

Abstract
The lateral part of the ventral bed nucleus of the stria terminalis (vlBNST) is a critical site for the antiaversive effects of noradrenergic drugs during opioid withdrawal. The objective of the present study is to identify the cellular action(s) of noradrenaline in the vlBNST after withdrawal froma5d treatment with morphine. The vlBNST is a heterogeneous cell group with multiple efferent projections. Therefore, neurons projecting to the midbrain were identified by retrograde transport of fluorescent microspheres injected in the ventral tegmental area (VTA). Whole-cell voltage clamp recordings of these neurons and of those sharing physiological properties were done in brain slices. Noradrenaline activated α1-adrenergic receptors to increase GABAA-IPSC frequency. Noradrenaline produced a similar increase in GABAA-IPSCs during acute opioid withdrawal, but this increase resulted from activation ofβ-adrenergic receptors, adenylyl cyclase, and protein kinase A, as well as α1-adrenergic receptors. Given that neurons in the vlBNST send an excitatory projection to the VTA, noradrenaline may reduce excitatory drive to mesolimbic dopamine cells. This mechanism might contribute to the withdrawal-induced inhibition of dopamine neurons and explain how noradrenergic drugs microinjected into the vlBNST reduce aversive aspects of opioid withdrawal.
Keywords: α1- and β-adrenergic receptors, withdrawal, electrophysiology, morphine, adenylyl cyclase, retrograde labeling
Introduction
The bed nucleus of the stria terminalis (BNST) is a cluster of small nuclei surrounding the caudal part of the anterior commissure within the extended amygdala (Alheid et al., 1995). The BNST projects to many different areas of the brain (e.g., somatomotor, central autonomic control, neuroendocrine) and has a complex and heterogeneous physiology (Ju and Swanson, 1989; Alheid et al., 1995; Dong et al., 2000; Egli and Winder, 2003; Dong and Swanson, 2004). The BNST plays a role in several aspects of stress-induced behaviors, including relapse to cocaine use, and contributes to the reinforcing effects of opioids (Henke, 1984; Casada and Dafny, 1991; Erb and Stewart, 1999; Walker et al., 2000; Erb et al., 2001).
Compulsive use of opioids results from their reinforcing properties and the highly aversive withdrawal syndrome that follows their cessation (Bechara et al., 1998; Schuckit, 2000). The withdrawal syndrome comprises somatic and aversive components, and adrenergic drugs reduce both (Grosz, 1972; Gold et al., 1978; Roehrich and Gold, 1987). The lateral part of the ventral BNST (vlBNST) is an important brain site in rats for the antiaversive actions of adrenergic drugs (Delfs et al., 2000). Lesions of noradrenergic inputs from A1-A2 nuclei or microinjection of adrenergic drugs (β-adrenergic receptor antagonists or α2-adrenergic receptor agonists) directly into the vlBNST decrease avoidance behavior during morphine withdrawal (Delfs et al., 2000). It is unknown how noradrenaline regulates synaptic transmission in the vlBNST to trigger aversion during withdrawal and how noradrenergic drugs reverse this effect.
The present study focused on identified vlBNST neurons innervating dopamine neurons of the ventral tegmental area (VTA), as well as those with similar physiological properties. In control conditions, noradrenaline activated α1-adrenergic receptors and likely depolarized local GABA neurons to increase GABAA-IPSCs. During acute morphine withdrawal, noradrenaline also increased GABAA-IPSCs, but now both α1- and β-adrenergic receptors were involved. The recruitment of β-adrenergic receptors occurred because of an increased adenylyl cyclase-protein kinase A (AC-PKA) pathway during acute morphine withdrawal. Given that the activity of dopamine neurons decreases during morphine withdrawal (Diana et al., 1995; Georges et al., 2003) and that the vlBNST sends excitatory projections to the VTA (Georges and Aston-Jones, 2002), noradrenaline may inhibit vlBNST projection neurons and decrease excitatory input to midbrain dopamine neurons during withdrawal.
Materials and Methods
Intracerebral microinjections. Sixteen rats (Sprague Dawley, 150-250 gm) were assigned to the identification of BNST neurons projecting to the VTA. These rats were anesthetized with ketamine (50 mg/kg, i.p.) and xylazine (5 mg/kg, i.p.). Fluorescent microspheres (100 nl; Molecular Probes, Eugene, OR) were microinjected in the VTA (λ, +2.5; lateral, +1.5; vertical, -7.5 mm). Acute brain slices were prepared 2-3 d later to visualize (confocal microscopy) and record from vlBNST neurons that were labeled after retrograde transport of the microspheres. No labeling was observed in the ventral BNST (vBNST) when microinjections were made dorsal to the VTA.
Morphine treatment. Rats were randomly assigned to naive, placebo, or morphine groups. The placebo treatment did not affect the measured parameters such that naive and placebo rats were pooled as the control group. All but naive rats were anesthetized with isoflurane and received placebo- or morphine-containing subcutaneous pellet (75 mg morphine base per pellet; National Institute on Drug Abuse) implants. The morphine treatment consisted of one pellet on day 1 and two pellets on days 3 and 5. Experiments were done on days 6 or 7. The morphine group received a single subcutaneous injection of morphine (10 mg/kg). This pretreatment eliminated mortality induced by the initial pellet implantation. The placebo group received a saline injection.
