

Abstract
Background
Migraine attacks occur spontaneously in those who suffer from the condition, but migraine-like attacks can also be induced artificially by a number of substances. Previously published evidence makes the meninges a likely source of migraine related pain. This article investigates the effect of several vasodilators on meningeal arteries in order to find a connection between the effect of a substance on a meningeal vessel and its ability to artificially induce migraine.
Methods
A myograph setup was used to test the vasodilator properties of the substances acetylcholine (ACh), sodium nitroprusside (SNP), sildenafil, prostaglandin E2 (PGE2), pituitary adenylate cyclase activating peptide-38 (PACAP-38), calcitonin gene-related peptide (CGRP) and NaCl buffer on meningeal arteries from human and rat. An unpaired t-test was used to statistically compare the mean Emax(%) at the highest concentration of each substance to the Emax(%) of NaCl buffer.
Results
In the human experiments, all substances except PACAP-38 had an Emax (%) higher than the NaCl buffer, but the difference was only significant for SNP and CGRP. For the human samples, clinically tested antimigraine compounds (sumatriptan, telcagepant) were applied to the isolated arteries, and both induced a significant decrease of the effect of exogenously administrated CGRP. In experiments on rat middle meningeal arteries, pre-contracted with PGF2α, similar tendencies were seen. When the pre-contraction was switched to K+ in a separate series of experiments, CGRP and sildenafil significantly relaxed the arteries.
Conclusions
Still no definite answer can be given as to why pain is experienced during an attack of migraine. No clear correlation was found between the efficacy of a substance as a meningeal artery vasodilator in human and the ability to artificially induce migraine or the mechanism of action. Vasodilatation could be an essential trigger, but only in conjunction with other unknown factors. The vasculature of the meninges likely contributes to the propagation of the migrainal cascade of symptoms, but more research is needed before any conclusions can be drawn about the nature of this contribution.
Keywords: Vasodilation, Middle meningeal artery, Migraine, CGRP
Background
The dura mater and its vasculature, the middle meningeal artery (MMA) and the venous system have for decades been central to many hypotheses aimed to explain migraine pathophysiology [1,2]. Ray and Wolff showed in the 1940s that direct stimulation of the dura mater may result in headache with migraine-like qualities [3]. Further research during the last two decades has demonstrated that sensitization and local inflammation in the dura mater can be elicited by various agents and stimulation paradigms [4]. The local administration of an “inflammatory soup” or the direct stimulation at different parts of the dura mater may result not only in vasoactive responses but sometimes also in mast cell degranulation and in plasma extravasation. Histamine, a major amine released from mast cells, is known to induce a migraine-like pain upon infusion into test subjects previously diagnosed with migraine [5]. Some migraine-associated symptoms, including the characteristic pain, could therefore stem from the activation of meningeal mast cells and aseptic inflammation initiated by histamine release [6]. However, local administration of calcitonin gene-related peptide (CGRP), which is a sensory neuropeptide in the dura mater and a known migrainogenic substance [7], results only in vasodilatation [8]. Despite the fact that mast cells have CGRP receptors [9,10], no activation or sensitization of meningeal nociceptors could be detected upon topical or systemic administration of this compound in rats [8].
Apart from CGRP and histamine, other substances that upon intravenous infusion can provoke migraine-like attacks in humans previously diagnosed with migraine are prostaglandin E2 (PGE2), pituitary adenylate cyclase activating peptide-38 (PACAP-38), epoprostenol (prostacyclin, PGI2), sildenafil and nitric oxide (NO) [11-15]. The only common factor among these substances is their vasodilator properties, but still there exist several other vasodilators which do not induce migraine-associated symptoms upon infusion. Vasoactive intestinal polypeptide (VIP) and carbachol, both associated with the parasympathetic system, are noted examples, although their ability to dilate meningeal arteries in vivo remains to be verified [16,17]. This shows that factors apart from the vasodilatation have to be taken into consideration. There is still controversy regarding whether vasodilatation directly contributes to any of the migraine-related symptoms or whether this is just a side-phenomenon. Several hypotheses exist that downplay the role of vasodilatation, including the observation of prodromal aura symptoms preceding any activity in the vasculature [18], that the vasoconstrictor triptan drugs are not an effective treatment in all migraine patients and that the extravascular tissue surrounding the dural arteries could be too rigid to allow for any significant expansion of the vessel diameter [19].
We have recently investigated the vasoactive effects of several substances on cerebral arteries from human and rat in vitro in order to find common denominators that would allow us to understand more about the possible factors behind the migraine-associated symptoms [20]. However, cerebral arteries are different from meningeal arteries regarding several morphological aspects, including receptor expression, anatomical origin (internal carotid artery (ICA) versus external carotid artery (ECA)) and the lack of a blood–brain barrier (BBB) in the meningeal arteries [21]. Nevertheless, both vascular regions receive sensory input from the first division of the trigeminal ganglion [22]. Therefore it is possible that the two artery types contribute to migraine differently if there indeed is a vascular component.
The aim of the present study is to evaluate whether there is a connection between the ability of a substance to dilate meningeal arteries and the previously reported migrainogenic properties of each substance. It is useful to verify whether vasodilatation of the meningeal arteries indeed is the trigger for the migraine-like pain triggered by these substances. Since the meningeal arteries lack BBB properties, systemic drugs are freely diffusible to endothelial and smooth muscle receptors to elicit a vasomotor response. A secondary goal of this study is to compare the functional aspect of the middle meningeal artery (MMA) between the species because rat is often used in mechanistic studies due to the difficulty in obtaining human tissue samples.
Methods
The experimental protocol (M11104) was approved by the Animal Protocol Review committee at the University of Lund. All human artery procedures were carried out strictly according to national laws and guidelines and approved by the Ethical Committee at the University of Lund (LU-818-01) and the local Ethics Committee at the Erasmus MC, Rotterdam.
