

Abstract
Although multiple different procedures to characterize the epitopes recognized by antibodies have been developed, site-directed mutagenesis remains the method of choice to define the energetic contribution of antigen residues to binding. These studies are useful to identify critical residues and to delineate functional maps of the epitopes. However, they tend to underestimate the roles of residues that are not critical for binding on their own, but contribute to the formation of the target epitope in an additive, or even cooperative, way. Mapping antigenic determinants with a diffuse energetic landscape, which establish multiple individually weak interactions with the antibody paratope, resulting in high affinity and specificity recognition of the epitope as a whole, is thus technically challenging. The current work was aimed at developing a combinatorial strategy to overcome the limitations of site-directed mutagenesis, relying on comprehensive randomization of discrete antigenic regions within phage-displayed antigen libraries. Two model antibodies recognizing epidermal growth factor were used to validate the mapping platform. Abrogation of antibody recognition due to the introduction of simultaneous replacements was able to show the involvement of particular amino acid clusters in epitope formation. The abundance of some of the original residues (or functionally equivalent amino acids sharing their physicochemical properties) among the set of mutated antigen variants selected on a given antibody highlighted their contributions and allowed delineation of a detailed functional map of the corresponding epitope. The use of the combinatorial approach could be expanded to map the interactions between other antigens/antibodies.
Keywords: EGF, library, phage display, randomization, site-directed mutagenesis
Introduction
Antibody (Ab) diversity has been exploited to obtain molecules able to establish multiple interactions with any potential target antigen (Ag). Binding of a given target by different monoclonal antibodies (mAbs) can result in divergent, and even opposite, biochemical and biological effects, depending on the precise nature of each interaction.1,2 Both binding strength (affinity) and the ability to recognize a particular region, i.e., an epitope, within the whole antigen (fine specificity) determine the usefulness of each antibody.3,4 Epitope mapping procedures, aimed at identifying the recognized epitope(s), include both structural and functional approaches.5 Structural methods like X-ray crystallography result in a picture of the Ag/Ab complex,6 where the Ag residues in close contact with the Ab can be readily located, giving rise to the definition of structural epitope. On the other hand, the functional epitope comprises the subset of Ag residues that are engaged in interactions with the Ab, making an energetic contribution to binding.7
Functional epitope mapping has largely relied on Ag mutagenesis procedures8 that allow the identification of mutational hotspots in the context of the whole native Ag resulting in abrogation of recognition. These techniques are usually laborious and time-consuming due to the need for manipulation of multiple mutated Ag variants. Alternative methods based on synthetic/recombinant Ag fragments9,10 help to locate the antigenic regions recognized by each Ab, but have limited usefulness for mapping conformational epitopes that depend upon the global Ag architecture. The huge combinatorial diversity and high throughput potential of phage-displayed random peptide libraries have been exploited to select peptides that resemble the nominal epitope in terms of recognition by a given Ab.11 The identification of the actual epitopes based on the selected peptide motifs is hampered by the difficulties in establishing an unequivocal correspondence between peptides and regions on the Ag surface.12 Selected peptides can establish different interactions with the Ab compared with the original Ag.13
Epitope mapping procedures based on extensive mutagenesis of the phage-displayed Ag have been optimized in our laboratory, combining the high throughput potential of phage display14 and Kunkel mutagenesis,15 with the intrinsic advantage of manipulating the whole correctly folded target Ag. Relevant antigenic regions are comprehensively explored by site-directed randomization, giving rise to profiles of tolerated/non-tolerated mutations and revealing the existence of critical residues. Even though this approach has been useful to study the molecular details of the epitopes recognized by several mAbs and to understand their biological effects,3,4 two major drawbacks still limit its scope. The method is focused on the identification of residues having critical individual contributions to binding and could tend to underestimate the additional effects of other residues that also belong to the functional epitope(s), resulting in incomplete pictures of the antigenic determinants. This limitation is illustrated by the identification of a few critical epitope residues, which cannot explain the cross-reactivity profile against the whole Ag, for the anti-IL-2 antibody JES6–1A12.4 More importantly, when recognition of an epitope by a given mAb is the net result of multiple simultaneous and very weak interactions, there is a risk that no single critical amino acids can be defined depending on the sensitivity of the screening method, and site-directed mutagenesis could totally fail to reveal the epitope location.
The current work was aimed at developing a combinatorial mutagenesis scanning approach based on simultaneous randomization of several positions within a relevant antigenic region. The method, instead of defining individual critical residues, revealed an array of amino acids (aa) that contribute to epitope formation as a whole. The strategy was validated with two model mAbs (CB-EGF.1 and CB-EGF.2) recognizing epidermal growth factor (EGF).16 Site-directed mutagenesis at the positions identified as major functional contributors within the CB-EGF.1 epitope confirmed the better suitability of the combinatorial approach to characterize epitopes like this one, having a diffuse energetic landscape. The combinatorial approach could be extended to map other Ag/Ab pairs.
Results
The epitopes recognized by anti-EGF antibodies were shown to be conformation-sensitive
The first clue about the nature of the epitopes recognized by CB-EGF.1 and CB-EGF.2 mAbs was provided by experiments disrupting EGF disulfide bonds. The treatment resulted in a severe antigenicity loss (by more than 90%) toward both antibodies (Fig. 1A) in an enzyme-linked immunosorbent assay (ELISA). The importance of proper folding of the target Ag led to the subsequent evaluation of the antigenicity of phage-displayed EGF, both native and denatured, since this the mapping approach under development would rely on the use of this Ag format. Proper display of both epitopes on filamentous phages was confirmed (Fig. 1B), and the drastic antigenicity loss upon denaturation accurately reproduced the results with the soluble Ag, indicating that both forms of the molecule are equivalent. The attachment of a c-myc tag to phage-displayed EGF served the additional purpose of providing a positive control (the anti-tag 9E10 mAb), which ruled out any non-specific ELISA interference of the reduction/alkylation treatment.
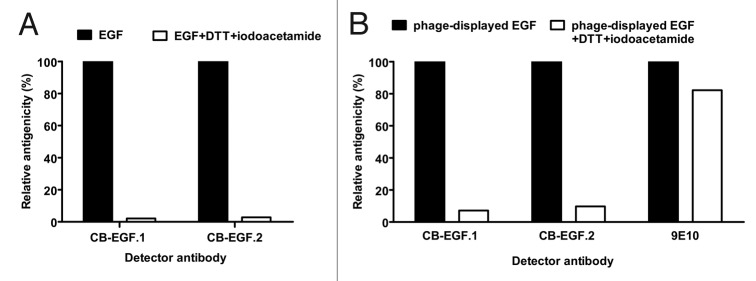
Figure 1. Conformation-sensitivity of the epitopes recognized by CB-EGF.1 and CB-EGF.2 mAbs. Microtiter plates coated with either recombinant EGF (A) or phage-displayed EGF fused to a c-myc tag (B) were sequentially treated with dithiothreitol (DTT) and iodoacetamide to disrupt disulfide bonds. CB-EGF.1 and CB-EGF.2 mAbs, as well as the control anti-c-myc tag 9E10 mAb, were added to the plates. Bound antibodies were detected with anti-mouse IgG polyclonal Abs conjugated to horseradish peroxidase. Relative antigenicity of treated EGF toward each mAb was calculated, using the signal on untreated control coated wells as the reference of maximal antigenicity (100%).