Slice preparation and electrophysiology. Rats were anesthetized with halothane and their brains were rapidly removed. Coronal slices (250 μm) containing the BNST were prepared in a physiological solution containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 6 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 d-glucose at 15°C. Slices were incubated at 34°C for 30 min and transferred to a chamber that was constantly perfused (1.5 ml/min) with physiological solution maintained at 34°C and equilibrated with 95%O2-5%CO2. Slices from morphine-treated rats maintained for 1 hr in normal physiological solution were considered withdrawn. In some cases, morphine (1 μm) in the physiological solution avoided spontaneous withdrawal of treated slices. To have comparable conditions, control slices were also kept in morphine (1 μm) for 1 hr. The addition of naloxone (1 μm) during the recordings precipitated morphine withdrawal. Whole-cell voltage-clamp recordings were made using microelectrodes filled with a solution containing (in mm): 70 K+ gluconate, 80 KCl, 1 EGTA, 5 HEPES, 2 MgATP, and 0.3 GTP. GABAA-IPSCs were evoked by local fiber stimulation with bipolar electrodes. Electrodes were placed in the vBNST, 100-500 μm medial from the recorded neuron, and paired electrical stimuli (0.1 msec duration; 50 msec interval) were applied at 0.1Hz. GABAA-IPSCs were pharmacologically isolated by adding NBQX (2,3-dihydroxy-6-nitro-7-sulfonylbenzo[f]quinoxaline) (5 μm) and MK-801 ((+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate) (10 μm) to block AMPA and NMDA receptor-dependent postsynaptic currents, respectively. Spontaneous GABAA-IPSCs were recorded at 10 kHz and digitally filtered at 1 kHz. The amplitude and frequency of spontaneous GABAA-IPSCs were determined from 30 sec recording episodes. Miniature GABAA-IPSCs were recorded at 10 kHz with tetrodotoxin (0.5 μm) in the perfusing solution. The amplitude and frequency of miniature GABAA-IPSCs were measured. At the end of each experiment, picrotoxin (100 μm) was added and completely abolished all evoked or spontaneous postsynaptic currents.
Drugs. Stock solutions of NBQX (50 mm), prazosin (1 mm), clonidine (1 mm), yohimbine (1 mm), forskolin (100 mm), H89 (10 mm), and MK-801 (10 mm) were prepared in DMSO (100%). Drugs were further dissolved in the physiological solution at the desired concentration. DMSO concentration never exceeded 0.001%. Stock solutions of tetrodotoxin (1 mm), -(-) noradrenaline (10 mm), propranolol (10 mm), DAMGO (d-Ala2-N-Me-Phe4-Glycol5-enkephalin) (1 mm), morphine (10 mm), and naloxone (10 mm) were prepared in water and dissolved in the physiological solution at the desired concentration.
Results
Neurons of the vlBNST projecting to the VTA had characteristic physiological properties
Fluorescent microspheres injected into the VTA were retrogradely transported to the vlBNST. Labeled neurons were visualized in brain slices, and whole-cell voltage-clamp recordings were made from 29 labeled neurons under confocal microscopy (Fig. 1). These neurons were <30 μm in diameter and displayed characteristic resting membrane properties with a small membrane capacitance and inwardly rectifying potassium conductance (Table 1, Fig. 1D,E). They responded to noradrenaline with a small current in voltage clamp (Fig. 1F) or a small hyperpolarization in current clamp (Fig. 1G). In contrast, a subset of neurons that were not labeled displayed different physiological characteristics. These neurons had a larger membrane capacitance, time-dependent inward rectification, and responded to noradrenaline with a large inward current (Table 1). These neurons were also distinguished by their size of >30 μm in diameter. These unlabeled and physiologically distinct neurons were not included in this study. Only labeled neurons (n = 29) or those sharing the physiological properties with labeled neurons (n = 238) were included in this study.
Figure 1.
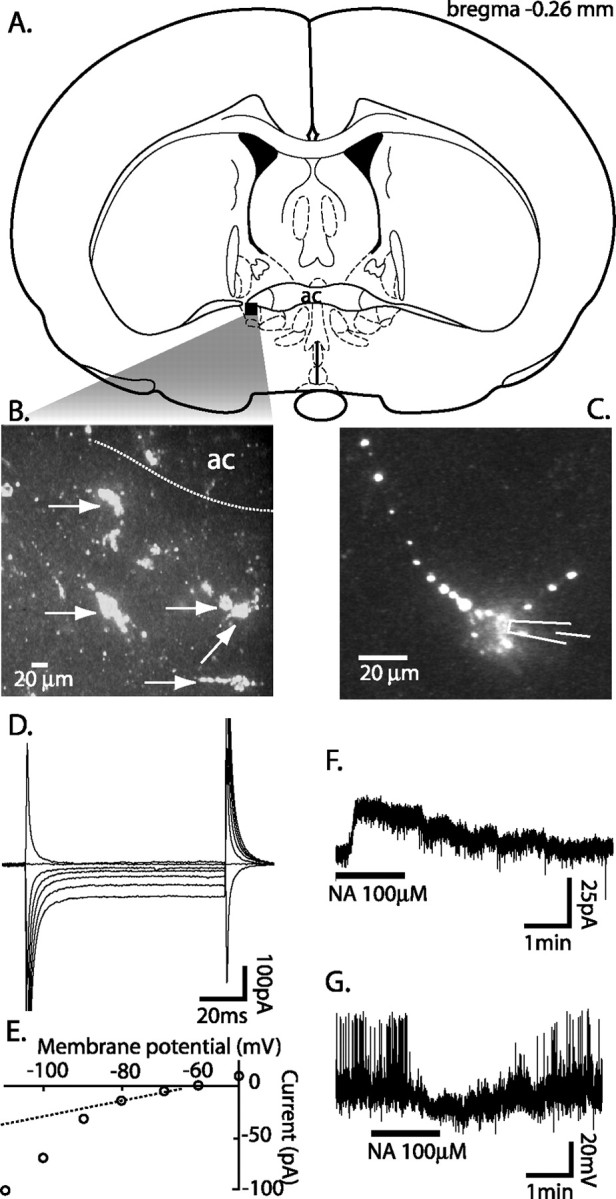
Anatomical (A-C) and electrophysiological (D-G) identification of neurons in the vlBNST projecting to the VTA. A, Recordings were restricted to the vlBNST where most labeling was observed (adapted from Paxinos and Watson, 1998). B, Neurons (arrows) in the vlBNST filled with fluorescent microspheres transported retrograde to their cell bodies after being microinjected in the VTA. C, Focus on one labeled neuron showing individual fluorescent beads in both the cell body and processes. D, Superimposed steady-state currents in response to voltage steps, ranging from +10 to -50 mV, applied from rest (-60 mV). E, The current-voltage plot revealed an inwardly rectifying potassium conductance observed in labeled neurons. Bath application of noradrenaline produced small outward currents in the voltage-clamp configuration (F) and a corresponding hyperpolarization in the current-clamp conditions (G). In this particular neuron, noradrenaline also blocked the spontaneous firing (G). ac, Anterior commissure.
