (Figure 2B).
Figure 2.
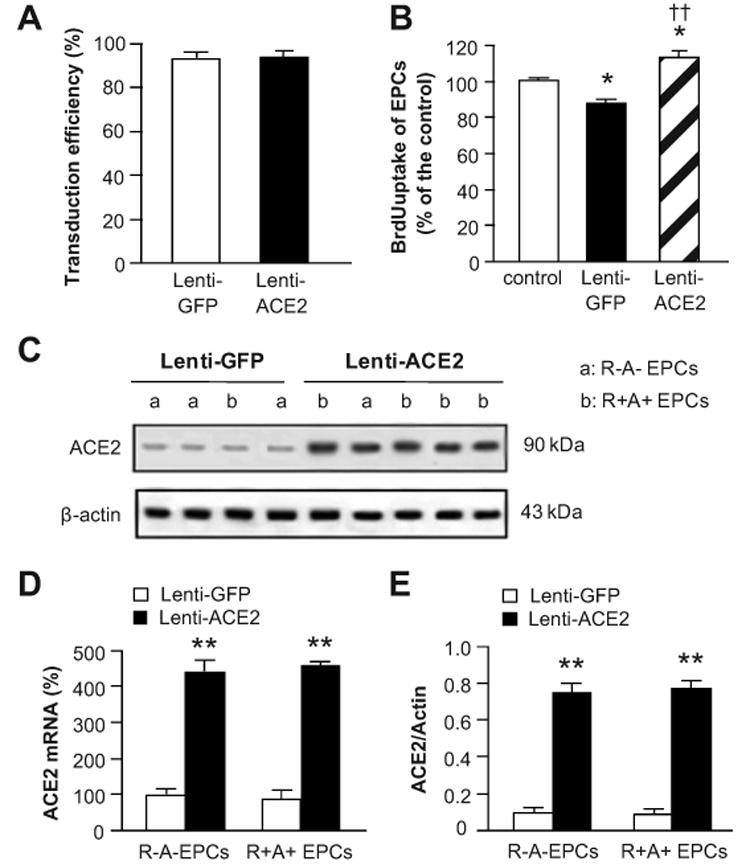
Transduction efficiency, proliferation rate and ACE2 expression in EPCs. A, Transduction efficiency of lentivirus in EPCs. B, Proliferation rate of control, and EPCs transuded with Lenti-GFP or Lenti-ACE2. *P<0.05 versus control; ††P<0.01 versus Lenti-GFP. C, Representative Western blot bands showing the ACE2 expression in EPCs. Summarized data of ACE2 expression in EPCs at mRNA (D) and protein (E) levels. **P<0.01 versus Lenti-GFP, n=6/group. Control: untransduced EPCs.
Lenti-ACE2 Transduction Increases ACE2 Expression in EPCs
The basal levels of ACE2 mRNA and ACE2 protein expression were not different in the EPCs derived from R-A- and R+A+ mice. Lenti-ACE2 transduction induced a four-fold up-regulation of ACE2 in EPCs at both mRNA (P<0.01; Figure 2D) and protein levels (P<0.01; Figure 2C and E).
Transduction of EPCs with Lenti-ACE2 Increases eNOS Expression and Decreases Nox2, 4 Expressions
At basal, the level of eNOS expression was lower (0.24 ± 0.02 versus 0.18 ± 0.01, P<0.05; Figure 3A), whereas levels of Nox2 (0.36 ± 0.03 versus 0.48 ± 0.03, P<0.05; Figure 3B) and Nox4 (0.34 ± 0.02 versus 0.46 ± 0.03, P<0.01; Figure 3C) were higher in the EPCs generated from R+A+ mice. Transduction of Lenti-ACE2 increased eNOS, and decreased Nox2 and Nox4 expressions in EPCs from both R+A+ (by 61%, 27%and 28%, respectively) and R-A- (by 62%, 44%and 50%, respectively) mice (P<0.01). In addition, DX600 (ACE2 inhibitor) was able to partially block those effects of Lenti-ACE2 on EPCs (P<0.05 or 0.01; Figure 3).
Figure 3.