The screening of phage-displayed EGF fragments highlighted two different broad antigenic regions for CB-EGF.1 and CB-EGF.2 mAbs
Due to the technical complexity of a comprehensive mutagenesis scanning of the whole Ag surface (related to limitations in library size), the first step of our mapping strategy was aimed at locating the putative antigenic region recognized by each mAb. The intrinsic architecture of EGF (formed by three loops)17 and the conformation-sensitivity of the interactions under study led to the design of a set of five EGF fragments that covered the full protein length and kept some essential structural features of the molecule (pep1-pep5). Three of the fragments corresponded to loop A, loop B and loop C, respectively, (including the disulfide bonds defining each loop), while the two other fragments comprised the extra-loop N-terminal and C-terminal segments (Fig. 2). Screening of phage-displayed EGF fragments would reveal broad antigenic regions containing the target epitopes recognized by the mAbs. Cys residues forming disulfide bonds with other Cys located outside of a given fragment were replaced by Ser, in order to avoid epitope disruption due to non-natural intra- and intermolecular bonds involving unpaired Cys. A second set of five Cys-free EGF fragments overlapping with the previous ones, formed by protruding segments not corresponding to any of the entire loops (pep6-pep10), was also tested (Fig. 2) to obtain additional information.
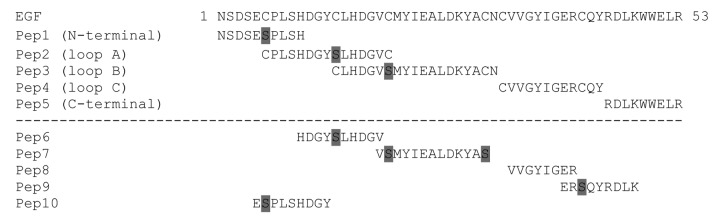
Figure 2. Phage-displayed EGF fragments designed to identify the antigenic regions recognized by anti-EGF mAbs. The first set of fragments (pep1-pep5) covers the entire EGF sequence and keeps important structural elements. The fragments correspond to the three EGF loops (loop A, loop B and loop C) including the two Cys defining each loop, as well as to the extra-loop N-terminal and C-terminal segments. Additional unpaired Cys within each fragment were replaced by Ser (shaded in gray). The second set (pep6-pep10) includes protruding fragments not corresponding to any entire loop, where all the Cys were substituted by Ser (shaded in gray).
ELISA screening of phage-displayed EGF fragments located the epitopes recognized by CB-EGF.1 and CB-EGF.2 mAbs within loop A and loop B, respectively (Fig. 3). Moderate recognition of pep10 (comprising the N-terminal end of loop A) by CB-EGF.1 pointed to a major role of this segment in the interaction, although the difference in reactivity compared with pep 2 (representing the whole loop A) precluded narrowing the antigenic region (Fig. 3).
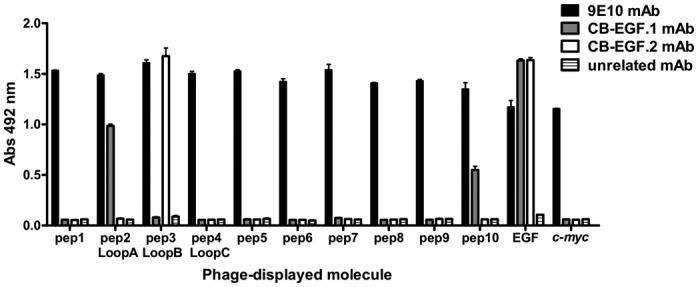
Figure 3. Recognition of phage-displayed EGF fragments. Polyvinyl chloride microtiter plates were coated with anti-EGF mAbs (CB-EGF.1 and CB-EGF.2), the anti-c-myc tag 9E10 mAb or an unrelated antibody. Phages displaying the different EGF fragments (1011 viral particles/mL) were added to the plates. Phage-displayed EGF and phages displaying only the c-myc tag fused to M13 PVIII were used as controls. Bound phages were detected with an anti-M13 mAb conjugated to horseradish peroxidase.
Focused combinatorial mutagenesis scanning of phage-displayed EGF revealed the differential contributions of smaller segments within loop A to CB-EGF.1 epitope formation
A more detailed analysis of CB-EGF.1 epitope was achieved through mutagenesis of loop A. Even though the initial evidence highlighting this region came from studies with phage-displayed EGF fragments, the subsequent scanning was performed on the full-length phage-displayed EGF. This procedure allowed the evaluation of the contributions of discrete regions in their natural context (the whole native Ag). Randomization was chosen as the mutagenesis strategy (instead of alanine or homolog scanning) because characterizing the effects of multiple conservative and non-conservative substitutions at each position would reveal more information about the nature of the interactions under investigation. Loop A length (15 aa) precluded randomization of the whole fragment in a single library containing all the theoretically possible combinations. The antigenic region (C6-C20) was thus divided in two segments, each of which could be comprehensively randomized. Cys residues were not changed in the libraries in order to maintain EGF architecture.
The replacement of P7-Y13 by random aa mixtures gave rise to a first library of 2.2 x 1010 EGF mutated variants, while randomization of positions corresponding to L15-V19 resulted in a second library of 109 members. Non-selected clones from both libraries produced a significant proportion of EGF variants (100% and 42%, respectively) that were not recognized by CB-EGF.1 mAb despite being properly displayed on phages (as assessed with the anti-c-myc tag 9E10 mAb) and fully reactive with a second anti-EGF mAb (CB-EGF.2). Specific loss of recognition by CB-EGF.1 implied that both segments (P7-Y13 and L15-V19) have a functional contribution to the formation of its epitope. High sequence divergence from the original Ag among the above described CB-EGF.1-negative variants (Figs. 4A and 4B) confirmed successful randomization. Most of them contained no aa identical to the original ones within the targeted segments.

Figure 4. Sequence divergence between EGF mutated variants recognized by CB-EGF.1 and non-reactive variants. Residues in the segment P7-Y13 were substituted by a random aa mixture in a library. Sequence identity with the non-mutated EGF within the targeted segment was evaluated in two groups of EGF variants: non-selected phage-displayed molecules not recognized by CB-EGF.1 mAb (35 unique sequences) and positive variants selected on immobilized CB-EGF.1 (42 unique sequences). Composition differences between both groups of variants are shown in (A). A second library targeted the L15-V19 segment. Differences between negative (24 unique sequences) and positive variants (29 unique sequences) from this library are shown in (B). An additional analysis took into account not only strict aa conservation, but also conservative replacements, as indicators of sequence similarity to wt EGF. Results with P7-Y13 and L15-V19 libraries are shown in (C) and (D) respectively.