Obtaining the MMA
A. Fresh samples of MMA were obtained from the meninges of male Sprague–Dawley rats by the following procedure. The rats were anesthetized using CO2 and decapitated. The cranium was opened from the top and the brain removed, which exposed the dura mater. The cranium and dura mater were divided sagitally and each half was placed in a buffer solution composed of NaCl 119 mM, NaHCO3 15 mM, KCl 4.6 mM, MgCl2 1.2 mM, NaH2PO4 1.2 mM, CaCl2 1.5 mM and glucose 5.5 mM. The dura mater was carefully separated from the cranium and moved to a Petri dish, where it was submerged in buffer solution, stretched and suspended by needles. The MMA was removed from the dura mater by a dissecting microscope, cut into segments 1–2 mm long that were placed in parallel tissue baths of ice-cold bicarbonate buffer solution aerated with a gas composed of 95% O2 and 5% CO2, with a resulting pH of 7.4 [23].
B. Human samples of intracranial MMA branches were acquired from patients undergoing neurosurgery at the University Hospital of Lund and the Erasmus University Medical Center (Rotterdam, the Netherlands). The vessel segments themselves were from visually healthy tissue.
The artery samples were upon removal immediately placed in cold Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Invitrogen, Carlsbad, CA, USA) in Lund and in cold medium M199 (Gibco, Invitrogen, Carlsbad, CA, USA) in Rotterdam and immediately transported to the laboratory where they were prepared in an identical way to the rat MMA.
Myography
Each segment of MMA was mounted on a pair of thin metal wires (human: 40 μm, rat: 20/25 μm) in an arterial myograph. One wire was connected to a micrometer screw, allowing for fine adjustment of the vascular tone by varying the distance between the wires. The other wire was connected to a force displacement transducer, paired with an analogue-digital converter (ADInstruments, Oxford, UK). Data was recorded on a computer using a PowerLab unit (ADInstruments).
The aerated bicarbonate buffer was heated to +37°C and used to submerge the wires with the MMA-segment during the experiment. The segment was normalized, attaining 90% of the internal circumference that a fully relaxed vessel would have under a transmural pressure of 100 mmHg or 50 mmHg (see below). A reference value for the contractile capacity of the segment was determined by temporarily replacing part of the NaCl in the buffer solution with 60 mM K+.
The effects on the segment of the different vasoactive substances was tested by first recording the spontaneous resting diameter of the artery segment and define it as the maximum dilatation possible. Next, the segment was pre-contracted with 10-6 M prostaglandin F2α (PGF2α) to achieve a stable tension for the duration of the experiment. Finally, concentrations of the tested substances in the range of 10-10-10-5 M (the exact range varied between substances) were applied cumulatively and the vessel response was recorded. Because of the unstable response to PGF2α in the first rat experiments, we changed the protocol and used 30 mM K+ as a more stable contraction. In addition, the transmural pressure was lowered to 90% of 50 mmHg in these experiments.
Artery segments were omitted from the study if they failed to fulfill the inclusion criteria. Segments included in the final calculations were required to respond to initial testing with K+ and to the pre-contraction with a maximum contractile capacity of at least 1 mN for human arteries and 0.1 mN for rat arteries.
Studies with clinically tested anti migraine compounds
The vessel segments were precontracted with 30 mM K+. After precontraction the vessel segments were dilated with 4 nM CGRP. When a stable dilatation was achieved, sumatriptan or telcagepant was added to the arteries in concentrations corresponding to the Cmax (160 nM and 5 μM, for the 100 mg and 300 mg oral doses, respectively) [24-26], as well as in concentrations corrected for plasma protein binding. The Cmax for sumatriptan was corrected for 17.5% plasma protein binding [27], while the Cmax for telcagepant was calculated using a plasma protein binding of 96.4% [28].
Calculations
The maximum dilatory response (Emax(%)) for each tested substance was obtained for the vessel segments as the percentage tonus relaxed by the maximum concentration of substance from the PGF2α- or K+-induced pre-contraction towards the resting tonus sampled before the lowest concentration of substance was added. In the case of significant relaxation the negative logarithm of the concentration of substance required for dilatation of the vessel segments to half of their Emax(%). The pEC50 was obtained for the purpose of comparing the potency of the different substances, calculated using GraphPad Prism software (GraphPad Software, San Diego, California, USA).
If a test subject contributed with more than one vessel segment to the testing of a substance, all results obtained from that individual regarding that substance were pooled together into an average before further processing of data. The hypothesis was that each substance would induce vasodilatation and to determine the significance of the dilatation an unpaired t test was performed using the GraphPad Prism software (GraphPad Software, San Diego, California, USA.) comparing the average Emax(%) of the highest concentration of each substance to the average Emax(%) of NaCl buffer at the end of the session.
Figures were compiled where it is shown how the PGF2α/K+ precontraction (100%) during a session is restored towards the original resting baseline at different concentrations of each of the substances tested and NaCl buffer (Figures 1, 2, 3, 4, and 5). The contractile responses to telcagepant and sumatriptan were expressed as percentage of the previous relaxant response to 4 nM CGRP in the same segment (Figure 2). All data are presented as mean ± S.E.M. Statistical analysis and pEC50 calculations were performed using Graphpad Prism 5 software. Statistical significance was accepted at P < 0.05.
Figure 1.
