

Abstract
Podocytes are specialized kidney cells that form the kidney filtration barrier through the connection of their foot processes. Nephrin and Neph family transmembrane molecules at the surface of podocytes interconnect to form a unique type of cell-cell junction, the slit diaphragm, which acts as a molecular sieve. The cytoplasmic tails of Nephrin and Neph mediate cytoskeletal rearrangement that contributes to the maintenance of the filtration barrier. Nephrin and Neph1 orthologs are essential to regulate cell-cell adhesion and Rac-dependent actin rearrangement during Drosophila myoblast fusion. We hypothesized here that molecules regulating myoblast fusion in Drosophila could contribute to signaling downstream of Nephrin and Neph1 in podocytes. We found that Nephrin engagement promoted recruitment of the Rac exchange factor Dock1 to the membrane. Furthermore, Nephrin overexpression led to lamellipodia formation that could be blocked by inhibiting Rac1 activity. We generated in vivo mouse models to investigate whether Dock1 and Dock5 contribute to the formation and maintenance of the kidney filtration barrier. Our results indicate that while Dock1 and Dock5 are expressed in podocytes, their functions are not essential for the development of the glomerular filtration barrier. Furthermore, mice lacking Dock1 were not protected from LPS-induced podocyte effacement. Our data suggest that Dock1 and Dock5 are not the important exchange factors regulating Rac activity during the establishment and maintenance of the glomerular barrier.
Keywords: Dock1, Dock180, Dock5, Elmo, Rac, Rho GTPase, podocyte
Introduction
Glomerular filtration, the first step during urine formation, allows for the passage of water and solutes into the urinary space while retaining plasma proteins. The kidney filtration barrier is formed by three components: a fenestrated endothelium, a glomerular basement membrane and the podocytes.1 Podocytes are specialized epithelial cells of the kidney that extend microtubule-rich primary processes and actin-rich secondary foot processes that interdigitate and fold around the glomerular capillaries to form the so-called slit diaphragm that restricts the passage of macromolecules.2 Podocytes insult results in foot process effacement, disappearance of the slit diaphragm, and ultimately loss of blood proteins in the urine.2 Several studies highlight that the slit diaphragm is not only a unique type of cell-cell junction, but also an important signaling hub that dynamically regulates the cytoskeleton network of podocytes to maintain integrity and plasticity of the kidney filtration barrier.3 Nephrin and Neph1 molecules are the transmembrane cores of the slit diaphragm. Trans interaction between their extracellular domains generates the size exclusion molecular sieve that is anchored to the podocytes’ cytoskeleton via their cytoplasmic tail. In humans, mutation in the Nephrin gene, NPHS1, causes the Congenital Nephrotic Syndrome of the Finnish type characterized by massive proteinuria, highlighting the important contribution of this cell surface protein in podocytes function.4
Structures similar to the slit diaphragm are observed in model organisms. Notably, Drosophila nephrocytes express and rely on orthologs of Nephrin (sticks and stones; sns and hibris; hbs) and Neph1 (kirre) to achieve filtration of the hemolymphe.5-7 These Drosophila receptors also mediate myoblasts cell-cell adhesion and are fundamental components of the fly myoblast fusion machinery that regulates the formation of multi-nucleated muscle fibers.8-11 Mechanistically, trans interaction between the extracellular domains of kirre, expressed by founder myoblasts (Fm), and sns and hbs, both expressed by fusion competent myoblasts (Fcm), connects the cells membranes and activates signaling pathways that converge toward reorganization of the actin cytoskeleton which was shown to be essential for cell fusion.11 In Fm, kirre couples to the adaptor protein antisocial (ants) and acts upstream of the Rac GTPase to remodel the actin cytoskeleton.12 In Fcm, sns and hbs recruit the molecular adaptor Crk; this interaction is proposed to regulate Wasp-mediated actin polymerization by the Arp2/3 complex.13 Sns and hbs also mediate Rac activation and signaling through the Rac guanine exchange factor (GEF) myoblast city (mbc) and its protein partner dElmo.14,15 Until recently, it was unclear whether molecules identified as part of the fly myoblast fusion machinery would play similar roles in mammals. We identified the Rac GEF Dock1, the mammalian ortholog of mbc, as the first example of an important orchestrator of myoblast fusion in mammals: Dock1-null embryos exhibited a severe block in myoblast fusion.16 Similarly, inactivation of Rac1 and N-Wasp in mice revealed an essential role for these molecules during fusion.17,18 These studies highlight the important degree of conservation between the mechanism of myoblast fusion and cytoskeleton regulation in these cells between species. Nevertheless, Nephrin and Neph1 molecules are surprisingly not major regulators of myoblast fusion in mammals. Yet, signaling occurring on the cytosolic portion of Nephrin and Neph1 in podocytes controls the formation, maintenance and remodeling of foot processes through a significant regulation of the cytoskeleton, notably by controlling the activation status of Rho GTPases.1
Based on the central role of Nephrin and Neph1 in fly myoblast fusion, we tested here the hypothesis that signaling molecules of the myoblast fusion machinery are expressed in kidney podocytes to control the formation and maintenance of the filtration barrier. More specifically, we investigated if expression of Dock1 and Dock5 is essential in vivo for cytoskeletal regulation during the formation of the slit diaphragm or if Dock1 contributes to foot process effacement in disease. Our results with mutant mice suggest that Dock1 and Dock5 are not essential for the establishment and maintenance of the filtration barrier.
Results
The Drosophila myoblast fusion machinery is expressed in murine podocytes
In Drosophila, the orthologs of Nephrin and Neph promote myoblast cell-cell adhesion and fusion by activating signaling cascades that control cytoskeletal dynamics.11 Because Nephrin is a central cell-surface signaling protein in podocytes and is essential for the biology of these cells, we took advantage of a conditionally immortalized mouse podocyte cell line to investigate whether orthologs of Drosophila genes necessary for myoblast fusion in Drosophila are also expressed in podocytes.19,20 These podocyte progenitors proliferate rapidly when maintained under permissive conditions (33 °C; +INF-γ) while non-permissive conditions (37 °C; -INF-γ) induce their growth arrest and extensive remodeling of their cytoskeleton in a manner reminiscent of mature podocytes in vivo.19,20 Podocytes were cultured under permissive and non-permissive conditions and RNA was extracted at various time points to evaluate expression of molecules of the myoblast fusion machinery by RT-PCR. As expected, both undifferentiated and differentiated podocytes expressed Wt1 as a marker of the podocyte lineage (Fig. 1A). Both Neph1 and Neph2, but not Neph3, were also detectable and expression of the slit diaphragm protein Synaptopodin (Synpo) was induced upon INF-γ withdrawal, as previously reported,19 indicating optimal cell differentiation conditions (Fig. 1A).