Phage selection from the P7-Y13 library on CB-EGF.1 mAb resulted in the enrichment of EGF variants able to react with the selector antibody. Such positive variants were distinguished by the abundance of the original residues within the targeted segment (3–4 in most of them), in sharp contrast with the wide diversity found among negative variants (Fig. 4A). This result indicated the involvement of P7-Y13 segment in recognition by CB-EGF.1. Despite the increased representation of the original residues among the positive variants, all of them carried at least one substitution compared with wild-type (wt) EGF, and most exhibited several replacements, implying a certain degree of epitope tolerance to mutations. Antibody-driven appearance of residues with similar properties compared with the original ones was even more remarkable than strict aa conservation among positive variants (Fig. 4C). Although different systems have been proposed to measure the similarities between aa,18,19 and the concept of conservative mutation remains controversial,20 we used a rather simple physicochemical classification, grouping aromatic (F, W, Y), non-aromatic hydrophobic (I, L, M, V), basic (H, K, R) and acidic (D, E) residues.
Phage selection from the L15-V19 library rendered different results. Even though EGF variants reacting with CB-EGF.1 were concentrated after selection, the frequency of the original residues (and even of similar aa) in the targeted segment was not greatly increased among them in comparison with non-selected negative variants (Figs. 4B and 4D). The fact that mutated Ag molecules from the L15-V19 library could be segregated into CB-EGF.1-positive and -negative variants, but without a marked correlation between binding and conservation of sequence features, indicated that this segment modulates the antigenicity of the epitope instead of establishing direct interactions. Such a minor, but functionally important, role is consistent with the high reactivity of the whole phage-displayed EGF loop A (C6-C20) and the lower antigenicity of the shorter peptide E5-Y13 (Fig. 3).
Combinatorial mutagenesis scanning of EGF loop B revealed the contributions of two smaller segments to CB-EGF.2 recognition
The size of the CB-EGF.2 antigenic region (loop B), containing 19 residues (C14-N32), made a dissection necessary before performing combinatorial randomization. Because the C-terminal end of loop A overlaps with the N-terminal end of loop B, one of the libraries already used to characterize CB-EGF.1 epitope targeted the first segment (L15-V19) of the CB-EGF.2 antigenic region Mutagenesis within this segment, as previously described, affected recognition by the first antibody, leaving the reactivity to the second one unchanged. This information ruled out any influence of L15-V19 segment on the formation of the epitope recognized by CB-EGF.2.
The rest of loop B was divided in two additional segments, and each of them was comprehensively scanned in a combinatorial mutagenesis library. The first library was composed of 1.25 x 108 members with mutations in the segment M21-L26, while the other one targeted the segment D27-N32 (keeping C31 invariant) and contained 2.55 x 109 clones. Almost all non-selected phage-displayed EGF variants (100% and 93% in each library respectively) were not able to bind CB-EGF.2 (although recognized by the anti-c-myc tag and CB-EGF.1 mAbs). These CB-EGF.2-negative variants were distinguished by the absence or scarcity of the original residues within the targeted segments (Figs. 5A and 5B).

Figure 5. Sequence divergence between EGF mutated variants recognized by CB-EGF.2 and non-reactive variants. Residues in the segment M21-L26 were substituted by a random aa mixture in a library. Sequence identity with the non-mutated EGF within the targeted segment was evaluated in two groups of EGF variants: non-selected phage-displayed molecules not recognized by CB-EGF.2 mAb (20 unique sequences) and positive variants selected on immobilized CB-EGF.2 (43 unique sequences). Composition differences between both groups of variants are shown in (A). A second library targeted the D27-N32 segment. Differences between negative (24 unique sequences) and positive variants (43 unique sequences) from this library are shown in (B). An additional analysis took into account not only strict aa conservation, but also conservative replacements, as indicators of sequence similarity to wt EGF. Results with M21-L26 and D27-N32 libraries are shown in (C) and (D) respectively.
Phage selection from both libraries on CB-EGF.2 mAb rendered collections of EGF variants recognized by the selector antibody and showing an increase in the frequency of the original residues within each of the targeted segments over the non-selected CB-EGF.2-negative variants. Most positive variants selected from the M21-L26 library exhibited 2–3 residues identical to the original ones (Fig. 5A), while the majority of the positive variants from the D27-N32 library conserved 1–2 original residues within the mutated segment (Fig. 5B). Multiple mutations, even combined, were tolerated without disrupting the CB-EGF.2 epitope, indicating that none of the full segments was strictly required for binding. Even though some positive variants lacked all the original residues within one of the targeted segments, all of them were shown to have at least one aa sharing physicochemical properties with the original one, reinforcing the relevance of conservation of properties rather than strict residue conservation (Figs. 5C and 5D). The above described results indicated that the interactions contributing to recognition by CB-EGF.2 are distributed between the segments M21-L26 and D27-N32 within EGF loop B.
Antibody-driven conservation profiles of each original residue within the targeted segments delineated functional maps of both epitopes
Even though the analysis of global sequence divergence of EGF-derived mutated variants from the original Ag was useful to define the contribution of Ag segments to antigenicity, these data did not provide information about the particular interactions contributing to epitope/paratope binding. A closer examination of individual positions in the set of EGF mutated variants recognized by a given antibody revealed much more details and shed some light on the nature of the interactions behind recognition of the whole epitope. Residues prominently present in the pool of positive variants are most likely to have a functional role. Two criteria were defined for a residue to be considered as a contributor to epitope formation in the current work: 1) Its frequency among the Ab-selected positive variants should be higher (by more than 2-fold) than its frequency at the same position among non-selected phages displaying negative variants, indicating responsiveness to the selective pressure; and 2) Its abundance among the selected positive variants should be above 20% to avoid the confusing effects of the casual appearance of a few variants having the original residue after selection. Some original aa were particularly enriched among the positive variants (by more than 10-fold compared with the negative panel), and had a high likelihood of being major functional contributors to epitope formation.
The original residues within the targeted segment P7-Y13 were in general over-represented after selection on CB-EGF.1 mAb (Table 1). Several of them (P7, L8, S9, D11, G12, Y13) were considered to contribute to epitope formation according to the above described criteria. Although none of them was present in all of the CB-EGF.1-reactive variants, there was a marked increase (> 10x) in the abundance of D11, G12 and Y13 under CB-EGF.1 selective pressure. These data, together with the preponderance (more than 80%) of residues with shared chemical properties at positions 11 (negatively charged) and 13 (aromatic side chains) among positive variants (Table 1), determined the classification of D11 and Y13 as major functional contributors. High prevalence of G12 (unable to establish side chain interactions), and its replacement by residues with small side chains (volumes lower than 115 Å3) having diverse chemical characteristics, were interpreted as indicators of an accessory role of small aa at this position, allowing the interactions of the flanking major contributors. L8 was predominantly replaced by other hydrophobic residues among positive variants, reinforcing the relevance of physicochemical properties’ conservation for epitope formation. A similar analysis of variants from L15-V19 library resulted in the identification of a single additional contributor (G18), which was highly abundant and predominantly replaced by other small aa (Table 1). Its functional importance was thus associated to the absence of structural effects disturbing the epitope.