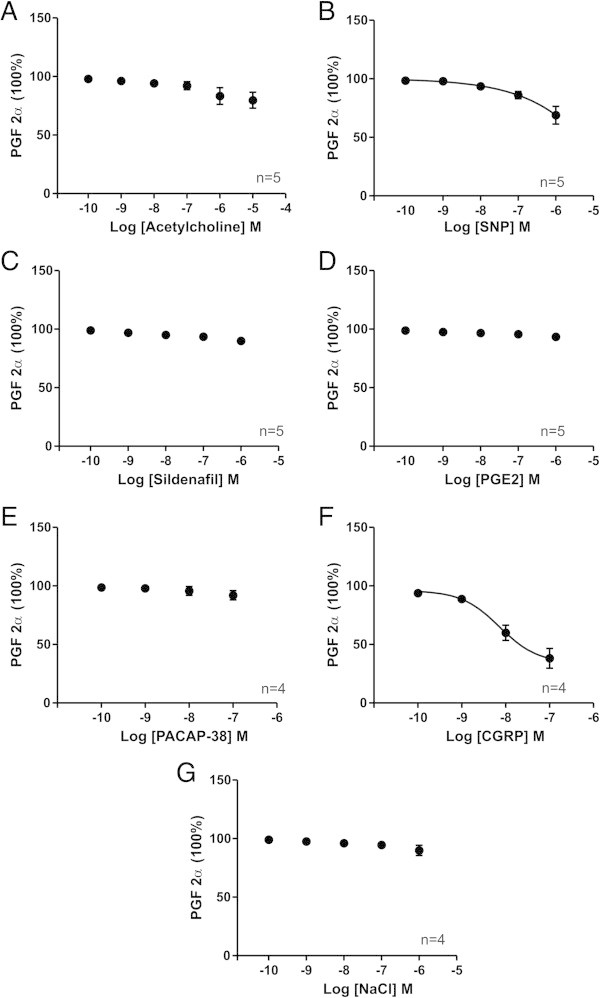
The effect of substances on human middle meningeal arteries precontracted with PGF2α. The relaxation relative to PGF2α induced tone in human meningeal arteries by increasing concentrations of each of the tested substances (A-F) and NaCl buffer (G). Values are given as mean ± SEM (n = 4 – 6), where PGF2α precontraction is set to be 100%.
Figure 2.
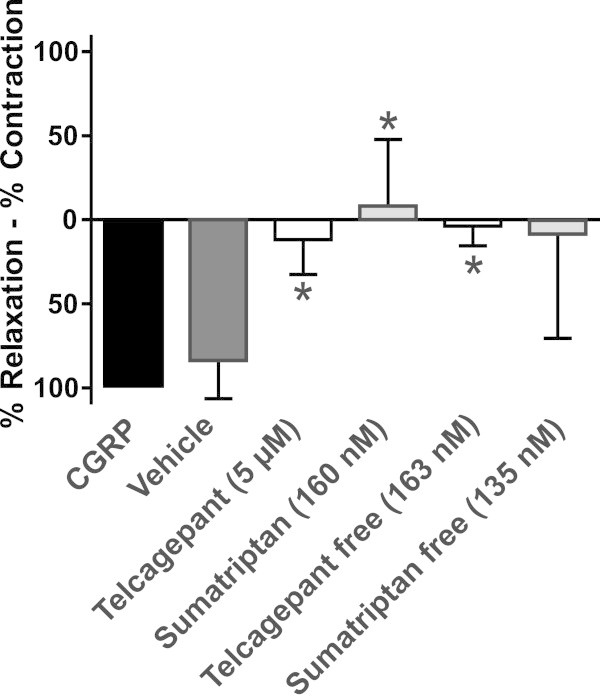
Effect of telcagepant and sumatriptan on CGRP induced relaxation of the middle meningeal artery. Arteries were precontracted with 30 mM K+, dilated with 4 nM CGRP and sumatriptan or telcagepant was subsequently added (* = p < 0.05). Values are given as % of the CGRP relaxation (mean ± SEM, n = 5).
Figure 3.
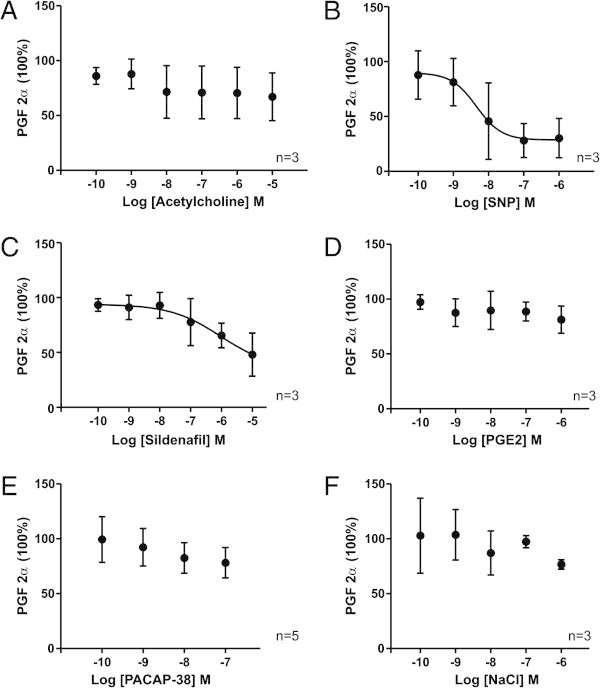
The effect of substances on rat middle meningeal arteries precontracted with PGF2α. The relaxation induced in rat meningeal arteries pre-contracted with PGF2α at increasing concentrations of each of the tested substances (A-E) and NaCl buffer (F). Values are given as mean ± SEM (n = 3 – 5), where PGF2α precontraction is set to be 100%.
Figure 4.
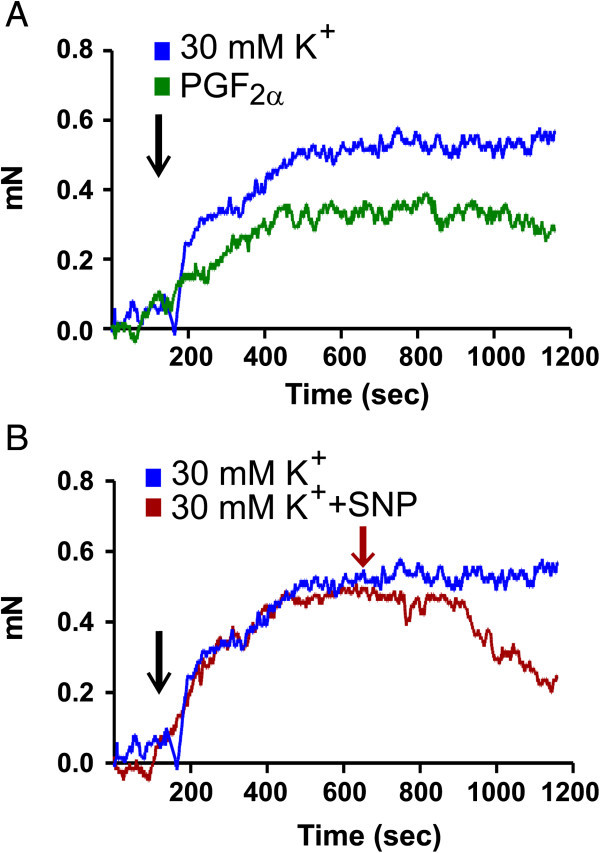
Comparison of 30 mM K+ and PGF2α on rat middle meningeal arteries, and sample relaxation. A) Sample contraction induced by of 30 mM K+ or PGF2α in rat meningeal artery. One can observe the slight positive drift for 30 mM K+ and slight the negative drift for PGF2α. B) Sample relaxation in an artery precontracted with 30 mM K+ and the cumulative relaxation of added sodium nitroprusside (SNP). Small plateaus were allowed before next concentration was added.