Figure 1. The Drosophila myoblast fusion machinery is expressed in mouse podocytes. (A) Mouse podocytes were differentiated in vitro for 7 or 14 d or left undifferentiated and expression of components of the Drosophila myoblast fusion machinery was evaluated by RT-PCR. (B) Dock1 is expressed in the kidney glomerulus. IHC analyses showing Dock1 (left), Wt1 (middle) and merge (right) expression in E16.5 Dock1+/+ and Dock1−/− glomerulus (Scale bar: 20 μm, 40x).
Expression of the Dock-A/B subgroups of the Dock GEFs family was examined. Dock1, Dock4, and Dock5 were expressed in podocytes regardless of whether they were differentiated or not, while expression of Dock2 and Dock3 was not detected (Fig. 1A). Mouse podocytes also expressed the Dock-A/B members-binding partner Elmo2, while expression of Elmo1 and Elmo3 was not detectable (Fig. 1A). The SH3 adaptors Crk and Crk-L, which form a physical complex with DOCK1,21 were also expressed in podocytes. In Drosophila myoblasts, Sns (Nephrin) connects to the adaptor protein Ants to promote cell-cell fusion.12 We observed that orthologs of Ants, Tanc1 and Tanc2, are both expressed in podocytes and their expression increased with differentiation time (Fig. 1A). Finally the Rho GTPase Rac1 and Rac3 were expressed while expression of Rac2 was not detected (Fig. 1A).
Since Dock1 was reported to play a similar role to mbc during myoblast fusion, we hypothesized that Dock1 is a candidate GEF to promote Rac activation and cytoskeleton dynamics downstream of Nephrin/Neph1 cell surface proteins in podocytes.16 We examined the expression pattern of Dock1 during kidney development in vivo by immunohistochemistry (IHC) analyses using a Dock1 specific antibody and found this GEF to be highly expressed in E16.5 mouse embryonic kidney (Fig. 1B). The absence of staining of the Dock1 antibody in Dock1-null embryos confirmed the specificity of the observed expression pattern (Fig. 1B). Co-staining with Wt1, a marker of the podocyte lineage, indicated that Dock1 is expressed in these cells. Notably, we could also detect strong expression of this GEF in the capillary loop of the developing glomerulus (Fig. 1B). Our previous studies demonstrated a role for Dock1 in endothelial cell motility.22 Collectively, these results indicate that molecules of the myoblast fusion machinery are found in podocytes and that Dock1 is expressed in mouse podocytes during development.
Dock1 is recruited to actin foci upon Nephrin clustering
Clustering of Nephrin molecules in cells was previously shown to promote phosphorylation of its cytoplasmic tail and induce signaling that mediates the formation of distinguishable actin comet tails at sites of clustering.23 We took advantage of this model to examine whether Dock1 could signal downstream of Nephrin molecules. CHO.K1 cells were transfected with a construct coding for the intracellular domain of Nephrin fused to CD16 (CD16-NephrinIC-Myc) and Flag-DOCK1. Clustering of Nephrin by an anti-CD16 antibody induced the formation of actin comet tails that were visualized by phalloidin staining (Fig. 2A; bottom). In non-treated cells, DOCK1 was distributed in the cytosplasm (Fig. 2A; top). Upon anti-CD16-mediated Nephrin clustering, CD16-NephrinIC-Myc and DOCK1 co-localized at site of actin foci (Fig. 2A; bottom) suggesting that DOCK1 might be required to mediate Nephrin signaling. Moreover, overexpression of CD16-NephrinIC-Myc in CHO.K1 promoted lamellipodia formation, which could be prevented by treatment of cells with a Rac1 inhibitor (Fig. 2B-C). These in vitro experiments suggest that Dock1 is a candidate GEF to mediate Rac1 activation downstream of Nephrin.
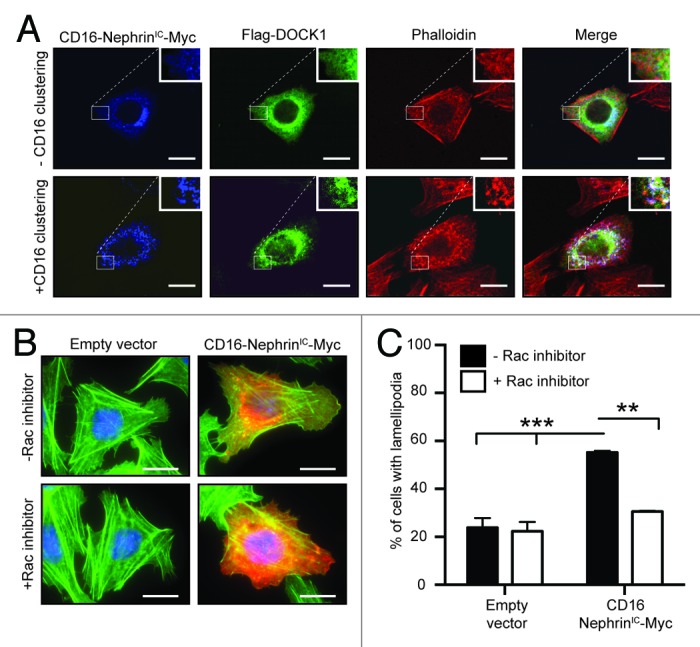
Figure 2. Dock1 is recruited to actin comet tails upon clustering of Nephrin molecules. (A) DOCK1 is recruited to actin comet tails upon clustering of chimeric CD16-Nephrin molecules. CHO.KI cells were transfected with CD16-NephrinIC-Myc and Flag-DOCK1 and clustering of Nephrin molecules was induced by an anti-CD16 antibody treatment. Immunofluorescence analyses showing the distribution of CD16-NephrinIC-Myc (blue), Flag-DOCK1 (green) and actin (red; phalloidin) molecules in non-treated and CD16-treated cells (Scale bar: 20 μm, 60x). (B) Expression of CD16-NephrinIC-Myc induces lamellipodia formation in cells through Rac activation. CHO.KI cells were transfected with CD16-NephrinIC-Myc and left untreated or treated with a Rac1 inhibitor. Immunofluorescence analyses showing expression of CD16-NephrinIC-Myc (red) and actin (green; phalloidin) (Scale bar: 20 μm, 60x). (C) Quantification of the % of cells with lamellipodia in B (n = 3).