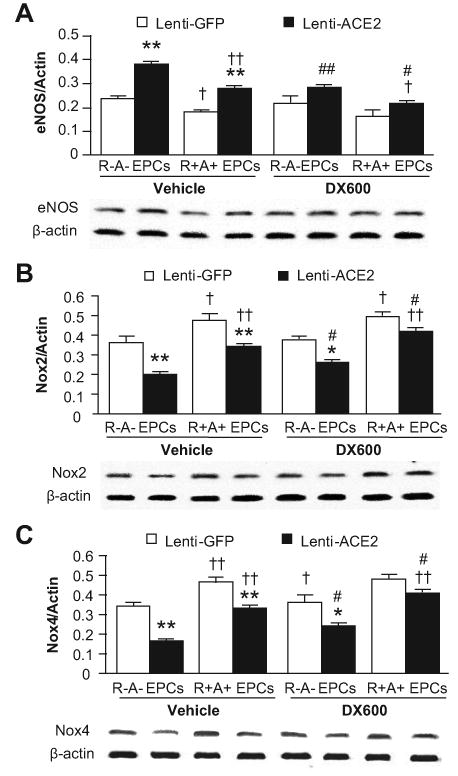
The effects of ACE2 over-expression on gene expression. The eNOS (A), Nox2 (B) and Nox4 (C) expression in EPCs. EPCs were transduced with Lenti-GFP or Lenti-ACE2, and then incubated with or without DX600 (1 μmol/L). The protein expression was normalized to the expression of β-actin. *P<0.05, **P<0.01 versus Lenti-GFP; †P<0.05, ††P<0.01 versus R-A- EPCs; #P<0.05 versus vehicle, n=6/group. eNOS: endothelial nitric oxide synthase; Nox: nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.
Transduction of EPCs with Lenti-ACE2 Increases NO and Decreases ROS Production
The NO production was higher (versus P<0.05; Figure 4A), whereas the ROS production was lower (P<0.05; Figure 4B) in the EPCs generated from R+A+ mice. Lenti-ACE2 transduction increased 25% NO production, and decreased 48% ROS production in EPCs derived from R-A- mice, and induced 32% increase of NO and 28% decrease of ROS production from R+A+ mice (P<0.01). Again, DX600 partially blocked those effects of Lenti-ACE2 on EPCs (P<0.05). In addition, APO (Nox inhibitor) decreased the ROS production, while L-NAME (eNOS inhibitor) blocked the NO production in Lenti-ACE2 primed EPCs (P<0.05; Figure 4).
Figure 4.

The effects of ACE2 over-expression on ROS and NO production of EPCs. The ROS (A) and NO (B) production in EPCs transduced with Lenti-GFP or Lenti-ACE2. Cells were incubated with DX600 (1 μmol/L), APO (10 μmol/L) or L-NAME (1 mmol/L). *P<0.05, **P<0.01 versus Lenti-GFP; †P<0.05, ††P<0.01 versus R-A- EPCs; #P<0.05 versus Lenti-ACE2, n=6/group. ROS: reactive oxygen species; NO: nitric oxygen.
Lenti-ACE2 Transduction Enhances EPC Function via Both eNOS and Nox Pathways
As shown in Figure 5, the abilities of EPC migration and tube formation were attenuated in EPCs from R+A+ mice by 31% and 26%, respectively (P<0.01 or 0.05). Lenti-ACE2 transduction enhanced those functions of EPCs from both R+A+ (by 47% and 52%, respectively) and R-A- (by 34% and 50%, respectively) mice (P<0.01). Moreover, DX600 abolished and L-NAME partially blocked Lenti-ACE2 induced enhancement of those functions in EPCs (P<0.01). APO further enhanced those functions in Lenti-ACE2 primed EPCs from R+A+ mice (P<0.05), but not in Lenti-ACE2 primed EPCs from R-A- mice.
Figure 5.
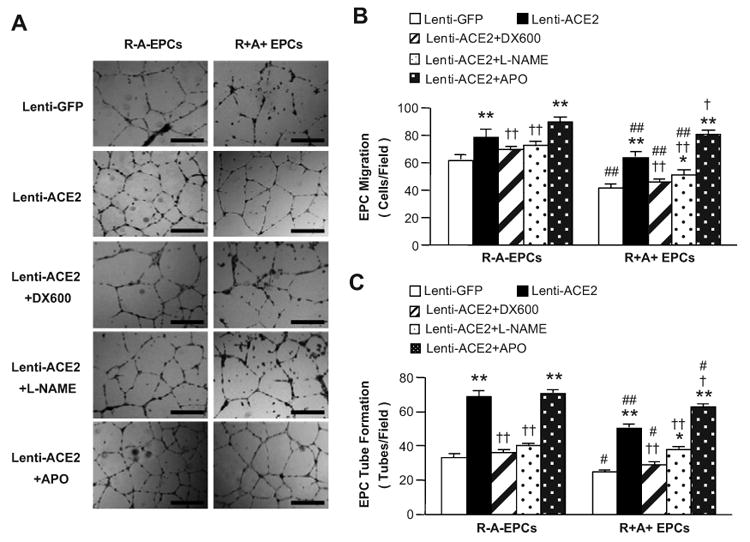
ACE2 over-expression enhances the function of EPCs. A, Representative images of EPC tube formation. Scale bar: 600 μm. The abilities of EPC migration (B) and tube formation (C) in EPCs transduced with Lenti-GFP or Lenti-ACE2. Cells were incubated with DX600 (1 μmol/L), APO (10 μmol/L) or L-NAME (1 mmol/L). *P<0.05, **P<0.01 versus Lenti-GFP; †P<0.05, ††P<0.01 versus Lenti-ACE2; #P<0.05, ##P<0.01 versus R-A- EPCs, n=6/group.