Table 1. CB-EGF.1 mAb-driven selection of the original residues from libraries of EGF mutated variants.
| Targeted segment | Original residue | Unselected phage-displayed mAb- negative EGF variants | Selected mAb-positive EGF variants |
|---|---|---|---|
| P7-Y13 | P7 |
20% Pro other: ACFGHIKLMNQRSTVY |
45.2% Pro (> 2x) other: DGHLMNRSTW |
| L8 |
11.4% Leu other: ADGMNPQSTVWY |
26.1% Leu (> 2x) 64.1% hydrophobic: ILMV other: AFHKPRTWY |
|
| S9 |
5.7% Ser other: ADEGHIKLMPRTVY |
31.0% Ser (> 5x) other: AFHILNPQRTVY |
|
| H10 |
5.7% His other: ADEGIKLNPQRTVWY |
11.9% His (> 2x) other: AFILNPQSTVW |
|
| D11 |
No Asp other: EFGHKLMNPQRSTVWY |
88.1% Asp (> 88x) 95.2% negatively charged: DE Other: ST |
|
| G12 |
2.9% Gly other: ADFHMLPQRSTV |
61.9% Gly (> 21x) other: ADNPS |
|
| Y13 |
2.9% Tyr other: ACDEGHIKLMNPQRSTVW |
45.2% Tyr (> 15x) 83.3% aromatic: FWY other: AHLS |
|
| L15-V19 | L15 |
8.3% Leu other: AEFHIKMPQSTV |
6.9% Leu other: AEGMNPQRSTVW |
| H16 |
4.2% His other: ADGLMNPQRSTVW |
17.2% His (> 4x) other: ADEGLMNPQRSTY |
|
| D17 |
No Asp other: AGHIKLMPQRSTVWY |
3.4% Asp (> 3x) other: ACEGHKLNPQRSVW |
|
| G18 |
No Gly other: AEHILNPQR |
31% Gly (> 31x) other: AHQRS |
|
| V19 |
4.2% Val other: ACEHKLNPRTY |
6.9% Val other: ADEGHLMNPQRSTWY |
Residues within the EGF P7-Y13 and L15-V19 segments were replaced by random aa mixtures in two separate libraries. Samples of non-selected CB-EGF.1-negative EGF variants and positive variants selected on CB-EGF.1 mAb were sequenced. The frequencies (%) of each original residue in both groups of variants are shown. The relative increase in the frequency of a given residue after selection is indicated between parentheses. The combined prevalence (%) of residues with shared physicochemical properties at some positions is represented. Other amino acids found at each position in both groups of variants are also shown. Residues that contribute to epitope formation are shaded in gray, whereas those classified as major functional contributors are highlighted in dark gray.
Five residues distributed within the two segments separately explored by combinatorial mutagenesis and selection on CB-EGF.2 mAb were shown to contribute to the formation of its epitope (Table 2). Y22, I23 and L26 were contained in the first targeted segment (M21-L26), whereas D27 and K28 belonged to the second one (D27-N32). The marked increase of their abundance (> 10x) under the CB-EGF.2 selective pressure highlighted I23, L26, D27 and K28 as major functional contributors. Some of them were frequently replaced by aa with shared physicochemical properties: hydrophobic residues were repeatedly found at positions 23 and 26, whereas positively charged aa predominated at position 28. D27 was only replaced by aa with small side chains (volumes lower than 115 Å3), implying that besides its own functional contribution, one important property at this position is the absence of steric hindrance effects on the interactions mediated by the surrounding contributors. It is noteworthy that the only feature absolutely conserved among positive variants was the presence of non-aromatic hydrophobic residues at position 26. All the remaining contributor positions showed some degree of variability, reinforcing the importance of combined aa effects in epitope formation.
Table 2. CB-EGF.2 mAb-driven selection of the original residues from a library of EGF mutated variants.
| Targeted segment | Original residue | Unselected phage-displayed mAb- negative EGF variants | Selected mAb-positive EGF variants |
|---|---|---|---|
| M21-L26 | M21 |
5.0% Met other: ACEGHIKPQRST |
No Met other: ADEGHIKLNQRSTVWY |
| Y22 |
5.0% Tyr other: ADEFKLMPRSTV |
44.1% Tyr (> 8x) other: AFHILMNQRST |
|
| I23 |
No Ile other: DGHLPQRSTY |
25.6% Ile (> 25x) 69.8% hydrophobic: ILV other: AHKQRSW |
|
| E24 |
No Glu other: ACDFGHILMNPTV |
7.0% Glu (> 7x) other: AHIKLMNPQRSTVY |
|
| A25 |
15% Ala other: EFGHILPQST |
27.9% Ala other: HLMPQVY |
|
| L26 |
No Leu other: ADGHKMPQRVWY |
69.8% Leu (> 69x) 100% hydrophobic: ILMV no other residues |
|
| D27-N32 | D27 |
4.2% Asp other: AEFILPQRST |
60.5% Asp (> 14x) other: AGNS |
| K28 |
No Lys other: AGHNPQRST |
32.6% Lys (> 32x) 76.8% positively charged: KR other: LMNSTV |
|
| Y29 |
No Tyr other: ACEGHIKPQST |
4.7% Tyr (> 4x) other: ADEGHIKLMNPQRST |
|
| A30 |
No Ala other: EHMNPQRSTV |
14.0% Ala (> 14x) other: DEFGHKLMNQSTVWY |
|
| N32 |
4.2% Asn other: ACDEGHPQRSTV |
4.7% Asn other: ADELMPQRSTY |
Residues within the M21-L26 and D27-N32 segments were replaced by random aa mixtures in two separate libraries. Samples of non-selected CB-EGF.2-negative EGF variants and positive variants selected on CB-EGF.2 mAb were sequenced. The frequencies (%) of each original residue in both groups of variants are shown. The relative increase in the frequency of a given residue after selection is indicated between parentheses. The combined prevalence (%) of residues with shared physicochemical properties at some positions is represented. Other amino acids found at each position in both groups of variants are also shown. Residues that contribute to epitope formation are shaded in gray, whereas those classified as major functional contributors are highlighted in dark gray.