Figure 5.
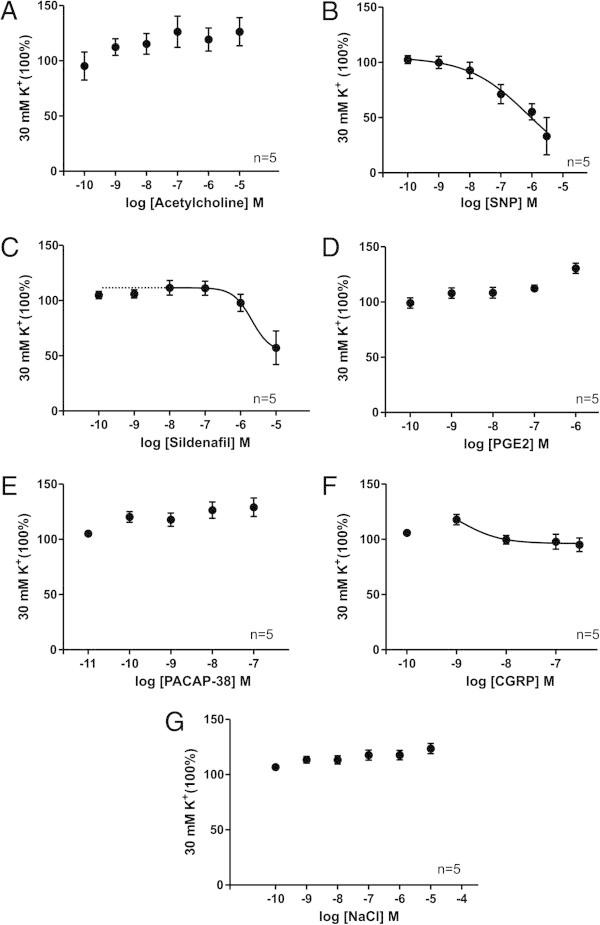
The effect of substances on rat middle meningeal arteries precontracted with K+. The relaxation induced in rat meningeal arteries pre-contracted with K+ at increasing concentrations of each of the tested substances (A-F) and NaCl buffer (G). Values are given as mean ± SEM (n = 5), where 30 mM K+ precontraction is set to be 100%).
Chemicals
The vasoactive substances used in the present study were: acetylcholine chloride (ACh), sodium nitroprusside (SNP), sildenafil, PACAP-38 (all purchased from Sigma Aldrich), PGE2 (Larodan fine Chemicals AB). The inhibitors used were the following: Telcagepant (MK-0974) (dissolved in DMSO and further diluted in H2O) (MSD, Whitehouse Station, NJ, USA), sumatriptan (dissolved in H2O) (Sigma Aldrich).
Results
Functional results from the MMA
Intracranial MMA segments from 6 humans were used to test the vasoactive properties of six different compounds including NaCl buffer but excluding CGRP. The vessels were divided into several segments of 1–2 mm in length and studied in parallel tissue baths. Not every segment was exposed to all compounds. Vessels from 4 additional human patients were used for the testing of CGRP.
Human MMA (100 mmHg, pre-contracted with PGF2α)
Of the compounds tested, SNP and CGRP on human artery showed significant dilatation compared to NaCl, with CGRP being the strongest vasodilator (Figure 1, Table 1). ACh showed a non-significant tendency of relaxation. PGE2, PACAP-38 and sildenafil showed no vasomotor activity distinct from the gradual loss of pre-contraction seen when using only NaCl buffer (Figure 1, Table 1). The very high concentration of sildenafil (10 μM) was not tested on human vessels.
Table 1.
Human MMA, 100 mmHg, pre-contracted with PGF 2α
| Compound | E max (%) | pEC 50 (M) | p - value |
|---|---|---|---|
| ACh (n = 6) |
17 ± 6 |
N.A. |
0.44 |
| SNP (n = 6) |
32 ± 6 |
~3.5 |
0.034 |
| Sildenafil (n = 6) |
11 ± 2 |
N.A. |
0.91 |
| PGE2 (n = 6) |
8 ± 1 |
N.A. |
0.62 |
| PACAP-38 (n = 5) |
9 ± 3 |
N.A. |
0.89 |
| CGRP (n = 4) |
62 ± 8 |
8.1 ± 0.19 |
0.0015 |
| NaCl (n = 4) | 10 ± 4 | N.A. |
p-value for comparing agonist versus the NaCl effect. N.A. = Not Applicable. Values are ± SEM, n given in parenthesis.
Human MMA and CGRP (100 mmHg, pre-contracted with 30 mM K+)
For human samples the clinically tested anti migraine compounds were applied to the isolated arteries that that had been precontracted with 30 mM K+. CGRP induced a significant vasodilatation of meningeal arteries (84 ± 10% of precontraction with 30 mM K+). The concentrations corresponding to the Cmax obtained after oral administration of 100 mg sumatriptan (160 nM) or 300 mg telcagepant (5 μM) both lead to a significant decrease of the effect of the exogenous administrated CGRP (-8 ± 18% and 8 ± 8%, respectively) (Figure 2). Also the concentrations of both sumatriptan and telcagepant corrected for plasma protein binding abolished the effect of exogenous CGRP (8 ± 36% for sumatriptan and 4 ± 7% for telcagepant, respectively) (Figure 2).