Infusion of Lenti-ACE2-EPCs Enhances the Efficacy in Decreasing Ischemic Injury and Neurologic Deficit Score
The infarct volume of MCAO-induced stoke was exaggerated in R+A+ mice (P<0.05; Figure 6A and B). Lenti-GFP-EPC transfusion decreased infarct volume in both R-A- (by 33%, P<0.01) and R+A+ mice (by 20%, P<0.05; Figure 6A and B) and in neurologic deficit score (P<0.05; Figure 6C). Furthermore, transfusion of Lenti-ACE2-EPCs was able to further decrease the infarct volume in R-A- (by 30%, P<0.05) and R+A+ mice (by 40%, P<0.05; Figure 6A and B) and further improve neurologic motor function (P<0.05 or 0.01; Figure 6C).
Figure 6.
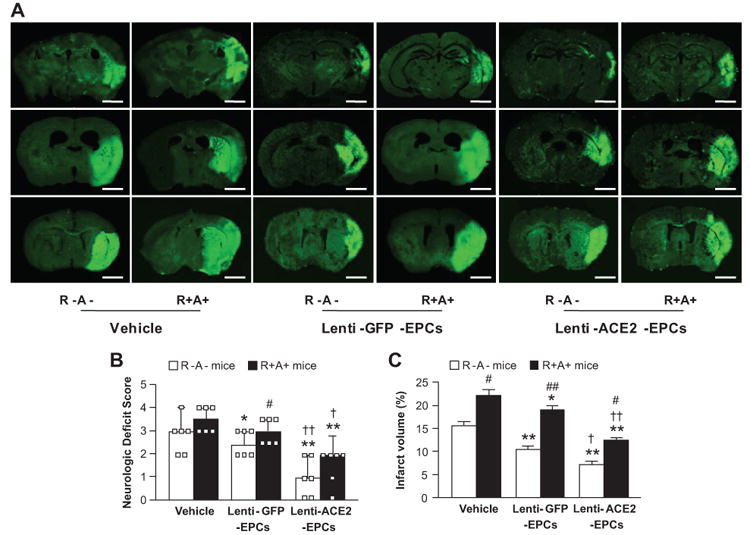
Effects of Lenti-ACE2-EPC infusion on infarct volume and neurologic deficit score. A, Representative pictures of Fluoro-J staining 7 days after EPC infusion. Scale bar: 2 mm. Summarized data on neurologic deficit scores (B) and infarct volume (C). Transfusion of Lenti-ACE2-EPCs has better efficacy than Lenti-GFP-EPCs in decreasing infarct volume and improving neurologic deficit. *P<0.05, **P<0.01 versus vehicle; †P<0.05, ††P<0.01 versus Lenti-GFP-EPCs; #P<0.05, ##P<0.01 versus R-A- mice, n=6/group.
Infusion of Lenti-ACE2-EPCs Enhances the Efficacy in Increasing cMVD in the Peri-Infarct Area
At basal, the cMVD in the peri-infarct area was decreased by 41% in R+A+ mice (P<0.05; Figure 7A and B). Infusion of Lenti-GFP-EPCs was able to increase the cMVD in peri-infarct area by 22% in R-A- mice and 30% in R+A+ mice (P<0.01; Figure 7B). Transfusion of Lenti-ACE2-EPCs could further induce a 17% increase in cMVD in the peri-infarct area in R-A- mice (P<0.01; Figure 7B), and 55% in R+A+ mice (P<0.05).
Figure 7.