Site-directed individual mutagenesis confirmed the relevance of combined interactions within CB-EGF.1 epitope
To compare the output of the exploration by combinatorial mutagenesis with conventional site-directed mutagenesis results, the two EGF positions identified as displaying major functional contributors to CB-EGF.1 epitope (11 and 13) were separately randomized, leaving the rest of the Ag unchanged. The two approaches did not provide exactly the same information. A few individual replacements (including drastic substitutions by Pro which could have disturbing effects due to structural constraints on the backbone and the replacement of D11 by the oppositely charged Lys/Arg) resulted in more than 50% antigenicity loss (Table 3), confirming the epitope location. On the other hand, the wide tolerance to multiple individual substitutions by residues with different physicochemical properties at these positions (Table 3) reinforced the notion that, although several residues within this model epitope contribute to binding energy, none of them could be classified as totally critical for recognition. Therefore their involvement would have been difficult to decipher by alanine-scanning and even by single-position randomization. In contrast, their roles were underscored by panning with the mAb multiply mutated molecules, where the set of contributors was preferentially selected as a whole, with some original residues compensating for the loss of other important (although not individually critical) interactions in each variant. Although our mapping approach could be useful for mAbs with different recognition modes, this case illustrated how combinatorial mutagenesis scanning could circumvent the limitations of site-directed mutagenesis for some of them.
Table 3. Effects of site-directed individual mutagenesis within CB-EGF.1 epitope.
| Targeted residue | Effect of replacements | |
|---|---|---|
| Tolerated | Non-tolerated | |
| D11 | A, E, H, M, Q, S, T, Y | K, L, P, R, V |
| Y13 | A, E, F, G, K, M, N, Q, R, S, V, W | D, P |
Residues previously identified as major functional contributors to the CB-EGF.1 epitope were individually replaced by random aa mixtures. Recognition of each phage-displayed variant by CB-EGF.1 mAb was evaluated by ELISA. Non-tolerated replacements resulted in less than 50% reactivity of the corresponding mutated variant compared with phage-displayed wt EGF. Tolerated substitutions were those found in recognized variants (keeping more than 50% reactivity toward the antibody).
Epitope mapping results were consistent with previous use and biological activity of anti-EGF antibodies
Although combinatorial mutagenesis led to consistent results that point to a robust identification of the epitopes recognized by CB-EGF.1 and CB-EGF.2 mAbs, an analysis of their correspondence with data emerging from other experimental settings would result in an independent line of evidence supporting the accuracy of our approach. The divergent locations of both epitopes on the EGF surface (Fig. 6) was totally consistent with the lack of competition between both mAbs16 that allowed their use as capture/detector antibodies in a sandwich ELISA to quantitate EGF levels.21

Figure 6. Locations of the epitopes recognized by CB-EGF.1 and CB-EGF.2 mAbs on EGF surface. EGF molecule (PDB code 1IVO_C) is represented as a cartoon with semi-transparent surface. CB-EGF.1 and CB-EGF.2 epitope residues are labeled and their side chains are shown as lines. EGF interface with the receptor (< 5Å) is colored yellow. CB-EGF.1 and CB-EGF.2 epitopes are highlighted in red and blue respectively. The resulting overlapping areas between each epitope and the EGF contact regions with receptor domains are shown in orange and green, and the rest of EGF is in gray. The figure was generated with Pymol.
Anti-EGF mAbs were shown to inhibit EGF-mediated activation of EGF receptor (EGF-R), as judged by ligand-induced phosphorylation inhibition, albeit with different neutralizing ability. mAb concentrations able to reduce EGF-R phosphorylation by 50% were 826 nmol/l for CB-EGF.1 and 153 nmol/l for CB-EGF.2. The identified epitopes recognized by both antibodies on EGF were shown to overlap with the EGF/EGF-R interface, providing a structural rationale for neutralization (Fig. 6). While several residues within CB-EGF.1 epitope are contained in the EGF interface with EGF-R domain III, the cluster of aa contributing to CB-EGF.2 epitope formation partially belongs to the interface with EGF-R domain I. Since EGF-R signaling requires the ligand to be accommodated between both domains, allowing the receptor to adopt the active extended conformation (dimerization-prone),17 inhibition of each individual interaction by mAbs (despite their divergent fine specificities) would result in the observed outcome: neutralization of EGF biological effects.
Discussion
The introduction of several simultaneous mutations in a single molecule is a bystander event when characterizing functional interfaces (including epitopes) by random mutagenesis. This is illustrated by the use of yeast and phage display to screen multiple Ag variants generated through error-prone polymerase chain reaction (PCR) in order to define the regions contributing to recognition by specific Abs.22-24 Although construction of libraries containing a huge diversity of variants (some of them displaying combinations of replacements) allows formal classification of these approaches as combinatorial, the rationale behind them is not focused on the relevance of additive/cooperative effects of several residues on binding. Molecules with more than one substitution are sometimes excluded from the analysis because of the uncertainty in assigning a role to one of the mutated residues.22 The library construction procedure itself precludes identification of relevant combined effects because the uncontrolled mutagenesis procedure tends to result in either single mutations or combinations of mutations targeting positions that are not necessarily located in the same antigenic region (neither in the primary sequence nor in the 3D structure) and are in general less likely to have a combined influence on binding. Mapping efforts exploiting these platforms have thus a different goal and a limited scope compared with the current work. While sequence space coverage by error-prone PCR can be very extensive, combinatorial randomization of a discrete antigenic region results in a more comprehensive local exploration of a functionally relevant area, including multiple conservative and non-conservative replacements at each position, as well as all their possible combinations. The putative region contributing to epitope formation is thus better characterized.
Other methods, like combinatorial alanine scanning,25 far from dismissing the information emerging from multiply mutated variants, rely on them to establish the relevance of individual residues. Combinatorial alanine scanning of phage-displayed proteins is thus technically closer to our mapping strategy. The difference resides in the fact that the published work, focused on a previously known structural interface, tried to dissect major functional contributors within it based on the analysis of the relative representations of Ala and the original residue at each position among mutated variants that retained the ability to engage in the interaction under study.26 The authors indeed highlighted the coincidence between the individual roles assigned to several critical residues through combinatorial screening and those determined by conventional single-position alanine scanning. The combinatorial approach was thus presented as a high throughput way to quickly analyze multiple positions, as opposed to the laborious site-directed mutagenesis, more than as a tool to study combinatorial effects on binding.
The same happened with quantitative saturation mutagenesis,27 which incorporates data emerging from randomization of targeted positions. This approach took advantage of the useful information about the nature of the interactions generated by evaluating the effects of multiple conservative and non-conservative substitutions. Library size constraints limited the number of positions that could be mutated in the same library in order to reach complete sequence coverage of the theoretical diversity, and dictated the need of constructing several libraries. Positions to be mutated were deliberately grouped in a way that tended to minimize the cooperative effects of replacements. Each library targeted residues that were as far apart as possible within the interface and included only one mutated position corresponding to a previously known critical residue. This approach was thus aimed at obtaining a comprehensive energetic landscape of the interface in a relatively simple way, but still focused on the individual roles of the original residues.