Rat MMA, (100 mmHg, pre-contracted with PGF2α)
SNP and sildenafil showed a tendency to relax the arteries but the observed dilatation was not statistically significant (Figure 3, Table 2). ACh, PGE2 and PACAP-38 did not display any discernible vasodilatation compared to NaCl (Figure 3, Table 2). The results from these rat experiments showed a larger spread compared to the experiments using human artery segments or a different protocol, and thus had a higher ratio of not passing the inclusion criteria.
Table 2.
Rat MMA, 100 mmHg, pre-contracted with PGF 2α
| Compound | E max (%) | pEC 50 (M) | p - value |
|---|---|---|---|
| ACh (n = 3) |
33 ± 22 |
N.A. |
0.68 |
| SNP (n = 3) |
70 ± 18 |
8.3 ± 0.7 |
0.066 |
| Sildenafil (n = 3) |
52 ± 20 |
~7 |
0.23 |
| PGE2 (n = 3) |
23 ± 12 |
N.A. |
0.82 |
| PACAP-38 (n = 4) |
22 ± 14 |
N.A. |
0.93 |
| NaCl (n = 3) | 23 ± 4 | N.A. |
p-value for comparing agonist versus the NaCl effect. N.A. = Not Applicable. Values are ± SEM, n given in parenthesis.
For the first experiments above, the same protocol as for the human vessels (100 mmHg and pre-contracted with PGF2α) was used to test all substances including NaCl on MMA segments from 5 rats. These vessels showed a large variation and had a high ratio of not passing the inclusion criteria of 0.1 mN. Therefore, the protocol was changed by lowering the transmural pressure to 50 mmHg and the experiments were repeated using an additional 5 rats. This markedly improved the success rate. In addition, the MMAs pre-contracted with 30 mM K+, did not have a negative drift over time (Figure 4A). A sample relaxation to a cumulative dose of SNP is also shown (Figure 4B).
Rat MMA, (50 mmHg, pre-contracted with 30 mM K+)
The results showed significant relaxation upon exposure to SNP, CGRP and sildenafil compared to the NaCl control group, SNP being the strongest vasodilatator (Figure 5, Table 3). PGE2, ACh and PACAP-38 showed no significant response compared to the control group (Figure 5, Table 3). These results correlate with the tendency seen for the above MMA.
Table 3.
Rat MMA, 50 mmHg, pre-contracted with 30 mM K +
| Compound | E max (%) | pEC 50 (M) | p - value |
|---|---|---|---|
| ACh (n = 5) |
-26 ± 13 |
N.A. |
0.85 |
| SNP (n = 5) |
67 ± 22; |
6.1 ± 1.3 |
0.015 |
| Sildenafil (n = 5) |
40 ± 17 |
5.7 ± 3.8 |
0.0069 |
| PGE2 (n = 5) |
-31 ± 5 |
N.A. |
0.311 |
| PACAP-38 (n = 5) |
27 ± 8 |
N.A. |
0.94 |
| CGRP (n = 5) |
-1 ± 6 |
~ 8 |
0.0015 |
| NaCl (n = 5) | -24 ± 5 | N.A. |
p-value for comparing agonist versus the NaCl effect. N.A. = Not Applicable. Values are ± SEM, n given in parenthesis.
Discussion
The addition of substances on human vessels was studied at a transmural pressure of 100 mmHg and pre-contracted with PGF2α. Two different approaches for the rat MMAs were used, and in this article the results from both methods are presented. Lowering of the transmural pressure from 100 mmHg to 50 mmHg greatly improved the success rate, and the more constant 30 mM K+ pre-contraction improved reproducibility. The gradual failing of the PGF2α pre-contraction and increase of the K+-induced pre-contraction was mitigated by comparing the vasomotor activity of the substances with the spontaneous activity observed when subjecting the vessel segments to only NaCl buffer. It is important to inform the reader, that the isolation and insertion of wires into the rat MMA, destroys the endothelial cell layer because of the small diameter of the rat MMA (around 100 μm in o.d.). The lack of endothelium can be seen in Figure 4A because ACh induces a contraction, most likely acting on ACh receptors on the smooth muscle cells. The endothelial function in the human vessels is not substantial either, as no significant relaxation to ACh is observed. The lack of full endothelial function and the lack of sympathetic tone in the vessels in this study could alter some of the effects of the substances added. For example, sildenafil has both endothelial-dependent and -independent effects [29], which could be different in human and rat.
The results from both types of experiments on rat tissue were consistent and showed that SNP, CGRP and sildenafil induced vasodilatation, while ACh, PGE2 and PACAP-38 had little effect. When used on human vessels, the same myography method yielded significant results for SNP and CGRP, where a strong dilatation was observed. An alternative method that has been used in rodents with some success is the cranial window method where substances can be applied topically through a thinned calvarium [30], but with this method quantification is difficult, as the concentration of active substance that reaches the vessel is difficult to control, hence pEC50 and Emax cannot reliably be calculated. Further, there are more factors (e.g., blood pressure) that can influence the results than when using isolated vessels in vitro.
For the experiments on human vessels, the greatest difference in response between MMA and the previously published data for MCA [20] was seen when subjecting the vessels to sildenafil and PACAP-38, which in both cases constituted a stronger dilatation in the MCA. This was surprising in the case of sildenafil since there are previous reports of rat MMA containing higher levels of phosphodieterase type 5 (PDE5) than reported for rat MCA [31]. If the same conditions were valid for human vessels, the higher concentration of the target enzyme in the MMA means that sildenafil should have had more effect there. However, the effect is also dependent on basal NO and cGMP levels, and we cannot rule out that there could be an influence of the age of the human compared to the rat vessels. In addition, the sympathetic tone in vivo could be counteracted by increased NO production, which would not occur in our setup. Therefore, sildenafil could be effective in vivo, but not in the myographs. It is hard to draw conclusions about the difference in Emax(%) of PACAP-38 due to previous conflicting observations of the activity of PACAP-38 in rat. Previously published in vitro experiments where human intracranial MMA was subjected to the substance suggests that PACAP-38 could indeed dilate human MMA (Emax(%) = 34 ± 12; pEC50 = 6.9 ± 0.1) [32] and that the absence of vasodilatation observed in the rat experiments in the present study is either due to lack of receptors or technical issues. However, there is no data suggesting an endothelial response to PACAP-38. This suggests that the rat is not a good model for studies of the PACAP-38 mechanisms.