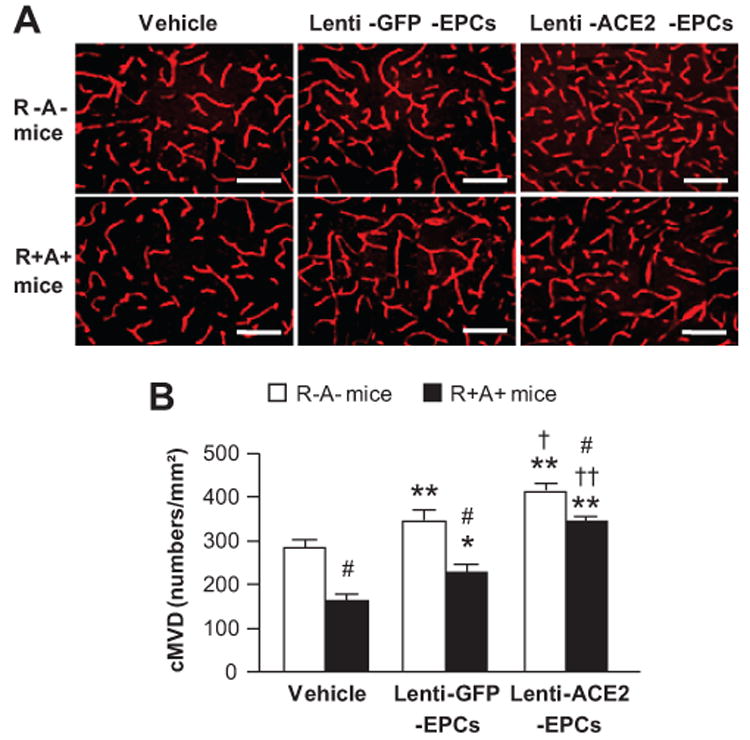
Effects of Lenti-ACE2-EPC infusion on cMVD in cerebral microvasculature in the peri-infarct area. A, Representative pictures of cMVD (CD31 immunostaining) on day 7 following MCAO and EPC infusion. Scale bar: 50 μm. B, Summarized data of cMVD. *P<0.05, **P<0.01 versus vehicle; †P<0.05, ††P<0.01 versus Lenti-GFP-EPCs; #P<0.05 versus R-A- mice, n=6/group. cMVD: cerebral microvascular density.
Infusion of Lenti-ACE2-EPCs Enhances the Efficacy in Promoting Angiogenesis in the Peri-Infarct Area
Figure 8A shows representative images of in vivo tracking of transfused EPCs (GFP+ cells) in the peri-infarct area in different groups on day 7 after MCAO surgery. The number of GFP+ cells was more in Lenti-ACE2-EPCs transfusion group than in Lenti-GFP-EPC transfusion group (P<0.01; Figure 8C).
Figure 8.
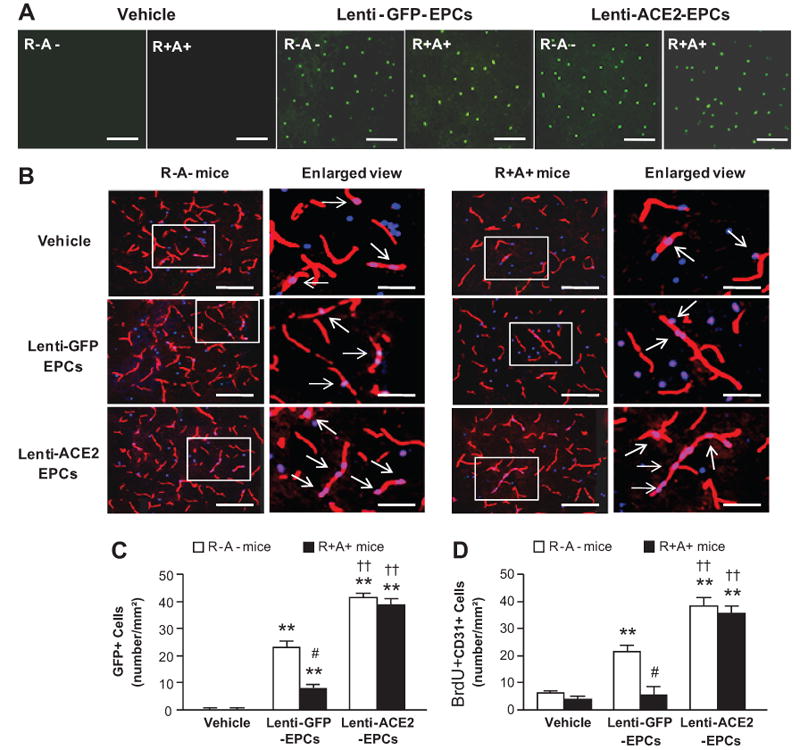
Effect of Lenti-ACE2-EPC infusion on angiogenesis in the peri-infarct area. A, Representative pictures of in vivo tracking of transfused EPCs 7 days after EPC infusion. Green: GFP for transfused EPCs. Scale bar: 50 μm. B, Representative pictures of angiogenesis (BrdU+CD31+ cells). Red: CD31 for vessel; Blue: BrdU for new generated cells; Pink: double staining. Scale bar: 50 μm. The enlarged view of double staining cells is in the left panel of each image. The arrows indicate the double staining cells. Scale bar: 20 μm. Histogram showing the number of GFP+ cells (C) and BrdU+CD31+ cells (D). **P<0.01 versus vehicle; ††P<0.01 versus Lenti-GFP-EPCs; #P<0.05 versus R-A- mice, n=6/group.
Figure 8B shows representative micrographs of angiogenesis (BrdU+CD31+ cells) in the peri-infarct area. Lenti-GFP-EPC transfusion promoted angiogenesis on day 7 in R-A- mice (by 30%; P<0.01). Transfusion of Lenti-ACE2-EPCs further promoted angiogenesis in both R-A- (by 81%; P<0.01) and R+A+ mice (by 300%; P<0.01; Figure 8D).