Table 1.
Summary of the physiological properties of neurons (projection neurons) frequently labeled by retrograde transport of fluorescent microspheres microinjected in the VTA compared with those (other neurons) that were never labeled
|
|
Projection neurons |
Other neurons |
|---|---|---|
| Membrance capacitance | 21.9 ± 1.2 pF | 54.4 ± 8 pF |
| Input resistance | 1090 ± 171 MΩ | 229 ± 25 MΩ |
| Rectification | IR | Ih |
| Response to noradrenaline |
VC, in or out <50 pA CC, hyperpolarization |
In >50 pA |
CC, Current clamp; VC, voltage clamp; IR, inward rectification; Ih, hyperpolarization-activated current.
Opioids reduced GABAA-IPSCs
The cellular targets of morphine were first investigated in the vlBNST. One of the most robust acute effects of morphine was to decrease GABAA-IPSCs. In control rats, morphine decreased the amplitude of evoked GABAA-IPSCs by 30 ± 6% (t = 5.3, paired Student's t test; p = 0.006, before vs after morphine; n = 15) (Fig. 2A,B). In slices from morphine-treated rats, morphine caused a larger inhibition of GABAA-IPSCs (52.5 ± 4%; t = 6.1, paired Student's t test; p = 0.0009, before vs after morphine; n = 15; t = 5.4, unpaired Student's t test; p = 0.0001, control vs morphine-treated; n = 15 per group) (Fig. 2A,B). Morphine (1 μm) produced no measurable postsynaptic current, suggesting that it reduced GABA release by a presynaptic mechanism.
Figure 2.

Chronic morphine sensitized GABA synapses to morphine in the vlBNST. Morphine (A, B) and the selective μ-opioid receptor agonist DAMGO (C) reduced electrically evoked (0.1 Hz) GABAA-IPSCs in slices from control and morphine-treated rats. Slices from morphine-treated rats were washed with morphine-free solution for 60 min such that they were in withdrawal. A, Each trace is the average of five evoked GABAA-IPSCs before (left traces), during superfusion with morphine (1 μm, 20 min; middle traces), and after the addition of naloxone (1 μm; right traces). B, Summary of the effect of 1 μm morphine on the amplitude of evoked GABAA-IPSCs from 15 neurons- groups (10 rats in each group). C, Dose-response curves of the effect of cumulative concentrations of the μ-opioid receptor agonist DAMGO on evoked GABAA-IPSCs. Inset shows representative traces, averaged from 5-10 evoked GABAA-IPSCs in a slice from a control rat, before (left), after DAMGO (1 μm; middle), and after reversing the effect of DAMGO with naloxone (1 μm; right). S2, Stimulus 2; S1, stimulus 1. Error bars indicate SEM.
The paired-pulse ratio of two IPSCs evoked at a 50 msec interval was determined to further evaluate a presynaptic site of action of morphine. In control rats, the paired-pulse ratio was a slight facilitation (1.1 ± 0.1) (Fig. 2A, top left trace), and this changed to a slight depression in treated slices during acute morphine withdrawal (0.9 ± 0.03) (Fig. 2A, bottom left trace). Morphine (1 μm) not only decreased the amplitude of the GABAA-IPSCs but also caused paired-pulse facilitation in withdrawn slices (2.6 ± 0.2) (Fig. 2A, bottom middle trace). This further suggests that morphine regulated GABA release by a presynaptic and more efficient mechanism during withdrawal.
Chronic morphine did not change the inhibition caused by cumulative concentrations of DAMGO (Fig. 2C). Neither the potency (morphine-treated, 15 ± 1.6 nm; control, 21 ± 1.7 nm; F(2,62) = -0.005; p > 0.05; n = 7 and 5) nor the efficacy (morphine-treated, 61 ± 4%; control, 60 ± 5%) of DAMGO changed after chronic morphine treatment. This differed from the increased efficacy of morphine (1 μm) observed during withdrawal. Given that DAMGO is a full agonist at μ-opioid receptors with a potentially large receptor reserve, it may be expected that a small change in sensitivity to DAMGO would be difficult to detect. An increase in sensitivity would be more easily observed with a partial agonist, such as morphine. Similarly, the shift in the cumulative response to DAMGO in locus ceruleus neurons after chronic morphine treatment was smaller than that for morphine (Christie et al., 1987).
In other brain areas, chronic morphine augmented the presynaptic AC-PKA pathway and amplified the maximum effect of morphine (Ingram et al., 1998). The next series of experiments were aimed at testing this hypothesis in the vlBNST.