Even though randomization was also chosen as the diversification strategy in our procedure, establishing a parallel with the previously described method, two major elements distinguished the current work. First, our procedure was aimed at locating previously unknown epitopes on the Ag surface in the absence of any structural data about the complexes. Although the pattern of tolerated/non-tolerated replacements provided valuable information about the molecular details of recognition by mAbs, the main goal of the screening was to reveal the involvement of each discrete antigenic region (EGF segment in the current cases) as a whole in binding, establishing the boundaries of the epitope(s). Second and most important, the positions to be mutated in the same library were grouped (in this case by primary sequence proximity) to maximize the chances of underscoring combined effects mediated by neighbor residues, because our strategy is directed to epitopes with a diffuse energetic landscape without prominent critical residues, which are particularly difficult to characterize by site-directed mutagenesis. The current approach is thus not only technically combinatorial (due of the use of large libraries), but also focused on combinatorial effects within binding determinants.
Another study of combined contributions to epitope formation used the procedure called alanine shaving,28 which consists of the substitution by alanine of several residues surrounding those already identified as critical for mAb binding. The goal was to measure the context dependence of recognition of major antigenic determinants. Unlike this example, our approach took advantage of the high throughput potential of phage display and the highly informative nature of randomization, and allowed de novo mapping of the epitope(s) without the need to identify individually critical residues.
A prerequisite for the use of our approach is the segregation of the Ag into discrete antigenic regions that could be comprehensively scanned by combinatorial mutagenesis. Even though the Ag surface could be divided into multiple regions, covering it as a whole, it is highly desirable to have some preliminary information highlighting particular sequences or clusters of residues to reduce the required mapping efforts. In the case of EGF, used as the model Ag, this knowledge came from ELISA screening of phage-displayed Ag fragments. Although the reproduction of naturally-occurring Cys-flanked loops made Ag fragmentation useful to start mapping of EGF epitopes, this is not the only way to define the region to be explored by combinatorial mutagenesis. Such initial information could in principle be derived from any other available experimental system, including evaluation of synthetic peptides, peptide library screening, cross-reactivity data with similar proteins of different origin, and competition assays with receptors or mAbs of known specificity. Predictions based on computer simulations could also be used. Independently of the completeness and reliability of such previous data, they could serve as the starting point and a comprehensive mutagenesis scanning of the putative antigenic region in the context of the whole native Ag would establish or discard its involvement in a definitive way.
The groups of positions to be mutated in the libraries in the case of our model Ag (EGF) were continuous stretches in the primary sequence, due its particular architecture, but small clusters of residues distant in the primary sequence and relatively close in the tertiary structure could equally be defined. The only limit to include residues in a combinatorial mutagenesis scanning is the number of positions that can be simultaneously randomized due to library size constraints, irrespectively of their relative locations.
Since the antigenic regions to be explored are defined without knowing the exact location of the antigenic determinant, the epitope contributors can be targeted in different libraries, precluding a complete scanning of their combined effects. This was the case with CB-EGF.2 mAb, for which contributors (and even major contributors) to epitope formation were identified within different libraries. This example illustrates that even in such a complex context, the epitope location could be deciphered, but also opens the avenue of constructing a new library targeting all the contributor positions, as well as their close neighbors, to achieve a comprehensive scanning of their combined influence and a refined functional picture of the epitope as a whole.
EGF had been previously displayed on filamentous phages for a variety of purposes, including transduction of genetic information into EGF-R+ cells,29 functional mapping and directed evolution of its interactions with cellular receptors,30-32 and engineering of novel EGF-derived targeting agents.33 Its choice as a model Ag in the current work was not only due to the availability of two anti-EGF mAbs that have been previously difficult to map by site-directed mutagenesis (unpublished results), but also to its intrinsic relevance as an antibody target. Immunization with an EGF-based vaccine leads to sequestration of circulating EGF by elicited antibodies, resulting in clinical benefit for cancer patients (mediated by tumor growth factor deprivation).34 Mapping approaches focused on EGF are thus promising beyond characterization of mAbs. Its usefulness could be extended to study therapeutic humoral responses.
In summary, our work led to the development of a combinatorial mutagenesis approach, validated with two mAbs against a relevant target Ag (EGF), which expands the available phage display-based functional epitope mapping platform to those antigenic determinants that are difficult to map through site-directed mutagenesis. Its use can be extended to other antibodies/antigens to understand their structure/function relationships.
Materials and Methods
Phage display of EGF and EGF fragments
EGF-coding gene (amplified by PCR) and the synthetic genes coding for EGF fragments were cloned into the phagemid vector pC89/c-myc using flanking EcoRI and BamHI restriction sites. All the genes were thus fused to the c-myc tag gene and to the M13 gene 8. The resulting genetic constructs were sequenced by Macrogen (Korea) and used to transform TG1 E.coli cells (K12_(lac-pro), supE, thi, hsdD5/F’ traD36, proA+B+, lacIq, lacZ_M15), from which phages displaying EGF and ten different EGF fragments as c-myc-labeled fusion proteins attached to the major phage coat protein PVIII were rescued with M13KO7 helper phage at a 50 mL scale.35
Assessment of the conformation sensitivity of EGF epitopes
Maxisorp microtiter plates were coated with recombinant EGF (at 10 µg/mL in coating carbonate buffer) overnight at 4 °C. After washing with phosphate-buffered saline (PBS), some coated wells were sequentially treated during 1h at room temperature (RT) in the dark, first with DTT (at 0.1 mol/L in PBS) and later with iodoacetamide (at 0.1 mol/L in PBS). Untreated control coated wells were simultaneously incubated with PBS. Plates were washed with PBS and blocked with skim powder milk at 4% (w/v) in PBS (M-PBS) during 1h at 37 °C. Both anti-EGF mAbs (CB-EGF.1 and CB-EGF.2) diluted at 10 µg/mL in M-PBS were incubated on the plates during 1h at 37 °C. Plates were washed with PBS containing 0.1% (v/v) Tween 20 (PBS-T) and anti-mouse IgG polyclonal antibodies conjugated to horseradish peroxidase (HRP), properly diluted in M-PBS, were added for 1h at 37 °C. Plates were washed again and the substrate solution (500 µg/mL ortho-phenylenediamine and 0.015% (v/v) hydrogen peroxide in 0.1 mol/L citrate-phosphate buffer, pH 5.0) was added. The reaction was stopped after 15 min, with 2.5 mol/L sulfuric acid. Absorbances were measured at 492 nm. Relative antigenicity (%) of treated EGF toward each mAb was calculated, taking the maximal signal obtained in untreated wells as the reference.
A similar experiment was performed on phage-displayed EGF. Polyvinyl chloride microtiter plates were coated with phages displaying EGF (1012 viral particles/mL in PBS) overnight at 4 °C. Sequential treatment with DTT and iodoacetamide was performed as previously described. The next steps (incubation with mAbs and detection of bound antibodies) also followed the protocol described above; except that all the incubations were done at RT instead of 37 °C and that the anti-c-myc tag 9E10 mAb was included as a control.