Generation of Podocyte-specific Dock1 knockout mice
Our data indicates that Dock1 is expressed in multiple cell types of the kidney glomerulus. Moreover, Dock1 knockout mice die at birth and present several developmental abnormalities.16,22 Like mbc mutants, Dock1-null animals display a block in myoblast fusion but also present cardiovascular defects.16,22 Consequently, to examine the role of Dock1 in the formation and/or maintenance of the kidney filtration barrier in vivo, we generated a mouse with conditional deletion of Dock1 expression by crossing Dock1 floxed (flx) animals (Fig. 3A) with Podocin-Cre+ (Pod-Cre+; also known as Nphs2-Cre+) transgenic mice that express the Cre recombinase specifically in podocytes.24,25 Using PCR analyses on mice tails and kidney, we confirmed that expression of the Cre transgene led to the recombination of the Dock1 flx allele in the kidney (Fig. 3B). Furthermore, Cre-mediated genetic ablation of Dock1 reduced the level of Dock1 protein expression in podocytes of Pod-Cre+Dock1flx/flx animals as verified by IHC analysis (Fig. 3C).

Figure 3. Podocyte-specific ablation of Dock1 expression. (A) Partial schematic representation of the Dock1 wt allele, the Dock1 flx allele and the Dock1 recombined (ko) allele after exposure to the Cre recombinase. Positions of PCR primers used for genotyping analyses are indicated. (B) PCR analyses using P1, P2, P3 and Cre primer sets on genomic DNA extracted from mice kidneys or tails of the indicated genotype. A recombined Dock1 flx allele (ko allele) is detected in the kidney of transgenic Pod-Cre+ mice. (C) IHC analyses showing the absence of Dock1 expression in the podocytes of Pod-Cre+Dock1flx/flx mice (Scale bar: 20 μm, 100x).
Loss of Dock1 does not impair podocyte development and kidney function
Based on the observation that DOCK1 can be recruited to Nephrin and that Nephrin induces Rac-dependent cytoskeletal changes (Fig. 2A), we investigated whether Dock1 could be involved in the development of the glomerular filtration barrier in vivo. Pod-Cre+Dock1flx/flx animals were born and present at weaning (P21) with the expected Mendelian ratio (Table 1) and survived into adulthood. Moreover, gross examinations suggest that Pod-Cre+Dock1flx/flx mice are indistinguishable from control littermates. Histological analyses demonstrated that the kidneys and glomeruli develop normally in the absence of Dock1 expression (Fig. 4A). To evaluate the level of proteinuria, urine was collected, using metabolic cages, and fractionated on SDS-PAGE next to an albumin standard (0,25 to 1,5 μg). At 3 mo of age, Pod-Cre+Dock1flx/flx mice did not develop proteinuria in comparison to control animals as revealed by the trace amount of albumin present in the urine of both genotypes (Fig. 4B). The level of proteinuria was further quantified and expressed as a ratio between urinary albumin and creatinine and these analyses confirmed that Pod-Cre+Dock1flx/flx do not develop proteinuria at 3 and 6 mo of age (Fig. 4C). Older mutant animals did not develop proteinuria, deterioration in heath or reduction in weight (data not shown). These findings indicate that Dock1 is not essential in podocytes for the formation and maintenance of the filtration barrier.
Table 1. Viability of P21 pups derived from Pod-Cre+Dock1+/flx and Pod-Cre-Dock1+/flx mating .
| Frequency | Pod-Cre-Dock1+/+ | PodCre-Dock1+/flx | Pod-Cre-Dock1flx/flx |
Pod-Cre+ Dock1+/+ |
Pod-Cre+ Dock+/flx |
Pod-Cre+ Dock1flx/flx |
| Expected | 12.5% | 25% | 12.5% | 12.5% | 25% | 12.5% |
| Observed P21 | 9% (n = 4) | 30% (n = 14) | 22% (n = 10) | 13% (n = 6) | 15% (n = 7) | 11% (n = 5) |
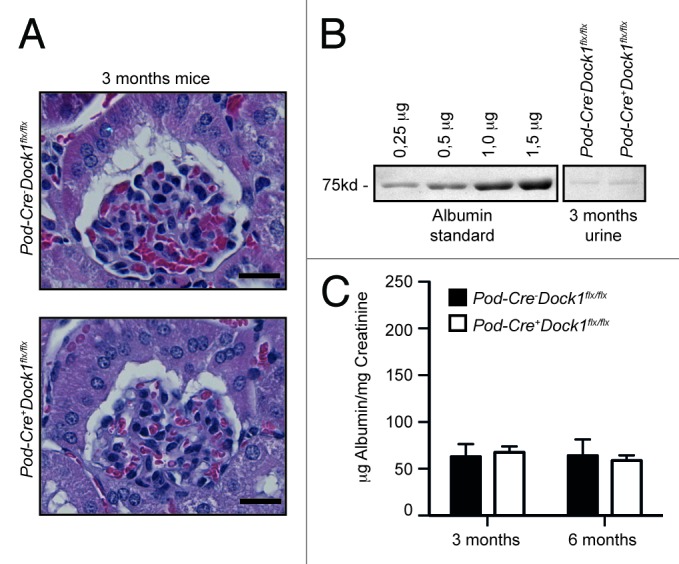
Figure 4.Pod-Cre+Dock1flx/flx mice display normal glomerular structure and function. (A) H&E-staining showing the normal glomerular development of 3 mo old Pod-Cre-Dock1flx/flx and Pod-Cre+Dock1flx/flx mice (Scale bar 20 μm, 40x). (B) Absence of proteinuria in the urine of 3-mo old Pod-Cre-Dock1flx/flx and Pod-Cre+Dock1flx/flx mice. Coomassie stained protein gel showing BSA standards (left) and the albumin content from the urine (right) of 3 mo old Pod-Cre-Dock1flx/flx and Pod-Cre+Dock1flx/flx mice. (C) Normal renal function in the absence of Dock1 expression in podocytes. Quantification of the average albumin-to-creatinine ratios in the urine of Pod-Cre-Dock1flx/flx and Pod-Cre+Dock1flx/flx mice at 3 and 6 mo after their birth (n = 5).