Augmented AC-PKA pathway and enhanced GABA release during acute morphine withdrawal
Chronic morphine treatment upregulates AC-PKA and results in rebound activity during opioid withdrawal that facilitates GABA release (Bonci and Williams, 1997; Chieng and Williams, 1998; Ingram et al., 1998; Jolas et al., 2000). To investigate acute morphine withdrawal, slices from morphine-treated and control rats were maintained in morphine (1 μm), and withdrawal was induced by the addition of naloxone (1 μm). Naloxone increased the amplitude of evoked GABAA-IPSCs in slices from control (Fig. 3A1, top traces) and morphine-treated (bottom traces) rats. The increase in IPSC amplitude was larger in slices from morphine-treated compared with control rats (morphine-treated, 160 ± 10%; control, 120 ± 20%; z = -5.7, Mann-Whitney; p < 0.0001; n = 10 and 14) (Fig. 3A2).
Figure 3.
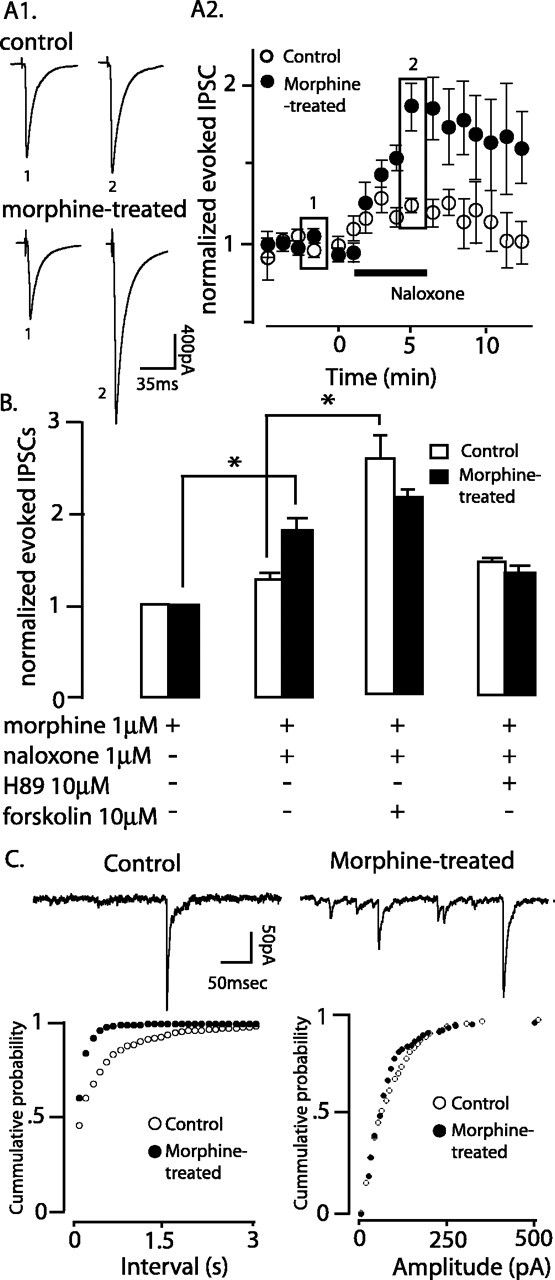
Acute morphine withdrawal increased evoked (A, B) and spontaneous miniature (C) GABAA-IPSCs through the AC-PKA pathway. A1, Traces represent the average of five evoked GABAA-IPSCs in the presence of morphine (1 μm) and at the peak effect of naloxone in slices from control (top) and morphine-treated rats (bottom). A2, Summary of the effect of acute naloxone-precipitated morphine withdrawal on evoked GABAA-IPSCs (n = 10 and 14, control andmorphine-treated,respectively). B, Summary of the effect of adenylyl cyclase blockade with H89 (10 μm; n = 5 and 7) and activation with forskolin (10 μm; n = 5 per group) on evoked GABAA-IPSCs during naloxone-precipitated morphine withdrawal. C, Top, representative recordings of miniature GABAA-IPSCs recorded in tetrodotoxin (0.5 μm) in slices from control (left trace) and morphine-treated (right trace) rats. Summary of the frequency (left) and amplitude (right) of miniature GABAA-IPSCs in slices from control (n = 9) and morphine-treated (n = 15) rats after acute morphine withdrawal. * p < 0.05. Error bars indicate SEM. Rectangles in A2 refer to 1 and 2 in A1.
In the presence of naloxone (1 μm), the activation of adenylyl cyclase with forskolin (10 μm) increased the IPSC amplitude more in slices from control than morphine-treated rats (morphine-treated, +29 ± 4%; control, +140 ± 15%; t = 6.5, unpaired Student's t test; p = 0.0001; n = 5 per group) (Fig. 3B). In fact, the increase caused by forskolin in control was similar to that caused by naloxone in morphine-treated slices (Fig. 3B). This raised the possibility that in withdrawal, the AC-PKA pathway was maximally active and therefore occluded the effect of forskolin. To further examine this possibility, naloxone was tested after inhibiting PKA with H89 (10 μm). In slices from morphine-treated rats, H89 completely abolished the naloxone-induced increase of evoked GABAA-IPSCs (+1.3 ± 0.1; t = 3.2, unpaired Student's t test; naloxone vs naloxone plus H89; p > 0.05; n = 10 and 5) (Fig. 3B), whereas it did not modify the effect of naloxone in control rats (Fig. 3B). This result indicates that withdrawal from morphine increased GABAA-IPSCs through an increase in excitability at the point of stimulation or an increase in the probability of release at the presynaptic terminal. Naloxone increased the frequency, but not the amplitude, of tetrodotoxin-insensitive (or miniature) GABAA-IPSCs, supporting the latter hypothesis (Fig. 3C). The increase in miniature GABAA-IPSC frequency was larger in slices from morphine-treated rats (morphine-treated, 270 ± 40%; control, 150 ± 10%, z = -2.7, Mann-Whitney; p = 0.006; n = 9 and 15). Thus, acute morphine withdrawal increased GABAA inhibition of vlBNST neurons projecting to the VTA.