Construction of libraries
Libraries were constructed as described.36 Briefly, single strand DNA corresponding to the genetic construct pC89/EGF-c-myc was purified from phages produced by the CJ236 E.coli strain (dut- ung- thi-1 relA1 spoT1 mcrA/pCJ105 (F’ camr)). Antisense mutagenic oligonucleotides were used to replace the segment of this original template to be targeted in each library by three stop codons (TAA)3 in order to obtain one suitable stop templates for the libraries. Single strand DNA corresponding to the stop templates was purified as described and used for large scale mutagenesis reactions with degenerate antisense oligonucleotides, able to replace each stop-containing segment by a stretch of NNK triplets, which coded for a random aa mixture at each of the targeted positions. TG1 E.coli cells were electroporated with the reaction products to give rise to the final libraries.
Phage selection on anti-EGF mAbs
Library phages were rescued with helper phage M13KO7 at a 300 ml scale as described.35 Immunotubes were coated with anti-EGF mAbs at 10 µg/mL in PBS overnight at 4 °C. Purified phages (5 x 1012 viral particles) and coated immunotubes were blocked with M-PBS during 1h at RT. Blocked phages were incubated on blocked immunotubes 1h at RT. Tubes were washed 20 times with PBS-T and twice with PBS. Bound phages were eluted with 100 mmol/L triethylamine during 10 min at RT, and neutralized with 1 mol/L Tris, pH 7.5. Exponentially growing TG1 E.coli cells were infected with the selected phages and used to amplify them at a 50 mL scale.35 Purified phages were the starting material for a new selection round. Three selection rounds were performed from each library. Infected TG1 colonies were also used to produce phages displaying the selected EGF variants (obtained after each selection round) and non-selected phages at a 96-well scale to test their recognition by anti-EGF mAbs in ELISA.
Site-directed mutagenesis
Individually mutated EGF variants were constructed by site-directed randomization using described procedures.36 Briefly, single strand DNA corresponding to the pC89/EGF-c-myc genetic construct was obtained from phages produced by the CJ236 E. coli strain and used as the template. Antisense mutagenic oligonucleotides introduced the degenerate randomization triplet NNK at the desired positions. TG1 E.coli cells were transformed with the reaction products to produce phages at a 96-well scale to perform ELISA screening of the new variants.
Phage ELISA screening of EGF, EGF fragments and EGF mutated variants
Polyvinyl chloride microtiter plates were coated with anti-EGF mAbs (CB-EGF.1 or CB-EGF.2), the anti-tag 9E10 mAb, or an unrelated mAb at 10 µg/mL in PBS. Plates were blocked for 1h at RT with M-PBS. Purified phages displaying either EGF or EGF fragments (properly diluted in M-PBS) and phage-containing supernatants rescued at a 96-well scale after library selection or site-directed randomization (diluted 1/5 in M-PBS) were incubated on coated/blocked plates during 1h at RT. After phage incubation, plates were washed with PBS-T and an anti-M13 mAb conjugated to HRP (GE Healthcare, USA), appropriately diluted in M-PBS, was added. Plates were incubated 1h at RT and washed. Substrate solution was added. The reaction was stopped after 15 min with 2.5 mol/L sulfuric acid. Absorbances were measured at 492 nm.
Molecular characterization of EGF mutated variants
EGF variants, recognized or not by the anti-EGF mAbs under study, were chosen for sequencing after ELISA screening. These variants were derived from the original unselected libraries, from selected phage pools and from EGF site-directed mutagenesis. Proper phage display (assessed with the anti-c-myc tag mAb) was a prerequisite to include them in the analysis. Exponentially growing E.coli XL-1 Blue cells (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac F´ proAB lacIqZ_M15 Tn10 Tetr) were infected with the corresponding phage-containing supernatants and used to purify plasmid DNA with the QIAprep Spin Miniprep kit (Qiagen, USA). Phagemid inserts were sequenced by Macrogen (Korea). Protein sequences of EGF variants were deduced from DNA sequences.
Inhibition of ligand-mediated EGF receptor activation by anti-EGF mAbs
A431 carcinoma cell line (EGF-R+) was grown in 6-well tissue culture plates in DMEM supplemented with 10% (v/v) fetal calf serum, penicillin (100 U/mL), streptomycin (100 µg/mL), L-glutamine (20 mmol/L), sodium pyruvate (10 mmol/L), HEPES (18 mmol/L), β-mercaptoethanol (20 µmol/L) and NaHCO3 (26 mmol/L) at 37 °C under 5% CO2 humidified atmosphere. Before the activation assay, cells were kept in the absence of fetal calf serum during 24h. After this period, cells were incubated with several concentrations of either anti-EGF mAbs or the unrelated control antibody (between 40 and 200 µg/mL) and EGF (100 ng/mL). After ten minutes incubation at 37 °C, cells were collected and lysed. Cell lysates (20 µg of total proteins) were run on SDS-PAGE and transferred to a nitrocellulose membrane. Phosphorylated EGF receptor was stained by western blot with an anti-phospho-EGF receptor antibody (Cell Signaling Technology, USA). Total EGF-R in the same samples was stained with a specific anti-EGF-R that is not dependent upon target phosphorylation status (Cell Signaling Technology, USA). Densitometric analysis of western blot bands was used to quantitate the level of receptor phosphorylation, which indicated the extent of ligand-induced activation. Phosphorylated receptor levels of different samples were calculated after normalizing taking into account the total EGF-R contents. Tyrosine kinase inhibitor AG1478 was used as a control of phosphorylation inhibition.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Yaquelin Marichal for her excellent technical assistance.