Loss of Dock5 does not impair glomerular function
Dock1, Dock2 and Dock5 are part of the Dock-A subgroup of Dock GEFs. While Dock2 expression is mostly restricted to cells of the hematopoietic lineages,26Dock5 was previously shown to act redundantly with Dock1 during the fine-tuning of muscle fiber formation.16 In addition, Dock5 was identified as one of the gene highly expressed in adult podocytes in comparison to its expression in the kidney cortex.27 To test the possibility that Dock2 or Dock5 GEFs could act redundantly with Dock1 during the establishment of the filtration barrier, we further confirmed their expression profiles in podocytes in vivo. Using Dock2-GFP knock-in mice, we confirmed our RT-PCR analyses by observing that Dock2 is not expressed in Podocalyxin-positive podocytes in vivo (Fig. 5A, top).28 In contrast, Dock2-GFP was readily detectable in the spleen, where an antibody against MOMA-1 marked resident macrophages in Dock2-GFP knock-in mice (Fig. 5A, bottom). As previously described,27 Dock5 expression was robustly detectable in adult mice podocytes as revealed by IHC using a Dock5 specific antibody (Fig. 5B). These data are in agreement with Dock5 mRNA expression pattern in cultured mouse podocyte cells in vitro (Fig. 1A). Dock5-positive podocytes were spatially localized in the glomerulus similarly to Wt1-positive podocytes (Fig. 5B).

Figure 5.Dock5 is not essential to glomerular function. (A) Dock2 is not expressed in the kidney. Dock2-GFP expression is absent in the kidney (top), but can be detected in the spleen (bottom) (Scale bar: 100 μm, 20x). (B) IHC analyses showing the expression of Dock5 and Wt1 in the podocytes of Pod-Cre+Dock1+/+Dock5+/+ mice (Scale bar: 25 μm, 100x). (C) Normal renal function in the absence of Dock5 expression in mice. Quantification of the average albumin-to-creatinine ratio in the urine of Pod-Cre+Dock1+/+Dock5+/+ and Pod-Cre+Dock1+/+Dock5−/− 2 mo old mice (n = 3). (D) Absence of proteinuria in the urine of P21 Pod-Cre-Dock1flx/flxDock5+/+ and Pod-Cre+Dock1flx/flxDock5−/− mice. Coomassie stained protein gel showing BSA standards (left) and the albumin content from the urine of a control mouse and 4 Pod-Cre+Dock1flx/flxDock5−/− mutant mice.
We previously demonstrated the absence of Dock5 protein expression in Dock5 mutant mice, which are viable and fertile and do not present gross developmental defects.16 To test the hypothesis that Dock5 could act redundantly with Dock1 in podocytes, we generated mice with deletion of these two GEFs by crossing Dock5−/− mice with Pod-Cre+Dock1flx/flx animals. We first tested if ablation of Dock5 expression alone affects glomerular filtration by testing the level of proteins in the urine of Pod-Cre+Dock1+/+Dock5−/− mice in comparison to Pod-Cre+Dock1+/+Dock5+/+ animals. Quantification of the ratio between urinary albumin and creatinine confirmed that deletion of Dock5 expression does not induce proteinuria (Fig. 5C). Pod-Cre+Dock1flx/flxDock5−/− animals were viable and mice were born and present at weaning (P21) with the expected Mendelian ratio (Table 2) and survived into adulthood (data not shown). Moreover, gross examination revealed that Pod-Cre+Dock1flx/flxDock5−/− mice were indistinguishable from their control littermates. To evaluate the level of proteinuria in double mutants, urine was collected and fractionated on SDS-PAGE next to an albumin standard (0,2 to 1,5 μg) (Fig. 5D). Pod-Cre+Dock1flx/flxDock5−/− mice did not develop proteinuria in comparison to control animals as revealed by the trace amount of albumin present in their urine (Fig. 5D). Moreover, Pod-Cre+Dock1flx/flxDock5−/− mice did not show deterioration in health or reduction in weight overtime (not shown). These findings indicate that Dock5 does not compensate for Dock1 loss in podocytes and that elimination of both Dock1 and Dock5 expression in these cells does not lead to aberrant formation nor contribute to maintenance of the glomerular filtration barrier.
Table 2. Viability of embryos and P21 pups derived from Pod-Cre+Dock1+/flxDock5−/− and Pod-Cre-Dock1+/flxDock5−/− mating .