Discussion
EPCs have emerged as a pivotal type of cells for maintaining endothelial homeostasis and replenishing injured ECs, and have been shown to substantially contribute to endothelial regeneration and functional restoration (7;26). It has been recently documented that Ang II reduces EPC number and induces EPC dysfunction in vitro and in vivo (9). In addition, studies indicated that ACE inhibitors and AT1 receptor blockers have protective effects on EPCs (27;28). A recent study showed that over-expressing ACE2 in blood vessels improves EC function (29). However, whether ACE2 counteracts the effects of Ang II on EPCs has not been explored.
In this study, we demonstrated that the abilities of migration and tube formation of BM derived EPCs from R+A+ mice are impaired. This result is consistent with previous studies showing that Ang II via AT1 receptor decreases the differentiation and accelerates the senescence of BM derived EPCs (11;30). Here, we also found that the eNOS expression and NO production are lower, whereas Nox2, 4 expression and ROS production are higher in the EPCs generated from R+A+ mice. These findings are supported by previous evidence demonstrating that the RAS plays a key role in modulating EC function through regulating NO and ROS production (31). Recently, it has been suggested that Ang II inhibits Akt-induced eNOS activation and NO release in ECs (32) and that Ang II infusion decreases NO production in aorta by causing eNOS uncoupling (33).
One of the major findings in the present study is that ACE2 over-expression in EPCs generated from both R-A- and R+A+ mice enhances their abilities of tube formation and migration. Interestingly, those effects are more evident in the EPCs from R+A+ mice and can be partially abolished by ACE2 inhibitor (DX600) suggesting that ACE2 might counteract the effect of Ang II on EPCs. We assume that the changes with ACE2 over-expression might mainly reflect the elevation of Ang (1-7) in addition to the reduction of Ang II. This is supported by a recent report showing that ACE2 over-expression in myocytes and fibroblasts induces Ang II reduction and Ang (1-7) elevation in cell lysates (34). DX600 is a large peptide which would not traverse the cell membrane. Since the presence of intracellular RAS peptidases (35), the incomplete reversal with DX600 would reflect that intracellular expression of ACE2 regulates the intracellular ratio of Ang II to Ang (1-7).
At the same time, our data show that ACE2 over-expression down-regulates the expression of Nox2/Nox4 and alleviates the ROS production, whereas, up-regulates the expression of eNOS and elevates the NO production. These findings are in agreement with other studies. ACE2/Ang (1-7) counterregulates Ang II induced Nox activation in ECs (36), and prevents ROS over-production and senescence in EPCs (37). The eNOS also has been reported to regulate the mobilization and function of EPCs (38). We speculate that the dual effects of ACE2 over-expression on stimulating NO and reducing ROS production may beneficial from the reduced formation of peroxynitrite. There is a study reported that ACE2 deficiency enhances Ang II-mediated peroxynitrite production (39). In addition, both the decreased ROS formation and enhanced ROS scavenging enzymes might account for the reduction of ROS production (40;41). To further confirm whether reduction of oxidative stress and up-regulation of eNOS associated with the ACE2 mediated improvement of EPC function, we conducted the pathway block studies. As we expected, the beneficial effects of ACE2 over-expression on EPCs were partially blocked by NOS inhibition and further enhanced by a Nox inhibitor. These data add new information to previous reports showing the protective role of ACE2 in cells. For examples, ACE2 over-expression in the human monocyte cell line attenuates Ang II-induced release of inflammatory factors (1). Transduction of cardiac fibroblasts with lenti-ACE2 results in a significant attenuation of both basal and hypoxia/re-oxygenation-induced collagen production and inflammatory cytokine production (16).