Noradrenaline activated GABAA-IPSCs in the vlBNST
In slices from control rats that were incubated in morphine (1 μm), noradrenaline (10 or 100 μm) produced the same increase in frequency of spontaneous GABAA-IPSCs before and after treatment with naloxone (1 μm) (in morphine, +10.9 ± 3.1 Hz; t = -3.3, paired Student's t test; p = 0.009; n = 10) (Fig. 4A,B) (in morphine and naloxone, +10.8 ± 3.1 Hz; t = -3.2, paired Student's t test; p = 0.01; n = 10) (Fig. 4A,C). Prazosin blocked the noradrenaline-induced increase in GABAA-IPSCs, indicating the contribution of α1-adrenergic receptors (Fig. 4C). Tetrodotoxin inhibited the increased frequency of IPSCs induced by noradrenaline, indicating that the action was dependent on the generation of action potentials (Fig. 4A). Noradrenaline did not change the mean amplitude of the spontaneous IPSCs (sIPSCs) in morphine (1 μm; before, 80 ± 12; in noradrenaline, 88 ± 13 pA; t = -0.5, paired Student's t test; p = 0.6; n = 10) or in naloxone (1 μm; before, 80 ± 14; in noradrenaline, 109 ± 19 pA; t = -1.5, paired Student's t test; p = 0.2; n = 10).
Figure 4.

Noradrenaline increased spontaneous GABAA-IPSCs in the vlBNST. A, Representative recording of spontaneous GABAA-IPSCs in a slice from a control rat. Inset, Noradrenaline-driven spontaneous GABAA-IPSCs at a larger time scale. B, Representative recordings of spontaneous GABAA-IPSCs in a slice from a morphine-treated rat after naloxone-precipitated withdrawal. C, Summary of the effect of naloxone-precipitated morphine withdrawal and of α1-, β-adrenergic receptor blockade on the noradrenaline-induced increase of GABAA-IPSCs. D, Effects of adenylyl cyclase blockade (H89, 10 μm) on the noradrenaline-induced increase of spontaneous GABAA-IPSCs in slices from control or morphine-treated slices spontaneously withdrawn (at least 60 min) from morphine. * p < 0.05, before versus after noradrenaline; ap < 0.05, before versus after naloxone 1 μm. Error bars indicate SEM.
Noradrenaline also increased the frequency of spontaneous GABAA-IPSCs in the presence of morphine after chronic treatment with morphine (+5.6 ± 1.9 Hz; t = -2.7, paired Student's t test; p = 0.02; before vs after noradrenaline; n = 10) (Fig. 4C). Similarly, noradrenaline increased the frequency of GABAA-IPSCs during naloxone-precipitated withdrawal (+8.6 ± 1.1 Hz; t = -4.9, paired Student's t test; p < 0.001; before vs after noradrenaline; n = 10) (Fig. 4B,C) (t = -1.5, unpaired Student's t test; noradrenaline before vs after naloxone; p = 0.15) (Fig. 4C). Propranolol (Fig. 4B,C) and the PKA inhibitor H89 (Fig. 4D) reduced the noradrenaline increase in GABAA-IPSCs during acute morphine withdrawal. These drugs had no effect in control slices, demonstrating the recruitment of β-adrenergic receptors and of the AC-PKA pathway during acute opioid withdrawal. Noradrenaline did not increase the mean amplitude of the sIPSCs in the presence of morphine (1 μm;66 ± 8 and 67 ± 9 pA; before vs after noradrenaline; t = 0.2, paired Student's t test; p = 0.9; n = 10). However, the amplitude of the sIPSCs was increased by noradrenaline in naloxone (1 μm;65 ± 17 and 91 ± 12 pA; before vs after noradrenaline; t = -2.5, paired Student's t test; p = 0.02; n = 10). Clonidine (α2-agonist; 3 μm; -12 ± 12%) and yohimbine (α2-antagonist; 0.1 μm; 0 ± 20%) had no significant effect, suggesting the lack of presynaptic α2-adrenergic receptors on GABA terminals. To summarize, noradrenaline activated GABAA-IPSCs in the vlBNST in control and during acute morphine withdrawal. During opioid withdrawal, the net effect of noradrenaline was the same but the mechanism changed.
Discussion
Normally, the noradrenaline concentration is low in the vlBNST of conscious rats (Pacak et al., 1995; Cecchi et al., 2002). Noradrenaline increases in the vlBNST during physiological (stress) or pathological (opioid withdrawal) conditions (Pacak et al., 1995; Fuentealba et al., 2000; Cecchi et al., 2002). Bath applying noradrenaline on brain slices from control rats likely mimics the rise in noradrenaline concentration in the vlBNST during stress. Likewise, noradrenaline applied to a morphine-withdrawn slices may reproduce the effect of noradrenaline in the vlBNST in withdrawal. Noradrenaline increased GABAA inhibition in control and acute morphine withdrawal conditions by activating α1-adrenergic receptors. During acute morphine withdrawal, however, GABA release increased through a hyperactive AC-PKA pathway revealing a contribution of G-coupled β-adrenergic receptors. This mechanism might explain the aversive effect of noradrenaline in the vlBNST during opioid withdrawal because it affected neurons of the vlBNST projecting to the VTA, and that blockade of β-adrenergic receptors in the vlBNST is antiaversive.