Previous in vivo studies exist where magnetic resonance angiography (MRA) was used to measure the diameter of the extracranial part of the MMA of live human test subjects before, during and after spontaneous and artificially induced episodes of migraine. Intravenous infusion of CGRP [33], PACAP-38 [34] and nitroglycerine [35] in healthy non-migraineurs were shown to increase the diameter of the extracranial MMA. These substances are known to induce a headache lacking the migraine-related characteristics in non-migraineurs and this type of pain coincided with the observed vascular dilatation. When CGRP was infused to migraineurs, the results were reproduced and an additional, migraine-like headache with delayed onset occurred [36]. On the other hand, spontaneous migraine attacks reported in patients were associated with a modest increase in diameter (a magnitude of 11.4-13.0%) of the MCA and the intracranial parts (cavernous and cerebral) of the ICA [37]. The intracranial basilar artery (BA) and the extracranial vessels of ECA, superficial temporal artery (STA), the cervical part of the ICA and the extracranial part of the MMA were not significantly dilated.
Migraine-aborting triptan drugs had a constricting effect on arteries outside of the BBB, like the MMA and the extracranial part of the ICA, but not on cerebral arteries including the intracranial part of the ICA. Regardless of whether an episode of migraine was triggered naturally or by an infused substance, the pain was mitigated and the extracranial MMA contracted from the use of triptan drugs. However, it should be noted that these studies did not include the intracranial branches of the MMA, which is the source of the vessel segments used presently in the in vitro studies, and it is still unknown how this part of the artery would be affected in vivo by a spontaneous episode of migraine or a therapeutic dose of sumatriptan. The question is relevant, because all arteries shown to significantly dilate during an episode of migraine are all intracranial. Intracranial meningeal arteries would be unique in that they are both intracranial and without BBB properties. They are thereby a type of artery that could possibly be both dilated during an episode of migraine and contracted by drugs. In the in vitro study presented here, the effect of two well-known clinically tested anti migraine compounds were applied to the isolated human MMAs. Both sumatriptan (5-HT receptor agonist) and telcagepant (a CGRP receptor antagonist) significantly reduced the CGRP induced vasodilation. This shows that the vasoactive clinically tested compounds can exert their effect directly on the MMA.
In vivo experiments in rat, using the cranial window method to directly view and apply substances to the intracranial portion of the MMA, have yielded similar results to the in vivo observations in human regarding the extracranial part of the vessel. The MMA of rats was significantly dilated by α- and βCGRP [38,39], sildenafil [31], VIP, PACAP-38, PACAP-27 [40], histamine [41] and ACh [42]. VIP is interesting in that it is not a migrainogenic substance [16]. It has not been established what effect VIP could have on human isolated MMA in vivo, but since VIP is known to dilate human MMA branches in vitro[32] one can assume that the in vivo effect would mirror that found in the rat. Vasodilatation without migraine-related pain speaks against the dilation of the MMA as a direct cause of the pain. As with the effect of other substances on meningeal arteries, there are conflicting reports regarding VIP. No significant dilatation was observed during in vitro experiments using the myography method on the MMA [43]. Using the pressurized arteriography method cerebral and MMA were found to relax to the addition of VIP and PACAP [44,45]. PACAP was more potent than VIP [44] while the reverse was seen in the cerebral artery [45].
When the vasomotor responses from our experiments are compared with the migrainogenic properties of the tested substances, there seems to be little correlation between the ability of a substance to induce migraine-like attacks and their vasoactive properties in human meningeal arteries. Sildenafil, PGE2 and PACAP-38 showed no tendency of dilatation of the arteries in the current study, despite having known migrainogenic properties. One may wonder why and how the migrainogenic substances induce headache if vasodilatation of meningeal or cerebral arteries is not the mechanism. Endothelial nitric oxide (NO) is not the key molecule since the intracellular pathways activated by the migrainogenic substances CGRP and PACAP-38 are not dependent of NO in the cranial vessels [20]. The remaining factors are the perivascular and dural nerve fibers. It has been shown before that the dura mater contains a rich supply of sensory substance P and CGRP fibers, and of sympathetic noradrenaline and neuropeptide Y, but only a minor amount of parasympathetic VIP and PACAP containing fibers [46]. It is tentative to suggest that local release or systemic administration of the various substances may modify the activity of the dural nerve fibers via specific receptors or mechanisms. This clearly deserves future attention.
Conclusion
The lack of correlation between the vasoactive and migrainogenic properties of the tested substances leads us to the conclusion that direct vasodilatation of intracranial meningeal arteries is most likely not the sole trigger of the artificial migraine-like pain experienced by migraine sufferers upon infusion of these substances into the blood stream. This opinion is based on the fact that several migrainogenic substances induced no significant vasodilatation when applied in vitro to human meningeal arteries. We can however not rule out that the cranial pain and migrainogenic properties associated with the infusion of these substances could have a relation to cranial vasodilation in vivo. The in vivo vasodilatory responses to the substances could be affected by the vascular tone, endothelial interplay and the activation of perivascular nerve fibers around the dural arteries or in the trigeminal ganglion.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived and designed the experiments: GG, SL, KAH, AMvdB, LE. Performed the experiments: GG, SL, KAH. Analyzed the data: GG, SL, KAH, AMvdB. Contributed reagents/materials/analysis tools: AMvdB, LE. Wrote the paper: GG, SL, KAH, AMvdB, LE. All authors read and approved the final manuscript.
Contributor Information
Gustaf Grände, Email: gustafgrande@hotmail.com.