To explore the function of EPCs over-expressing ACE2 in vivo, we investigated the therapeutic efficacy of Leni-ACE2 primed EPCs on experimental ischemic stroke mice. Transfused EPCs were tracked in vivo suggesting recruitment of transfused EPCs to the peri-infarct area. Our data demonstrate for the first time that transfusion of ACE2 primed EPCs is able to enhance the efficacy of non-primed EPCs in attenuating cerebral damage (decreasing the infarct volume, improving neurologic deficits) and promoting cerebral repair (increasing cMVD and angiogenesis). Angiogenesis is a vital component of wound repair. EPCs are believed to play an important role in maintaining endothelial integrity and vascular homeostasis, and to participate in angiogenesis which represents an important endogenous tissue repair mechanism. Introduction or mobilization of EPCs can restore tissue vascularization after ischemic stroke and re-establish endothelial integrity (13;42). In this study, we double stained the brain slides with CD31 and BrdU to measure the newly generated ECs for the index of angiogenesis, which is commonly used in the similar studies (24;25). Our results showed that infusion of Lenti-GFP-EPCs increased angiogenesis in R-A- mice, but not in R+A+ mice. Although the detailed mechanism has not been explored in this study, we tentatively attribute this to the high level of Ang II in the R+A+ mice (22), which impairs the function of EPC (9;43). Of note, we further discovered that infusion of EPCs over-expressing ACE2 promotes angiogenesis in both R-A- and R+A+ mice, which might benefit from the ACE2 catalyzing Ang II into Ang (1-7). This is supported by other reports showing that Ang (1-7) improves endothelial function (29) and ameliorates Ang II-Induced endothelial cell apoptosis (44). Although it is unclear whether the improvement of angiogenesis is exclusively mediated by EPC integration or paracrine effects, our in vivo results imply that over-expression of ACE2 in EPCs could enhance the beneficial effect of EPC-based therapy for ischemic stroke. Future studies are warranted to determine the fate of transfused EPC populations by analyzing the dynamics of transfused cells in living animals.
In summary, both the in vitro functional and in vivo therapeutic findings of present study demonstrate that over-expression of ACE2 in EPCs enhances EPC function via eNOS/NO and Nox/ROS signaling pathways. Transfusion of Lenti-ACE2 primed EPCs might provide a novel approach for treating ischemic stroke, especially in those with Ang II activation.
Perspectives
Our findings show that ACE2 enhances EPC function via regulating eNOS/NO and Nox/ROS pathways and improves EPC-based therapeutic effects on ischemic stroke. This study depicts the new aspect of ACE2 gene function and the underlying mechanisms. Our data provide a strong rationale for using ACE2 target to develop vasoprotective drugs and to enhance the efficacy of EPC-based therapy. In addition, further study on the effect of ACE2 on EPCs should provide important insights into the pathogenesis of vascular disease and development of novel therapeutic interventions.
Novelty and Significance.
What Is New?
This study demonstrates the protective role of ACE2 in EPC function in vitro and the beneficial effects of ACE2 primed EPCs on treating ischemic stroke in vivo.
What Is Relevant?
The present study supports the hypothesis that overexpressing ACE2 could improve EPC function and enhance the efficacy of EPC-based therapy for ischemic stroke via counteracting the detrimental effects of Ang II.
Summary
ACE2 over-expression enhances EPC function via modulating Nox/ROS and eNOS/NO signaling pathways, and improves the therapeutic efficacy of EPC transfusion for ischemic stroke by reducing ischemic injury and promoting recovery. Our findings provide novel insight into ACE2 as a potential target for new drug development.
Acknowledgments
N/A.
Sources of Funding: This work was supported by the National Heart, Lung, and Blood Institute (HL-098637, Y.C. and HL-093567, M.M., HL-56921, M.K.R.), and the National Natural Science Foundation of China (NSFC, # 81271214, #81270195, #81070878).
Footnotes
Conflict(s) of Interest/Disclosure(s): The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Guo YJ, Li WH, Wu R, Xie Q, Cui LQ. ACE2 overexpression inhibits angiotensin II-induced monocyte chemoattractant protein-1 expression in macrophages. Arch Med Res. 2008;39:149–154. doi: 10.1016/j.arcmed.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107:1482–1494. doi: 10.1111/j.1471-4159.2008.05723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferreira AJ, Raizada MK. Genomic and proteomic approaches for targeting of angiotensin-converting enzyme2 for cardiovascular diseases. Curr Opin Cardiol. 2008;23:364–369. doi: 10.1097/HCO.0b013e328303b79b. [DOI] [PubMed] [Google Scholar]