Noradrenaline in the vlBNST; a role in stress and withdrawal
Our observations indirectly suggest that noradrenaline depolarized local GABA neurons to trigger an increase in GABAA-IPSCs. The fact that tetrodotoxin blocked the noradrenaline-induced increase in spontaneous IPSCs suggests the activation of action potentials in cell bodies in the slices and perhaps in the vlBNST itself. Furthermore, a second population of neurons in the vlBNST (that were never labeled with the tracer) responded to noradrenaline in voltage clamp with inward currents (Table 1). These cells could be local GABA interneurons with α1-, β-, or both adrenergic receptors (depending on the conditions) and could be responsible for the increase in GABAA-IPSCs.
The increase in GABA-IPSCs induced by noradrenaline might be relevant in stress behaviors. Indeed, immobilization stress releases noradrenaline in the vlBNST, which, through α1-adrenergic receptors, increases corticosterone levels (Cecchi et al., 2002). The BNST projects to the paraventricular nucleus of the hypothalamus (PVH) (Herman et al., 1994), and there is a possibility that our recordings included these neurons. In the PVH, the activity of CRF neurons increases corticosterone blood concentrations during stress. Consequently, the noradrenaline-induced GABA inhibition that we observed in control rats might influence the PVH and, consequently, corticosterone levels. Our results showed that noradrenaline triggers GABAA inhibition of vlBNST neurons sharing physiological properties with those innervating the VTA. This supports the idea that the BNST is a key brain structure that links stress and drug abuse (Erb and Stewart, 1999; Erb et al., 2000, 2001; Shalev et al., 2001, 2003; Wang et al., 2001; Leri et al., 2002).
Acute opioid withdrawal is a condition in which understanding the effect of noradrenaline in the vlBNST might have important therapeutic implications. Noradrenaline and noradrenergic nuclei of the brain contribute to the symptoms of withdrawal from drugs of abuse (Maldonado, 1997). Noradrenergic nuclei are hyperactive during withdrawal and release noradrenaline in their efferent targets (Aghajanian, 1978; Akaoka and Aston-Jones, 1991; Baraban et al., 1995; Delfs et al., 2000). As a result, noradrenaline release occurs in many areas of the brain, including the vlBNST (Pellegrini-Giampietro et al., 1988; Done et al., 1992; Rossetti et al., 1993; Kosten, 1994; Fuentealba et al., 2000). The noradrenaline released in the vBNST originates from brainstem A1-A2 noradrenergic regions and underlies conditioned place aversion of opioid withdrawal (Delfs et al., 2000). The present study extends these findings to the cellular level by showing that noradrenaline triggers GABA inhibition in the vlBNST during acute opioid withdrawal. The inhibition produced by noradrenaline during withdrawal was similar to control conditions. However, a significant effect of β-adrenergic receptors was found only after chronic morphine treatment. Noradrenaline also caused a small increase in the amplitude of spontaneous IPSCs only during withdrawal. The mechanism underlying this small increase could involve an increase in excitability of interneurons, an increase in probability of GABA release, or even an increase in postsynaptic sensitivity. Regardless of the mechanism, the increase in GABA inhibition mediated by noradrenaline could be the target for β-antagonists in the vBNST during withdrawal.
Chronic morphine, withdrawal, and the probability of GABA release
The increase of the AC-PKA pathway induced by chronic morphine occurs in many brain sites (Bonci and Williams, 1997; Jolas and Aghajanian, 1997; Chieng and Williams, 1998; Ingram et al., 1998). In the vlBNST, this enhanced pathway rebounded during withdrawal and facilitated GABA release because PKA inhibition reduced the augmented GABAA-IPSC frequency. The upregulation of adenylyl cyclase may also account for the increased response to β-receptor stimulation.
Finally, the acute withdrawal at GABA synapses in the vlBNST showed specificity. In preliminary experiments, acute withdrawal from chronic morphine did not change the frequency of spontaneous AMPA-EPSCs (control, 1 ± 0.1 Hz vs withdrawal, 0.7 ± 0.1 Hz; preliminary observations). Also, in the second neuron population (presumably interneurons), naloxone increased the amplitude of the evoked GABAA-IPSCs to the same extend in slices from control and morphine-treated rats (+54 ± 4 and + 54 ± 2%, respectively; n = 4 per group). Thus, GABA synapses on projection neurons were selectively affected by the chronic morphine treatment.
α1- and β-adrenergic receptors in withdrawal
In the present study, both α1- and β-adrenergic receptor antagonists blocked the noradrenaline-induced increase in IPSCs in the vBNST during withdrawal. Studies in animals and human drug users showed that β-antagonists reduce symptoms of morphine and cocaine withdrawal (Grosz, 1972; Harris and Aston-Jones, 1993a,b; Delfs et al., 2000). The few studies on the effects of α1-antagonists in withdrawal showed that phentolamine, phenoxybenzamine, and prazosin diminish some of the somatic symptoms of withdrawal in animal studies (Cicero et al., 1974; van der Laan, 1985). Our observations suggest that the combination of α1- and β-blockade could be a therapeutic possibility to control the symptoms of acute opioid withdrawal. Clonidine, an α2-agonist, reduces the symptoms of withdrawal in humans (Gold et al., 1978), and in rats completely abolishes aversion when microinjected in the vBNST (Delfs et al., 2000). In the present study, clonidine was ineffective. Because clonidine inhibits the release of noradrenaline through activation of presynaptic α2-autoreceptor, it was ineffective in the brain slice preparation because there is little or no release of endogenous noradrenaline.