| Frequency |
Pod-Cre-Dock1+/+ Dock5−/− |
Pod-Cre-Dock1+/flx Dock5−/− |
Pod-Cre-Dock1flx/flx Dock5−/− |
Pod-Cre+ Dock1+/+ Dock5−/− |
Pod-Cre+ Dock1+/flx Dock5−/− |
Pod-Cre+ Dock1flx/flx Dock5−/− |
| Expected | 12.5% | 25% | 12.5% | 12.5% | 25% | 12.5% |
| Observed E18.5 | 15% (n = 13) | 21% (n = 18) | 15% (n = 13) | 9% (n = 8) | 19% (n = 16) | 20% (n = 17) |
| Observed P21 | 7% (n = 3) | 34% (n = 15) | 21% (n = 9) | 7% (n = 3) | 21% (n = 9) | 11% (n = 5) |
Loss of Dock1 does not protect against LPS-induced proteinuria
A common response to podocyte insult includes a profound remodeling of the actin cytoskeleton, which induces foot processes to fuse and efface, a process that mimics lamellipodia formation and spreading in cultured cells.2 Foot process effacement has important clinical manifestations as it results in the malfunctioning of the slit diaphragm and the lost of blood proteins in the urine. Intraperitoneal injection of low-dose LPS was described as a powerful model to induce transient foot process effacement and proteinuria in mice.20 Moreover, several proteins were successfully identified as contributing to cytoskeleton rearrangement and therefore their inactivation in mice would protect them from LPS-induced proteinuria.29 We used the LPS model to test if Dock1 contributes to foot process effacement. Upon LPS injection, the general glomerular histology of control and Pod-Cre+Dock1flx/flx animals was not affected as reported for this model (Fig. 6A).29 To evaluate the level of proteinuria, mouse urine was collected before and after LPS injection and fractionated on SDS-PAGE next to an albumin standard (0,2 to 1,5 μg). (Fig. 6B). Both, Pod-Cre+Dock1+/+ and Pod-Cre+Dock1flx/flx mice developed proteinuria after LPS injection as revealed by the large amount of albumin present in their urine (Fig. 6B). The level of proteinuria before (t = 0) and 28 h after LPS injection (t = 28) was further quantified and expressed as a ratio between urinary albumin and creatinine; this confirmed that both Pod-Cre+Dock1+/+ and Pod-Cre+Dock1flx/flx animals develop similar proteinuria after LPS injection (Fig. 6C). These findings indicate that Dock1 does not contribute to foot process effacement and that inactivation of Dock1 in podocytes does not protect mice from LPS-induced proteinuria as previously observed for other Rac pathway activators or regulators.29
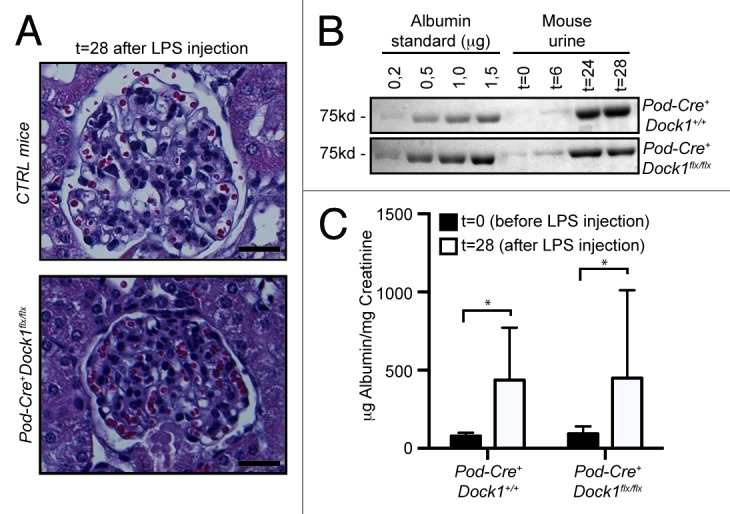
Figure 6. Loss of Dock1 expression in podocytes does not protect against LPS-induced proteinuria. (A) H&E-staining showing the glomerulus of 6 wk old Control and Pod-Cre+Dock1flx/flx female mice 28 h (t = 28) after LPS injection (Scale bar 20 μm, 40x). (B) LPS injection induces significant proteinuria in 6 wk old Pod-Cre+Dock1+/+ and Pod-Cre+Dock1flx/flx mice. Coomassie stained protein gel showing BSA standards (left) and the albumin content in the urine (right) collected from Pod-Cre+Dock1+/+ and Pod-Cre+Dock1flx/flx mice before (t = 0) and 6, 24 and 28 h (t = 6, t = 24, t = 28) after LPS injection. (C) Quantification of the average albumin-to-creatinine ratio in the urine of Pod-Cre-Dock1flx/flx and Pod-Cre+Dock1flx/flx before (t = 0) and 28 h (t = 28) after LPS injection.
Discussion
Podocytes have a highly dynamic and complex actin cytoskeleton that must be tightly regulated for their ability to exert glomerular filtration.2 We presented evidence that the Drosophila molecular machinery, involved in the control of the actin cytoskeleton during myoblast fusion, is expressed in mouse podocytes and that Nephrin clustering promotes recruitment of the Rac GEF Dock1 to the membrane where it colocalizes with actin comets. Nephrin engagement promotes the phosphorylation of its cytoplasmic tails that induces Nck recruitment, a crucial step to promote the formation of these actin structures.23 A direct interaction between Dock1 and Nck-2 has been reported.30 These in vitro evidences suggested that Dock1 could promote signaling downstream of Nephrin. However, our in vivo data clearly indicates that expression of Dock1 is not required for the formation and maintenance of the filtration barrier by podocytes. Moreover, we observed that expression of the closely related GEF Dock5 is also dispensable and that it does not compensate for the lack of Dock1 expression in mice. This was particularly surprising as complete ablation of Dock1 and Dock5 in mice completely prevented embryonic development such that no Dock1−/−Dock5 −/− mutants were ever recovered at E14.5.16 Similarly, Neph1 crosslinking promoted the recruitment of Grb2, another adaptor protein that can binds Dock1,31,32 to induce actin polymerization in cells.33 Much like we observed here for Dock1 and Dock5, podocyte-specific inactivation of Grb2 did not interfere with the formation of a functional glomerular filtration barrier in vivo.34 The expression data in mouse podocytes also revealed expression of Dock4 and a previous study identified this GEF as one of the gene highly expressed in podocytes in comparison to the rest of the kidney cortex.27 Therefore, it will be interesting in the future to generate a mouse model for Dock4 to address its biological role in vivo and more specifically to investigate its role in podocyte.
Two recent independent studies highlighted that expression of Rac1 is dispensable for podocytes-mediated glomerular filtration; these studies further substantiate our conclusions here that elimination of two Rac1 GEFs in podocytes does not induce proteinuria as a result of a defect in podocyte function.35,36 Altogether, these results are intriguing since podocytes need to first spread on the glomerular membrane of endothelial capillaries before extending primary and secondary processes. The expression profile in a mouse podocyte cell line suggests that Rac3 expression is increased upon differentiation of podocytes in vitro. It will be interesting to address in the future the role of this GTPase in mice and evaluate its potential redundancy with Rac1 during development. The role of the other prototypic Rho GTPases was also recently addressed in mice. While expression of RhoA is dispensable for podocytes physiological role, inactivation of Cdc42 leads to congenital nephropathy and massive proteinuria quickly after birth.35,36 Therefore, it will be important to identify the GEF(s) needed to specifically activate Cdc42 during the establishment of the glomerular filtration barrier.