- 4.Zhang C, Zhao YX, Zhang YH, Zhu L, Deng BP, Zhou ZL, Li SY, Lu XT, Song LL, Lei XM, Tang WB, Wang N, Pan CM, Song HD, Liu CX, Dong B, Zhang Y, Cao Y. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci U S A. 2010;107:15886–15891. doi: 10.1073/pnas.1001253107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 6.Sierra C, Coca A, Schiffrin EL. Vascular mechanisms in the pathogenesis of stroke. Curr Hypertens Rep. 2011;13:200–207. doi: 10.1007/s11906-011-0195-x. [DOI] [PubMed] [Google Scholar]
- 7.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 8.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 9.Endtmann C, Ebrahimian T, Czech T, Arfa O, Laufs U, Fritz M, Wassmann K, Werner N, Petoumenos V, Nickenig G, Wassmann S. Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regeneration. Hypertension. 2011;58:394–403. doi: 10.1161/HYPERTENSIONAHA.110.169193. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Liu Q, Wang N, Xu J. Correlation of different NADPH oxidase homologues with late endothelial progenitor cell senescence induced by angiotensin II: effect of telmisartan. Intern Med. 2011;50:1631–1642. doi: 10.2169/internalmedicine.50.5250. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Huang C, Zhang W, Sigmund CD, Chen Y. Bone marrow derived endothelial progenitor cells are reduced and dysfunctional in human renin and angiotensinogen double transgenic mice. Arterioscier Thromb Vasc Biol. 2008 Jun;:e76. [Google Scholar]
- 12.Lemarie CA, Shbat L, Marchesi C, Angulo OJ, Deschenes ME, Blostein MD, Paradis P, Schiffrin EL. Mthfr deficiency induces endothelial progenitor cell senescence via uncoupling of eNOS and downregulation of SIRT1. Am J Physiol Heart Circ Physiol. 2011;300:H745–H753. doi: 10.1152/ajpheart.00321.2010. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Chen S, Chen Y, Zhang C, Wang J, Zhang W, Liu G, Zhao B, Chen Y. Circulating endothelial progenitor cells and cellular membrane microparticles in db/db diabetic mouse: possible implications in cerebral ischemic damage. Am J Physiol Endocrinol Metab. 2011;301:E62–E71. doi: 10.1152/ajpendo.00026.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner N, Priller J, Laufs U, Endres M, Bohm M, Dirnagl U, Nickenig G. Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler Thromb Vasc Biol. 2002;22:1567–1572. doi: 10.1161/01.atv.0000036417.43987.d8. [DOI] [PubMed] [Google Scholar]
- 15.Huentelman MJ, Zubcevic J, Katovich MJ, Raizada MK. Cloning and characterization of a secreted form of angiotensin-converting enzyme 2. Regul Pept. 2004;122:61–67. doi: 10.1016/j.regpep.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Grobe JL, Der SS, Stewart JM, Meszaros JG, Raizada MK, Katovich MJ. ACE2 overexpression inhibits hypoxia-induced collagen production by cardiac fibroblasts. Clin Sci (Lond) 2007;113:357–364. doi: 10.1042/CS20070160. [DOI] [PubMed] [Google Scholar]
- 17.Bocker W, Docheva D, Prall WC, Egea V, Pappou E, Rossmann O, Popov C, Mutschler W, Ries C, Schieker M. IKK-2 is required for TNF-alpha-induced invasion and proliferation of human mesenchymal stem cells. J Mol Med (Berl) 2008;86:1183–1192. doi: 10.1007/s00109-008-0378-3. [DOI] [PubMed] [Google Scholar]
- 18.Huentelman MJ, Grobe JL, Vazquez J, Stewart JM, Mecca AP, Katovich MJ, Ferrario CM, Raizada MK. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp Physiol. 2005;90:783–790. doi: 10.1113/expphysiol.2005.031096. [DOI] [PubMed] [Google Scholar]
- 19.Mustapha NM, Tarr JM, Kohner EM, Chibber R. NADPH Oxidase versus Mitochondria-Derived ROS in Glucose-Induced Apoptosis of Pericytes in Early Diabetic Retinopathy. J Ophthalmol. 2010;2010:746978. doi: 10.1155/2010/746978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima H, Urano Y, Kikuchi K, Higuchi T, Hirata Y, Nagano T. Fluorescent Indicators for Imaging Nitric Oxide Production. Angew Chem Int Ed Engl. 1999;38:3209–3212. doi: 10.1002/(sici)1521-3773(19991102)38:21<3209::aid-anie3209>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 21.Iida S, Baumbach GL, Lavoie JL, Faraci FM, Sigmund CD, Heistad DD. Spontaneous stroke in a genetic model of hypertension in mice. Stroke. 2005;36:1253–1258. doi: 10.1161/01.str.0000167694.58419.a2. [DOI] [PubMed] [Google Scholar]
- 22.Chen S, Li G, Zhang W, Wang J, Sigmund CD, Olson JE, Chen Y. Ischemia-induced brain damage is enhanced in human renin and angiotensinogen double-transgenic mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1526–R1531. doi: 10.1152/ajpregu.91040.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foubert P, Matrone G, Souttou B, Lere-Dean C, Barateau V, Plouet J, Le Ricousse-Roussanne S, Levy BI, Silvestre JS, Tobelem G. Coadministration of endothelial and smooth muscle progenitor cells enhances the efficiency of proangiogenic cell-based therapy. Circ Res. 2008;103:751–760. doi: 10.1161/CIRCRESAHA.108.175083. [DOI] [PubMed] [Google Scholar]