Sieneke Labruijere, Email: s.korremans-labruijere@erasmusmc.nl.
Kristian Agmund Haanes, Email: khaa0039@regionh.dk.
Antoinette MaassenVanDenBrink, Email: a.vanharen-maassenvandenbrink@erasmusmc.nl.
Lars Edvinsson, Email: Lars.Edvinsson@med.lu.se.
Acknowledgement
The technical assistance from Elisabeth Nilsson is greatly appreciated.
Funding
Dr. Antoinette MaassenVanDenBrink was supported by the Netherlands Organization for Scientific Research (Vidi grant 917.11.349). Prof. Lars Edvinsson was supported by the Swedish Reseach Council (grant no 5958) and a Centre of Excellence grant from the Lundbeck foundation, Denmark.
References
- Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;15(7):679–690. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- Levy D. Migraine pain and nociceptor activation–where do we stand? Headache. 2010;15(5):909–916. doi: 10.1111/j.1526-4610.2010.01670.x. [DOI] [PubMed] [Google Scholar]
- Ray BS, Wolff HG. Experimental studies on headache-pain-sensitive structures of the head and their significance in headache. Arch Surg-Chicago. 1940;15(4):813–856. doi: 10.1001/archsurg.1940.01210040002001. [DOI] [Google Scholar]
- Burstein R, Jakubowski M, Rauch SD. The science of migraine. J Vestib Res. 2011;15(6):305–314. doi: 10.3233/VES-2012-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen LH, Christiansen I, Iversen HK, Jansen-Olesen I, Olesen J. The effect of nitric oxide synthase inhibition on histamine induced headache and arterial dilatation in migraineurs. Cephalalgia. 2003;15(9):877–886. doi: 10.1046/j.1468-2982.2003.00586.x. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007;15(1–2):166–176. doi: 10.1016/j.pain.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia. 2010;15(10):1179–1186. doi: 10.1177/0333102410368444. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;15(5):698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- Ottosson A, Edvinsson L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalalgia. 1997;15(3):166–174. doi: 10.1046/j.1468-2982.1997.1703166.x. [DOI] [PubMed] [Google Scholar]
- Messlinger K, Lennerz JK, Eberhardt M, Fischer MJM. CGRP and NO in the trigeminal system: mechanisms and role in headache generation. Headache. 2012;15(9):1411–1427. doi: 10.1111/j.1526-4610.2012.02212.x. [DOI] [PubMed] [Google Scholar]
- Antonova M, Wienecke T, Olesen J, Ashina M. Prostaglandin E(2) induces immediate migraine-like attack in migraine patients without aura. Cephalalgia. 2012;15(11):822–833. doi: 10.1177/0333102412451360. [DOI] [PubMed] [Google Scholar]
- Wienecke T, Olesen J, Ashina M. Prostaglandin I2 (epoprostenol) triggers migraine-like attacks in migraineurs. Cephalalgia. 2010;15(2):179–190. doi: 10.1111/j.1468-2982.2009.01923.x. [DOI] [PubMed] [Google Scholar]
- Kruuse C, Thomsen LL, Birk S, Olesen J. Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain. 2003;15(Pt 1):241–247. doi: 10.1093/brain/awg009. [DOI] [PubMed] [Google Scholar]
- Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M. PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain. 2009;15(Pt 1):16–25. doi: 10.1093/brain/awn307. [DOI] [PubMed] [Google Scholar]
- Olesen J, Iversen HK, Thomsen LL. Nitric oxide supersensitivity: a possible molecular mechanism of migraine pain. Neuroreport. 1993;15(8):1027–1030. doi: 10.1097/00001756-199308000-00008. [DOI] [PubMed] [Google Scholar]
- Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008;15(3):226–236. doi: 10.1111/j.1468-2982.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- Schytz HW, Wienecke T, Olesen J, Ashina M. Carbachol induces headache, but not migraine-like attacks, in patients with migraine without aura. Cephalalgia. 2010;15(3):337–345. doi: 10.1111/j.1468-2982.2009.01929.x. [DOI] [PubMed] [Google Scholar]
- Kelman L. The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache. 2004;15(9):865–872. doi: 10.1111/j.1526-4610.2004.04168.x. [DOI] [PubMed] [Google Scholar]
- Shevel E. Middle meningeal artery dilatation in migraine. Headache. 2009;15(10):1541–1543. doi: 10.1111/j.1526-4610.2009.01495.x. [DOI] [PubMed] [Google Scholar]
- Grande G, Nilsson E, Edvinsson L. Comparison of responses to vasoactive drugs in human and rat cerebral arteries using myography and pressurized cerebral artery method. Cephalalgia. 2013;15(3):152–159. doi: 10.1177/0333102412468340. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Tfelt-Hansen P. The blood–brain barrier in migraine treatment. Cephalalgia. 2008;15(12):1245–1258. doi: 10.1111/j.1468-2982.2008.01675.x. [DOI] [PubMed] [Google Scholar]
- Edvinsson L. Tracing neural connections to pain pathways with relevance to primary headaches. Cephalalgia. 2011;15(6):737–747. doi: 10.1177/0333102411398152. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;15(1):19–26. doi: 10.1161/01.RES.41.1.19. [DOI] [PubMed] [Google Scholar]
- Lacey LF, Hussey EK, Fowler PA. Single dose pharmacokinetics of sumatriptan in healthy volunteers. Eur J Clin Pharmacol. 1995;15(6):543–548. doi: 10.1007/BF00193709. [DOI] [PubMed] [Google Scholar]
- Plosker GL, McTavish D. Sumatriptan. A reappraisal of its pharmacology and therapeutic efficacy in the acute treatment of migraine and cluster headache. Drugs. 1994;15(4):622–651. doi: 10.2165/00003495-199447040-00006. [DOI] [PubMed] [Google Scholar]
- Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, Leibensperger H, Froman S, Assaid C, Lines C, Koppen H, Winner PK. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet. 2008;15(9656):2115–2123. doi: 10.1016/S0140-6736(08)61626-8. [DOI] [PubMed] [Google Scholar]
- Scott AK. Sumatriptan clinical pharmacokinetics. Clin Pharmacokinet. 1994;15(5):337–344. doi: 10.2165/00003088-199427050-00002. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Linde M. New drugs in migraine treatment and prophylaxis: telcagepant and topiramate. Lancet. 2010;15(9741):645–655. doi: 10.1016/S0140-6736(10)60323-6. [DOI] [PubMed] [Google Scholar]
- Teixeira CE, Jin L, Priviero FB, Ying Z, Webb RC. Comparative pharmacological analysis of Rho-kinase inhibitors and identification of molecular components of Ca2+ sensitization in the rat lower urinary tract. Biochem Pharmacol. 2007;15(4):647–658. doi: 10.1016/j.bcp.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DJ, Hargreaves RJ, Hill RG, Shepheard SL. Sumatriptan inhibits neurogenic vasodilation of dural blood vessels in the anaesthetized rat–intravital microscope studies. Cephalalgia. 1997;15(4):525–531. doi: 10.1046/j.1468-2982.1997.1704525.x. [DOI] [PubMed] [Google Scholar]
- Kruuse C, Gupta S, Nilsson E, Kruse L, Edvinsson L. Differential vasoactive effects of sildenafil and tadalafil on cerebral arteries. Eur J Pharmacol. 2012;15(2–3):345–351. doi: 10.1016/j.ejphar.2011.10.037. [DOI] [PubMed] [Google Scholar]
- Chan KY, Baun M, de Vries R, van den Bogaerdt AJ, Dirven CM, Danser AH, Jansen-Olesen I, Olesen J, Villalon CM, MaassenVanDenBrink A, Gupta S. Pharmacological characterization of VIP and PACAP receptors in the human meningeal and coronary artery. Cephalalgia. 2011;15(2):181–189. doi: 10.1177/0333102410375624. [DOI] [PubMed] [Google Scholar]
- Asghar MS, Hansen AE, Kapijimpanga T, van der Geest RJ, van der Koning P, Larsson HB, Olesen J, Ashina M. Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology. 2010;15(17):1520–1526. doi: 10.1212/WNL.0b013e3181f9626a. [DOI] [PubMed] [Google Scholar]
- Amin FM, Asghar MS, Guo S, Hougaard A, Hansen AE, Schytz HW, van der Geest RJ, de Koning PJ, Larsson HB, Olesen J, Ashina M. Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia. 2012;15(2):140–149. doi: 10.1177/0333102411431333. [DOI] [PubMed] [Google Scholar]
- Schoonman GG, Bakker D, Schmitz N, van der Geest RJ, van der Grond J, Ferrari MD, van Buchem MA. Magnetic resonance angiography of the human middle meningeal artery: implications for migraine. J Magn Reson Imaging. 2006;15(4):918–921. doi: 10.1002/jmri.20708. [DOI] [PubMed] [Google Scholar]
- Asghar MS, Hansen AE, Amin FM, van der Geest RJ, Koning P, Larsson HB, Olesen J, Ashina M. Evidence for a vascular factor in migraine. Ann Neurol. 2011;15(4):635–645. doi: 10.1002/ana.22292. [DOI] [PubMed] [Google Scholar]
- Amin FM, Asghar MS, Hougaard A, Hansen AE, Larsen VA, de Koning PJ, Larsson HB, Olesen J, Ashina M. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol. 2013;15(5):454–461. doi: 10.1016/S1474-4422(13)70067-X. [DOI] [PubMed] [Google Scholar]
- Petersen KA, Birk S, Doods H, Edvinsson L, Olesen J. Inhibitory effect of BIBN4096BS on cephalic vasodilatation induced by CGRP or transcranial electrical stimulation in the rat. Br J Pharmacol. 2004;15(6):697–704. doi: 10.1038/sj.bjp.0705966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhl L, Edvinsson L, Olesen J, Jansen-Olesen I. Effect of two novel CGRP-binding compounds in a closed cranial window rat model. Eur J Pharmacol. 2007;15(1–2):117–124. doi: 10.1016/j.ejphar.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Boni LJ, Ploug KB, Olesen J, Jansen-Olesen I, Gupta S. The in vivo effect of VIP, PACAP-38 and PACAP-27 and mRNA expression of their receptors in rat middle meningeal artery. Cephalalgia. 2009;15(8):837–847. doi: 10.1111/j.1468-2982.2008.01807.x. [DOI] [PubMed] [Google Scholar]
- Akerman S, Williamson DJ, Kaube H, Goadsby PJ. The role of histamine in dural vessel dilation. Brain Res. 2002;15(1):96–102. doi: 10.1016/S0006-8993(02)03485-6. [DOI] [PubMed] [Google Scholar]
- Kowacs F, Williamson DJ, Goadsby PJ. Neurogenic vasodilation of dural blood vessels is not mediated by cholinergic transmission in the anaesthetised rat. Eur J Pharmacol. 2004;15(1–3):133–137. doi: 10.1016/j.ejphar.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Baun M, Hay-Schmidt A, Edvinsson L, Olesen J, Jansen-Olesen I. Pharmacological characterization and expression of VIP and PACAP receptors in isolated cranial arteries of the rat. Eur J Pharmacol. 2011;15(1):186–194. doi: 10.1016/j.ejphar.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Syed AU, Koide M, Braas KM, May V, Wellman GC. Pituitary adenylate cyclase-activating polypeptide (PACAP) potently dilates middle meningeal arteries: implications for migraine. J Mol Neurosci. 2012;15(3):574–583. doi: 10.1007/s12031-012-9851-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdling A, Sheykhzade M, Maddahi A, Bari F, Edvinsson L. VIP/PACAP receptors in cerebral arteries of rat: characterization, localization and relation to intracellular calcium. Neuropeptides. 2013;15(2):85–92. doi: 10.1016/j.npep.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Uddman R. Adrenergic, cholinergic and peptidergic nerve fibres in dura mater–involvement in headache? Cephalalgia. 1981;15(4):175–179. doi: 10.1046/j.1468-2982.1981.0104175.x. [DOI] [PubMed] [Google Scholar]