Several studies now point toward Rac1 activity as playing a toxic role in podocytes such that it contributes to foot process effacement, a pathological state associated with an increase in cell motility. In a RhoGDIα mutant mouse, Rac1 was shown to be hyper-activated and these animals developed proteinuria which was reversible by administration of a Rac inhibitor.37,38 In addition, the inducible expression of an active form of the GTPase in podocytes induces the rapid unset of proteinuria.39 Moreover, a mutation that prevents Rac1 inactivation in the ARHGAP24 was identified in patients with focal segmental glomerulosclerosis.40 Using protamine sulfate injection as an experimental model to induced foot process effacement in mice, it was observed that loss of Rac1 expression as well as the inactivation of its upstream signaling intermediates Crk1/2 in podocytes could counteract foot process effacement.36,41 In a model where mice were challenged with LPS to induce foot process effacement, podocytes lacking expression of vitronectin, uPar or β3 integrin failed to adopt a motile phenotype and mice were protected from proteinuria.29 Dock1 was an attractive candidate to mediate Rac1 activation in podocytes since a previous study in breast cancer cell lines showed that signaling downstream of vitronectin/uPar/β3 integrin induces the recruitment of CrkII/Dock1 by p130Cas and was required to promote breast cancer migration.42 Nevertheless, our study using LPS injection in mice indicates that Dock1 is not the GEF regulating Rac1-mediated foot process effacement at least in this model of transiently induced proteinuria. Altogether, these studies further highlight the importance of identifying activators and effectors of Rac1 in podocytes since they could represent important therapeutic targets to prevent effacement of foot processes in several kidney diseases.
Materials and Methods
Animal care and mouse strains
Mice with a floxed Dock1 allele and Dock5 total mutant mice were previously described.16,24 Dock1flx/flxDock5−/− mice were generated by inter-crossing Dock1flx/flx mice with Dock5 mutants. Inactivation of Dock1 in podocytes was generated by crossing Dock1flx/flx animals with Podocin-Cre+ transgenic mice (Pod-Cre+; also known as Nphs2-Cre) mice.25 All mice were maintained on a mixed genetic background. Genomic DNA from mice tails and kidneys was extracted using standard procedures and mice were genotyped by PCR (see Table S1 for Primers). All mice were housed in a Specific Pathogen Free (SPF) Facility at the IRCM. Mouse experiments were approved by the IRCM Animal Care Committee and complied with the Canadian Council of Animal Care guidelines.
Cell culture
Podocytes were culture as previously reported.19,43 Briefly, cells were grown on dishes coated with 0.1 mg/ml Collagen type I and maintained undifferentiated in growth medium [10% FBS (Gibco), 100 U/ml Pen/Strep (Wisent), RPMI 1640 (Gibco)] supplemented with 50 U/ml INF-γ (for the first two passages) or 10 U/ml INF-γ (for the following passages) (#11276905001; Roche) in an incubator set at 33 °C. Podocyte differentiation was induced by splitting the cell at low density, removing INF-γ from the growth media and putting the cells in an incubator set at 37 °C. CHO.K1 cells were maintained in F-12 media (Gibco) containing 10% FBS (Gibco) and 100 U/ml Pen/Strep (Wisent).
CD16 Clustering experiment and immunocytochemistry
1.5x105 CHO.K1 cells were seeded in a 4-well Nunc Labtek glass chamber (BD Falcon). The following day, cells were transfected with CD16-NephrinIC-Myc (gift from Dr. A.J. Pawson) and Flag-DOCK1 (gift from Dr. M. Matsuda) plasmids using lipofectamine 2000 reagents (#11668–019; Invitrogen). Eighteen hours following transfection, clustering of Nephrin molecules was initiated by incubating cells with 1 μg/ml mouse anti-CD16 antibody for 15 min (#SC-19620; SantaCruz Biotechnology) as previously described.23 Cells were then washed with F-12 media and incubated with 1 μg/ml anti-mouse 350 antibody for 15 min (A-11045; Molecular Probes). Cells were washed 3 times with PBS and fixed in 4% Paraformaldehyde (PFA) for 10 min. Cells were washed again with PBS, permeabilized with PBST (0,1% Triton-X100 in PBS) and blocked with PBST 1% BSA for 1 h. Cells were stained using rabbit anti-Flag antibody diluted in PBST 1% BSA (#F7425; Sigma) for 1 h, washed 3 times with PBST and stained with anti-rabbit 488 antibody (1:1500; A-21441; Molecular Probes) for 30 min. Cells were washed 3 times with PBST and stained with Alexa Fluor 633 Phalloidin 633 (1:200; A-22284; Molecular Probes) for 20 min. Cells were washed again 3 times with PBST and slides were mounted using Mowiol reagent. Empty vector or CD16-Nephrin-IC-Myc transfected cells were treated with Rac1 inhibitor (#80602–892; EMD) for 30 min. Cells were then stained as described above.
RNA isolation and RT-PCR
Total RNA from undifferentiated mouse podocyte cells or cells differentiated for 7 or 14 d was extracted using TRIZOL reagent (#15596026; Invitrogen) and DNaseI (#18068015; Invitrogen) treatment was performed according to the recommended procedures. cDNAs were generated using Superscript II (#18064022; Invitrogen) and random primers (#48190011; Invitrogen) as recommended by the manufacturer. The expression of genes from the myoblast fusion machinery was tested by RT-PCR (see Table S1 for gene list and primer information) as previously described.24
Histology and Immunohistochemistry (IHC)
For histological analysis, kidneys were isolated and fixed in 4% PFA, embedded in paraffin, sectioned at 5 μm and stained with H&E. For IHC analysis of E16.5 embryonic kidney, tissues were embedded in OCT and sectioned at 5 μm. For IHC analyses, paraffin sections were deparaffinized in xylene and rehydrated in ethanol gradients while OCT sections were rinsed in PBS. Both paraffin and OCT sections were treated for antigen retrieval according to standard procedures in 10 mM sodium citrate buffer (pH6). Endogenous peroxidase activity was blocked on paraffin sections with 3% H2O2. Paraffin and OCT sections were permeabilized with IHC buffer (0,5% Triton X-100; 0,02% Tween-20 in PBS) and blocked for 1 h in IHC buffer containing 1% BSA. Primary antibodies were diluted in IHC buffer containing 1% BSA and incubated overnight. Sections were washed 3 times for 5 min with IHC buffer and incubated with either secondary or biotinylated secondary antibody diluted in IHC buffer containing 1% BSA for 1 h. Sections were washed 3 times with IHC buffer. OCT sections were mounted with Mowiol (#80058–440; VWR) and analyzed; paraffin sections were incubated with Streptavidin-HRP (1:1000; #554066; BD Biosciences) for 30 min. Paraffin sections were washed again 3 times with IHC buffer. The staining was revealed using DAB peroxidase substrate kit (#SK-4100; Vector Laboratories) and slides were counterstained with Hematoxylin. Antibodies used in IHC analysis include Dock1 (1:100; C19 #Sc-6167; Santa Cruz Biotechnology), Dock5 (1:100; from Dr Alan Hall), Wt1 (1:100; C19 #Sc-192; Santa Cruz Biotechnology), Podocalyxin (1:50; #MAB1556; R&D Systems) and MOMA-1 (1:200; #T-2011; BMA Biomedicals). Secondary antibodies used in IHC analyses include anti-goat 568 (1:1000; #A11057; Molecular Probes), anti-rabbit 488 (1:1000; #A21441; Molecular Probes), anti-goat biotinylated (1:150; #BA9500; Vector Laboratories) and anti-rabbit biotinylated (1:150; #BA1000; Vector Laboratories).