Significance of the noradrenergic inhibition of vlBNST projection neurons
Does the noradrenaline-induced increase of GABAA-IPSCs contribute to aversion during opioid withdrawal? GABA inhibits all types of neurons in the vBNST (Egli and Winder, 2003) that would reduce the efferent drive from the vBNST to projection areas, including excitation to dopamine neurons in the VTA. A decrease in the excitatory drive to dopamine neurons would contribute to the withdrawal-induced inhibition of these neurons and potentially play a role in the aversion associated with opioid withdrawal (Diana et al., 1995). Indeed, spontaneous and naloxone-precipitated withdrawal inhibits activity of VTA dopamine neurons (Diana et al., 1995; Georges et al., 2003) and hence, dopamine release in the nucleus accumbens (Acquas et al., 1991, 1992; Pothos et al., 1991; Rossetti et al., 1992; Kalivas, 1993).
The BNST plays a prominent role in processes ranging from stress and anxiety (Rainnie, 1999; Cecchi et al., 2002) to withdrawal (Delfs et al., 2000) and stress-induced relapse to psychostimulants and opioids (Erb et al., 2000, 2001). The present study identified a cellular action of noradrenaline in the vBNST during opioid withdrawal, which, through a projection to the VTA, suggests its contribution in the reward pathways.
Footnotes
This project was supported by the National Institutes of Health Grant DA08163. E.C.D. was a fellow of the Canadian Institutes of Health Research (MFE-49878). We thank Drs. Gregory P. Mark and Carlos A. Paladini for help with the microinjection procedure and Geneviève Paré for graphic assistance.
Correspondence should be addressed to J. T. Williams, Vollum Institute, Oregon Health Sciences University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: williamj@OHSU.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248198-07$15.00/0
References
- Acquas E, Carboni E, Di Chiara G (1991) Profound depression of mesolimbic dopamine release after morphine withdrawal in dependent rats. Eur J Pharmacol 193: 133-134. [DOI] [PubMed] [Google Scholar]
- Acquas E, Carboni E, de Ree RH, Da Prada M, Di Chiara G (1992) Extracellular concentrations of dopamine and metabolites in the rat caudate after oral administration of a novel catechol-O-methyltransferase inhibitor Ro 40-7592. J Neurochem 59: 326-330. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK (1978) Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature 276: 186-188. [DOI] [PubMed] [Google Scholar]
- Akaoka H, Aston-Jones G (1991) Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci 11: 3830-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alheid GF, de Olmos JS, Baltramino CA (1995) Amygdala and extended amygdala. In: The rat nervous system, Ed 2 (Paxinos G, ed), pp 495-578. San Diego: Academic.
- Baraban SC, Stornetta RL, Guyenet PG (1995) Effects of morphine and morphine withdrawal on adrenergic neurons of the rat rostral ventrolateral medulla. Brain Res 676: 245-257. [DOI] [PubMed] [Google Scholar]
- Bechara A, Nader K, van der Kooy D (1998) A two-separate-motivational-systems hypothesis of opioid addiction. Pharmacol Biochem Behav 59: 1-17. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT (1997) Increased probability of GABA release during withdrawal from morphine. J Neurosci 17: 796-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casada JH, Dafny N (1991) Restraint and stimulation of bed nucleus of the stria terminalis produce similar stress-like behaviors. Brain Res Bull 27: 207-212. [DOI] [PubMed] [Google Scholar]
- Cecchi M, Khoshbouei H, Javors M, Morilak DA (2002) Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience 112: 13-21. [DOI] [PubMed] [Google Scholar]
- Chieng B, Williams JT (1998) Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci 18: 7033-7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, North RA (1987) Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol 32: 633-638. [PubMed] [Google Scholar]
- Cicero TJ, Meyer ER, Smithloff BR (1974) Alpha adrenergic blocking agents: anti-nociceptive activity and enhancement of morphine-induced analgesia. J Pharmacol Exp Ther 189: 72-82. [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000) Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403: 430-434. [DOI] [PubMed] [Google Scholar]
- Diana M, Pistis M, Muntoni A, Gessa G (1995) Profound decrease of mesolimbic dopaminergic neuronal activity in morphine withdrawn rats. J Pharmacol Exp Ther 272: 781-785. [PubMed] [Google Scholar]
- Done C, Silverstone P, Sharp T (1992) Effect of naloxone-precipitated morphine withdrawal on noradrenaline release in rat hippocampus in vivo. Eur J Pharmacol 215: 333-336. [DOI] [PubMed] [Google Scholar]
- Dong H, Petrovich GD, Swanson LW (2000) Organization of projections from the juxtacapsular nucleus of the BST: a PHAL study in the rat. Brain Res 859: 1-14. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW (2004) Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol 468: 277-298. [DOI] [PubMed] [Google Scholar]
- Egli RE, Winder DG (2003) Dorsal and ventral distribution of excitable and synaptic properties of neurons of the bed nucleus of the stria terminalis. J Neurophysiol 90: 405-414. [DOI] [PubMed] [Google Scholar]
- Erb S, Stewart J (1999) A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci 19: RC35(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J (2000) Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology 23: 138-150. [DOI] [PubMed] [Google Scholar]
- Erb S, Salmaso N, Rodaros D, Stewart J (2001) A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 158: 360-365. [DOI] [PubMed] [Google Scholar]
- Fuentealba JA, Forray MI, Gysling K (2000) Chronic morphine treatment and withdrawal increase extracellular levels of norepinephrine in the rat bed nucleus of the stria terminalis. J Neurochem 75: 741-748. [DOI] [PubMed] [Google Scholar]