- 24.Shyu WC, Lin SZ, Yang HI, Tzeng YS, Pang CY, Yen PS, Li H. Functional recovery of stroke rats induced by granulocyte colony-stimulating factor-stimulated stem cells. Circulation. 2004;110:1847–1854. doi: 10.1161/01.CIR.0000142616.07367.66. [DOI] [PubMed] [Google Scholar]
- 25.Kawada H, Takizawa S, Takanashi T, Morita Y, Fujita J, Fukuda K, Takagi S, Okano H, Ando K, Hotta T. Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation. 2006;113:701–710. doi: 10.1161/CIRCULATIONAHA.105.563668. [DOI] [PubMed] [Google Scholar]
- 26.Werner N, Nickenig G. Endothelial progenitor cells in health and atherosclerotic disease. Ann Med. 2007;39:82–90. doi: 10.1080/07853890601073429. [DOI] [PubMed] [Google Scholar]
- 27.Muller P, Kazakov A, Jagoda P, Semenov A, Bohm M, Laufs U. ACE inhibition promotes upregulation of endothelial progenitor cells and neoangiogenesis in cardiac pressure overload. Cardiovasc Res. 2009;83:106–114. doi: 10.1093/cvr/cvp123. [DOI] [PubMed] [Google Scholar]
- 28.Steinmetz M, Brouwers C, Nickenig G, Wassmann S. Synergistic effects of telmisartan and simvastatin on endothelial progenitor cells. J Cell Mol Med. 2010;14:1645–1656. doi: 10.1111/j.1582-4934.2009.00829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rentzsch B, Todiras M, Iliescu R, Popova E, Campos LA, Oliveira ML, Baltatu OC, Santos RA, Bader M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52:967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 30.Kobayashi K, Imanishi T, Akasaka T. Endothelial progenitor cell differentiation and senescence in an angiotensin II-infusion rat model. Hypertens Res. 2006;29:449–455. doi: 10.1291/hypres.29.449. [DOI] [PubMed] [Google Scholar]
- 31.Silva PM. From endothelial dysfunction to vascular occlusion: role of the renin-angiotensin system. Rev Port Cardiol. 2010;29:801–824. [PubMed] [Google Scholar]
- 32.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002;90:E58–E65. doi: 10.1161/01.res.0000012569.55432.02. [DOI] [PubMed] [Google Scholar]
- 34.Dong B, Yu QT, Dai HY, Gao YY, Zhou ZL, Zhang L, Jiang H, Gao F, Li SY, Zhang YH, Bian HJ, Liu CX, Wang N, Xu H, Pan CM, Song HD, Zhang C, Zhang Y. Angiotensin-converting enzyme-2 overexpression improves left ventricular remodeling and function in a rat model of diabetic cardiomyopathy. J Am Coll Cardiol. 2012;59:739–747. doi: 10.1016/j.jacc.2011.09.071. [DOI] [PubMed] [Google Scholar]
- 35.Gwathmey TM, Shaltout HA, Rose JC, Diz DI, Chappell MC. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension. 2011;57:620–626. doi: 10.1161/HYPERTENSIONAHA.110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampaio WO, Henrique de CC, Santos RA, Schiffrin EL, Touyz RM. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension. 2007;50:1093–1098. doi: 10.1161/HYPERTENSIONAHA.106.084848. [DOI] [PubMed] [Google Scholar]
- 37.Qian C, Schoemaker RG, van Gilst WH, Roks AJ. The role of the renin-angiotensin-aldosterone system in cardiovascular progenitor cell function. Clin Sci (Lond) 2009;116:301–314. doi: 10.1042/CS20080157. [DOI] [PubMed] [Google Scholar]
- 38.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 39.Jin HY, Song B, Oudit GY, Davidge ST, Yu HM, Jiang YY, Gao PJ, Zhu DL, Ning G, Kassiri Z, Penninger JM, Zhong JC. ACE2 deficiency enhances angiotensin II-mediated aortic profilin-1 expression, inflammation and peroxynitrite production. PLoS One. 2012;7:e38502. doi: 10.1371/journal.pone.0038502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, Navar LG, Lazartigues E. ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS One. 2011;6:e22682. doi: 10.1371/journal.pone.0022682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang XH, Wang YH, Wang JJ, Liu YC, Deng W, Qin C, Gao JL, Zhang LY. Role of angiotensin-converting enzyme (ACE and ACE2) imbalance on tourniquet-induced remote kidney injury in a mouse hindlimb ischemia-reperfusion model. Peptides. 2012;36:60–70. doi: 10.1016/j.peptides.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 42.Lapergue B, Mohammad A, Shuaib A. Endothelial progenitor cells and cerebrovascular diseases. Prog Neurobiol. 2007(83):349–362. doi: 10.1016/j.pneurobio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Yang HY, Bian YF, Zhang HP, Gao F, Xiao CS, Liang B, Li J, Zhang NN, Yang ZM. Angiotensin-(1-7) Treatment Ameliorates Angiotensin II-Induced HUVEC Apoptosis. Clin Exp Pharmacol Physiol. 2012 doi: 10.1111/1440-1681.12016. In Press. [DOI] [PubMed] [Google Scholar]