Baseline urine proteinuria analysis and LPS mouse model
For baseline proteinuria analyses, mouse urine was collected in metabolic cages at different time points. The urine albumin content was determined by running 10 μl of urine on SDS-PAGE gels. Gels were then stained with Coomassie blue and protein bands were quantified according to the BSA standard using the Fiji software. The model of intraperitoneal injection of LPS was used to induce foot process effacement and transient proteinuria in mice exactly as previously described.20,29 Briefly, urine from 6 wk old female mice was collected (t = 0) before they were injected intraperitoneally with 10 μg per gram of mouse weight using a LPS solution (1 mg/ml in sterile PBS; #tlrl-eblps; Invivogen). Urine was collected at 6, 24 and 28 h (t = 6, t = 24, t = 28) after LPS injection. At t = 28, mice were euthanized and the kidneys were collected and processed for histological analyses. The albumin content in the urine from LPS-injected mice was quantified by fractionating 5 μl of urine on SDS-PAGE gels, staining them with Coomassie blue and quantifying the bands using Fiji software. The albumin-creatinine ratio was determined by using creatinine ELISA kits (#C500701; Cayman Chemical).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Tomoko Takano (McGill University) for helpful discussions and Dr M. Patel for critical reading of the manuscript. We are grateful to Drs Tony Pawson (CD16-Nephrin-Myc construct), Alan Hall (anti-Dock5 polyclonal antibody), Michiyuki Matsuda (Flag-DOCK1 construct) and Susan E. Quaggin (Podocin-Cre (Nphs2-Cre) transgenic mice) for generous gifts of reagents. We also acknowledge Dr Peter Mundel for providing the immortalized mouse podocyte cell line. Côté JF is a recipient of a Senior Investigator award from the Fonds de Recherche du Québec-Santé (FRQ-S). This work was funded by an operating grant from the Canadian Institutes of Health Research (CIHR: MOP-77591) to Côté JF.
Supplemental Material
Supplemental material may be found here: http://www.landesbioscience.com/journals/smallgtpases/article/27430
Glossary
Abbreviations:
- ants
antisocial
- BSA
bovine serum albumin
- Cdc42
cell division cycle 42
- Crk
v-crk avian sarcoma virus CT10 oncogene homolog
- Crkl
v-crk sarcoma virus CT10 oncogene homolg (avian)-like
- Dock1
dedicator of cytokinesis 1
- Dock2
dedicator of cytokinesis 2
- Dock3
dedicator of cytokinesis 3
- Dock4
dedicator of cytokinesis 4
- Dock5
dedicator of cytokinesis 5
- E
embryonic day
- Fbs
fetal bovine serum
- Fcm
fusion competent myoblast
- Flx
Floxed allele
- Fm
founder myoblast
- GEF
guanine exchange factor
- Grb2
growth factor receptor bound protein 2
- Hbs
hibris
- IHC
Immunohistochemistry
- INF
interferon
- LPS
lipopolysaccharide
- Mbc
myoblast city
- Nck
non-catalytic region for tyrosine kinase adaptor protein 1
- NPHS1
Nephrosis1, congenital, Finnish type (Nephrin)
- Nphs2
nephrosis 2, podocin
- N-Wasp
Wiskott-Aldrich syndrome-like
- Pod-Cre+
Podocin-Cre+ transgenic mouse
- Rac1
RAS-related C3 botulinum substrate 1
- Rac2
RAS-related C3 botulinum substrate 2
- Rac3
RAS-related C3 botulinum substrate 3
- Sns
sticks and stones
- Synpo
synaptopodin
- Tanc1
tetratricopeptide repeat, ankyrin repeat and coiled-coil containing 1
- Tanc2
tetratricopeptide repeat, ankyrin repeat anc coiled-coil containing 2
- Wt1
Wilms tumor 1 homolog
- −/−
Knockout
- +/+
Wild-type
Footnotes
Previously published online: www.landesbioscience.com/journals/smallgtpases/article/27430
References
- 1.Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm--from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9:587–98. doi: 10.1038/nrneph.2013.169. [DOI] [PubMed] [Google Scholar]
- 2.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–37. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm--from a thin grey line to a complex signalling hub. Nat Rev Nephrol. 2013;9:587–98. doi: 10.1038/nrneph.2013.169. [DOI] [PubMed] [Google Scholar]
- 4.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, et al. Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–82. doi: 10.1016/S1097-2765(00)80057-X. [DOI] [PubMed] [Google Scholar]
- 5.Weavers H, Prieto-Sánchez S, Grawe F, Garcia-López A, Artero R, Wilsch-Bräuninger M, Ruiz-Gómez M, Skaer H, Denholm B. The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature. 2009;457:322–6. doi: 10.1038/nature07526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhuang S, Shao H, Guo F, Trimble R, Pearce E, Abmayr SM. Sns and Kirre, the Drosophila orthologs of Nephrin and Neph1, direct adhesion, fusion and formation of a slit diaphragm-like structure in insect nephrocytes. Development. 2009;136:2335–44. doi: 10.1242/dev.031609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Na J, Cagan R. The Drosophila nephrocyte: back on stage. J Am Soc Nephrol. 2013;24:161–3. doi: 10.1681/ASN.2012121227. [DOI] [PubMed] [Google Scholar]
- 8.Bour BA, Chakravarti M, West JM, Abmayr SM. Drosophila SNS, a member of the immunoglobulin superfamily that is essential for myoblast fusion. Genes Dev. 2000;14:1498–511. [PMC free article] [PubMed] [Google Scholar]
- 9.Shelton C, Kocherlakota KS, Zhuang S, Abmayr SM. The immunoglobulin superfamily member Hbs functions redundantly with Sns in interactions between founder and fusion-competent myoblasts. Development. 2009;136:1159–68. doi: 10.1242/dev.026302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strünkelnberg M, Bonengel B, Moda LM, Hertenstein A, de Couet HG, Ramos RG, Fischbach KF. rst and its paralogue kirre act redundantly during embryonic muscle development in Drosophila. Development. 2001;128:4229–39. doi: 10.1242/dev.128.21.4229. [DOI] [PubMed] [Google Scholar]