- Georges F, Aston-Jones G (2002) Activation of ventral tegmental area cells by the bed nucleus of the stria terminalis: a novel excitatory amino acid input to midbrain dopamine neurons. J Neurosci 22: 5173-5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges F, Rajkowski J, Aston-Jones G (2003) Responses of midbrain dopamine neurons to morphine, morphine dependence, and withdrawal. Soc Neurosci Abstr 29: 110.2. [Google Scholar]
- Gold MS, Redmond Jr DE, Kleber HD (1978) Clonidine blocks acute opiate-withdrawal symptoms. Lancet 2: 599-602. [DOI] [PubMed] [Google Scholar]
- Grosz HJ (1972) Narcotic withdrawal symptoms in heroin users treated with propranolol. Lancet 2: 564-566. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G (1993a) Beta-adrenergic antagonists attenuate somatic and aversive signs of opiate withdrawal. Neuropsychopharmacology 9: 303-311. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G (1993b) Beta-adrenergic antagonists attenuate withdrawal anxiety in cocaine- and morphine-dependent rats. Psychopharmacology (Berl) 113: 131-136. [DOI] [PubMed] [Google Scholar]
- Henke PG (1984) The bed nucleus of the stria terminalis and immobilization-stress: unit activity, escape behaviour, and gastric pathology in rats. Behav Brain Res 11: 35-45. [DOI] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE, Watson SJ (1994) Involvement of the bed nucleus of the stria terminalis in tonic regulation of paraventricular hypothalamic CRH and AVP mRNA expression. J Neuroendocrinol 6: 433-442. [DOI] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18: 10269-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolas T, Aghajanian GK (1997) Opioids suppress spontaneous and NMDA-induced inhibitory postsynaptic currents in the dorsal raphe nucleus of the rat in vitro. Brain Res 755: 229-245. [DOI] [PubMed] [Google Scholar]
- Jolas T, Nestler EJ, Aghajanian GK (2000) Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience 95: 433-443. [DOI] [PubMed] [Google Scholar]
- Ju G, Swanson LW (1989) Studies on the cellular architecture of the bed nuclei of the stria terminalis in the rat: I. Cytoarchitecture. J Comp Neurol 280: 587-602. [DOI] [PubMed] [Google Scholar]
- Kalivas PW (1993) Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Brain Res Rev 18: 75-113. [DOI] [PubMed] [Google Scholar]
- Kosten TA (1994) Clonidine attenuates conditioned aversion produced by naloxone-precipitated opiate withdrawal. Eur J Pharmacol 254: 59-63. [DOI] [PubMed] [Google Scholar]
- Leri F, Flores J, Rodaros D, Stewart J (2002) Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci 22: 5713-5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R (1997) Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev 21: 91-104. [DOI] [PubMed] [Google Scholar]
- Pacak K, McCarty R, Palkovits M, Kopin IJ, Goldstein DS (1995) Effects of immobilization on in vivo release of norepinephrine in the bed nucleus of the stria terminalis in conscious rats. Brain Res 688: 242-246. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Pellegrini-Giampietro DE, Bacciottini L, Carla V, Moroni F (1988) Morphine withdrawal in cortical slices: suppression by Ca2+-channel inhibitors of abstinence-induced [3H]-noradrenaline release. Br J Pharmacol 93: 535-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothos E, Rada P, Mark GP, Hoebel BG (1991) Dopamine microdialysis in the nucleus accumbens during acute and chronic morphine, naloxone-precipitated withdrawal and clonidine treatment. Brain Res 566: 348-350. [DOI] [PubMed] [Google Scholar]
- Rainnie DG (1999) Neurons of the bed nucleus of the stria terminalis (BNST). Electrophysiological properties and their response to serotonin. Ann NY Acad Sci 877: 695-699. [DOI] [PubMed] [Google Scholar]
- Roehrich H, Gold MS (1987) Propranolol as adjunct to clonidine in opiate detoxification. Am J Psychiatry 144: 1099-1100. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Hmaidan Y, Gessa GL (1992) Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol 221: 227-234. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Longu G, Mercuro G, Gessa GL (1993) Extraneuronal noradrenaline in the prefrontal cortex of morphine-dependent rats: tolerance and withdrawal mechanisms. Brain Res 609: 316-320. [DOI] [PubMed] [Google Scholar]
- Schuckit MA (2000) Drug and alcohol abuse. A clinical guide to diagnosis and treatment., Ed 5. New York: Kluwer Academic/Plenum.
- Shalev U, Morales M, Hope B, Yap J, Shaham Y (2001) Time-dependent changes in extinction behavior and stress-induced reinstatement of drug seeking following withdrawal from heroin in rats. Psychopharmacology (Berl) 156: 98-107. [DOI] [PubMed] [Google Scholar]
- Shalev U, Robarts P, Shaham Y, Morales M (2003) Selective induction of c-Fos immunoreactivity in the prelimbic cortex during reinstatement of heroin seeking induced by acute food deprivation in rats. Behav Brain Res 145: 79-88. [DOI] [PubMed] [Google Scholar]
- van der Laan JW (1985) Effects of alpha 2-agonists on morphine withdrawal behaviour: potentiation of jumping mediated by alpha 2-receptors. Naunyn Schmiedebergs Arch Pharmacol 329: 293-298. [DOI] [PubMed] [Google Scholar]
- Walker JR, Ahmed SH, Gracy KN, Koob GF (2000) Microinjections of an opiate receptor antagonist into the bed nucleus of the stria terminalis suppress heroin self-administration in dependent rats. Brain Res 854: 85-92. [DOI] [PubMed] [Google Scholar]
- Wang X, Cen X, Lu L (2001) Noradrenaline in the bed nucleus of the stria terminalis is critical for stress-induced reactivation of morphine-conditioned place preference in rats. Eur J Pharmacol 432: 153-161. [DOI] [PubMed] [Google Scholar]