- 11.Abmayr SM, Pavlath GK. Myoblast fusion: lessons from flies and mice. Development. 2012;139:641–56. doi: 10.1242/dev.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen EH, Olson EN. Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev Cell. 2001;1:705–15. doi: 10.1016/S1534-5807(01)00084-3. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Shilagardi K, Zhang S, Hong SN, Sens KL, Bo J, Gonzalez GA, Chen EH. A critical function for the actin cytoskeleton in targeted exocytosis of prefusion vesicles during myoblast fusion. Dev Cell. 2007;12:571–86. doi: 10.1016/j.devcel.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Erickson MR, Galletta BJ, Abmayr SM. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol. 1997;138:589–603. doi: 10.1083/jcb.138.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geisbrecht ER, Haralalka S, Swanson SK, Florens L, Washburn MP, Abmayr SM. Drosophila ELMO/CED-12 interacts with Myoblast city to direct myoblast fusion and ommatidial organization. Dev Biol. 2008;314:137–49. doi: 10.1016/j.ydbio.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurin M, Fradet N, Blangy A, Hall A, Vuori K, Côté JF. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc Natl Acad Sci U S A. 2008;105:15446–51. doi: 10.1073/pnas.0805546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasyutina E, Martarelli B, Brakebusch C, Wende H, Birchmeier C. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc Natl Acad Sci U S A. 2009;106:8935–40. doi: 10.1073/pnas.0902501106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruenbaum-Cohen Y, Harel I, Umansky KB, Tzahor E, Snapper SB, Shilo BZ, Schejter ED. The actin regulator N-WASp is required for muscle-cell fusion in mice. Proc Natl Acad Sci U S A. 2012;109:11211–6. doi: 10.1073/pnas.1116065109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 20.Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L, Rastaldi MP, Calvaresi N, Watanabe H, Schwarz K, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–7. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–6. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanematsu F, Hirashima M, Laurin M, Takii R, Nishikimi A, Kitajima K, Ding G, Noda M, Murata Y, Tanaka Y, et al. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ Res. 2010;107:1102–5. doi: 10.1161/CIRCRESAHA.110.223388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, Huang H, Larose L, Li SS, Takano T, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–23. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 24.Laurin M, Huber J, Pelletier A, Houalla T, Park M, Fukui Y, Haibe-Kains B, Muller WJ, Côté JF. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci U S A. 2013;110:7434–9. doi: 10.1073/pnas.1213050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–16. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. 2001;412:826–31. doi: 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- 27.Brunskill EW, Georgas K, Rumballe B, Little MH, Potter SS. Defining the molecular character of the developing and adult kidney podocyte. PLoS One. 2011;6:e24640. doi: 10.1371/journal.pone.0024640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, et al. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol. 2006;174:647–52. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, Xie L, Henger A, Schmid H, Rastaldi MP, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 30.Tu Y, Kucik DF, Wu C. Identification and kinetic analysis of the interaction between Nck-2 and DOCK180. FEBS Lett. 2001;491:193–9. doi: 10.1016/S0014-5793(01)02195-0. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda M, Ota S, Tanimura R, Nakamura H, Matuoka K, Takenawa T, Nagashima K, Kurata T. Interaction between the amino-terminal SH3 domain of CRK and its natural target proteins. J Biol Chem. 1996;271:14468–72. doi: 10.1074/jbc.271.24.14468. [DOI] [PubMed] [Google Scholar]
- 32.Bisson N, James DA, Ivosev G, Tate SA, Bonner R, Taylor L, Pawson T. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat Biotechnol. 2011;29:653–8. doi: 10.1038/nbt.1905. [DOI] [PubMed] [Google Scholar]
- 33.Garg P, Verma R, Nihalani D, Johnstone DB, Holzman LB. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol Cell Biol. 2007;27:8698–712. doi: 10.1128/MCB.00948-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bisson N, Ruston J, Jeansson M, Vanderlaan R, Hardy WR, Du J, Hussein SM, Coward RJ, Quaggin SE, Pawson T. The adaptor protein Grb2 is not essential for the establishment of the glomerular filtration barrier. PLoS One. 2012;7:e50996. doi: 10.1371/journal.pone.0050996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott RP, Hawley SP, Ruston J, Du J, Brakebusch C, Jones N, Pawson T. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J Am Soc Nephrol. 2012;23:1149–54. doi: 10.1681/ASN.2011121206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, Vining C, Randolph A, Herbach N, Wanke R, et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013;84:920–30. doi: 10.1038/ki.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Doi T, Mizoguchi A, Matsuura N, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–80. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 38.Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–6. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 39.Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS, Akilesh S. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol. 2013;33:4755–64. doi: 10.1128/MCB.00730-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, Antignac C, Pollak M, Kopp JB, Winn MP, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest. 2011;121:4127–37. doi: 10.1172/JCI46458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.George B, Verma R, Soofi AA, Garg P, Zhang J, Park TJ, Giardino L, Ryzhova L, Johnstone DB, Wong H, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J Clin Invest. 2012;122:674–92. doi: 10.1172/JCI60070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol. 2008;182:777–90. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mundel P, Reiser J, Zúñiga Mejía Borja A, Pavenstädt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–58. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.