Glossary
Abbreviations:
- aa
amino acid
- Ab
antibody
- Abs
absorbance
- Ag
antigen
- DTT
dithiothreitol
- EGF
epidermal growth factor
- EGF-R
EGF receptor
- ELISA
enzyme-linked immunosorbent assay
- HRP
horseradish peroxidase
- IgG
immunoglobulin G
- mAb
monoclonal Ab
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
room temperature
- wt
wild-type
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/28395
References
- 1.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–7. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 2.Klein C, Lammens A, Schäfer W, Gorges S, Schwaiger M, Mössner E, Hopfner KP, Umaña P, Niederfellner G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationships to functional properties. MAbs. 2013;5:1–12. doi: 10.4161/mabs.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rojas G, Pupo A, Leon K, Avellanet J, Carmenate T, Sidhu S. Deciphering the molecular bases of the biological effects of antibodies against Interleukin-2: a versatile platform for fine epitope mapping. Immunobiology. 2013;218:105–13. doi: 10.1016/j.imbio.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Rojas G, Cabrera Infante Y, Pupo A, Carmenate T. Fine epitope specificity of antibodies against Interleukin-2 explains their paradoxical immunomodulatory effects. MAbs. 2013;6:273–85. doi: 10.4161/mabs.27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenspan NS, Di Cera E. Defining epitopes: It’s not as easy as it seems. Nat Biotechnol. 1999;17:936–7. doi: 10.1038/13590. [DOI] [PubMed] [Google Scholar]
- 6.Davies DR, Cohen GH. Interactions of protein antigens with antibodies. Proc Natl Acad Sci U S A. 1996;93:7–12. doi: 10.1073/pnas.93.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Regenmortel MH. Structural and functional approaches to the study of protein antigenicity. Immunol Today. 1989;10:266–72. doi: 10.1016/0167-5699(89)90140-0. [DOI] [PubMed] [Google Scholar]
- 8.Benjamin DC, Perdue SS. Site-directed mutagenesis in epitope mapping. Methods. 1996;9:508–15. doi: 10.1006/meth.1996.0058. [DOI] [PubMed] [Google Scholar]
- 9.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/S0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 10.Di Niro R, Ferrara F, Not T, Bradbury ARM, Chirdo F, Marzari R, Sblattero D. Characterizing monoclonal antibody epitopes by filtered gene fragment phage display. Biochem J. 2005;388:889–94. doi: 10.1042/BJ20041983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felici F, Castagnoli L, Musacchio A, Jappelli R, Cesareni G. Selection of antibody ligands from a large library of oligopeptides expressed on a multivalent exposition vector. J Mol Biol. 1991;222:301–10. doi: 10.1016/0022-2836(91)90213-P. [DOI] [PubMed] [Google Scholar]
- 12.Perosa F, Favoino E, Vicenti C, Guarnera A, Racanelli V, De Pinto V, Dammacco F. Two structurally different rituximab-specific CD20 mimotope peptides reveal that rituximab recognizes two different CD20-associated epitopes. J Immunol. 2009;182:416–23. doi: 10.4049/jimmunol.182.1.416. [DOI] [PubMed] [Google Scholar]
- 13.Saphire EO, Montero M, Menendez A, van Houten NE, Irving MB, Pantophlet R, Zwick MB, Parren PWHI, Burton DR, Scott JK, et al. Structure of a high-affinity “mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J Mol Biol. 2007;369:696–709. doi: 10.1016/j.jmb.2007.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith GP, Petrenko VA. Phage Display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freyre M, Vazquez J, Duarte C, Ferra E, Lopez I, Arteaga N, Alfaro A, Gavilondo J. Anticuerpos monoclonales que reconocen los factores de crecimiento epidérmico humano y murino. Interferon y Biotecnologia. 1989;6:32–43. [Google Scholar]
- 17.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim J-H, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–87. doi: 10.1016/S0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Nielsen R, Hasegawa M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol Biol Evol. 1998;15:1600–11. doi: 10.1093/oxfordjournals.molbev.a025888. [DOI] [PubMed] [Google Scholar]
- 19.Yampolsky LY, Stoltzfus A. The exchangeability of amino acids in proteins. Genetics. 2005;170:1459–72. doi: 10.1534/genetics.104.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jonson PH, Petersen SB. A critical view on conservative mutations. Protein Eng. 2001;14:397–402. doi: 10.1093/protein/14.6.397. [DOI] [PubMed] [Google Scholar]
- 21.Mainet D, Caccamo F, Duarte C. A fast and sensitive noncompetitive streptavidin-biotin enzyme immunoassay for human epidermal growth factor. Biotecnol Apl. 1993;10:176–82. [Google Scholar]
- 22.Chao G, Cochran JR, Wittrup KD. Fine epitope mapping of anti-epidermal growth factor receptor antibodies through random mutagenesis and yeast surface display. J Mol Biol. 2004;342:539–50. doi: 10.1016/j.jmb.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 23.Levy R, Forsyth CM, LaPorte SL, Geren IN, Smith LA, Marks JD. Fine and domain-level epitope mapping of botulinum neurotoxin type A neutralizing antibodies by yeast surface display. J Mol Biol. 2007;365:196–210. doi: 10.1016/j.jmb.2006.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jespers L, Jenné S, Lasters I, Collen D. Epitope mapping by negative selection of randomized antigen libraries displayed on filamentous phage. J Mol Biol. 1997;269:704–18. doi: 10.1006/jmbi.1997.1077. [DOI] [PubMed] [Google Scholar]
- 25.Morrison KL, Weiss GA. Combinatorial alanine-scanning. Curr Opin Chem Biol. 2001;5:302–7. doi: 10.1016/S1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 26.Weiss GA, Watanabe CK, Zhong A, Goddard A, Sidhu SS. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci U S A. 2000;97:8950–4. doi: 10.1073/pnas.160252097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pál G, Kouadio J-LK, Artis DR, Kossiakoff AA, Sidhu SS. Comprehensive and quantitative mapping of energy landscapes for protein-protein interactions by rapid combinatorial scanning. J Biol Chem. 2006;281:22378–85. doi: 10.1074/jbc.M603826200. [DOI] [PubMed] [Google Scholar]
- 28.Jin L, Wells JA. Dissecting the energetics of an antibody-antigen interface by alanine shaving and molecular grafting. Protein Sci. 1994;3:2351–7. doi: 10.1002/pro.5560031219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larocca D, Jensen-Pergakes K, Burg MA, Baird A. Receptor-targeted gene delivery using multivalent phagemid particles. Mol Ther. 2001;3:476–84. doi: 10.1006/mthe.2001.0284. [DOI] [PubMed] [Google Scholar]
- 30.Souriau C, Gracy J, Chiche L, Weill M. Direct selection of EGF mutants displayed on filamentous phage using cells overexpressing EGF receptor. Biol Chem. 1999;380:451–8. doi: 10.1515/BC.1999.059. [DOI] [PubMed] [Google Scholar]
- 31.Stortelers C, van der Woning SP, Jacobs-Oomen S, Wingens M, van Zoelen EJJ. Selective formation of ErbB-2/ErbB-3 heterodimers depends on the ErbB-3 affinity of epidermal growth factor-like ligands. J Biol Chem. 2003;278:12055–63. doi: 10.1074/jbc.M211948200. [DOI] [PubMed] [Google Scholar]
- 32.van der Woning SP, van Rotterdam W, Nabuurs SB, Venselaar H, Jacobs-Oomen S, Wingens M, Vriend G, Stortelers C, van Zoelen EJJ. Negative constraints underlie the ErbB specificity of epidermal growth factor-like ligands. J Biol Chem. 2006;281:40033–40. doi: 10.1074/jbc.M603168200. [DOI] [PubMed] [Google Scholar]
- 33.Bach M, Hölig P, Schlosser E, Völkel T, Graser A, Müller R, Kontermann RE. Isolation from phage display libraries of lysine-deficient human epidermal growth factor variants for directional conjugation as targeting ligands. Protein Eng. 2003;16:1107–13. doi: 10.1093/protein/gzg145. [DOI] [PubMed] [Google Scholar]
- 34.Rodríguez PC, Rodríguez G, González G, Lage A. Clinical development and perspectives of CIMAvax EGF, Cuban vaccine for non-small-cell lung cancer therapy. MEDICC Rev. 2010;12:17–23. doi: 10.37757/MR2010.V12.N1.4. [DOI] [PubMed] [Google Scholar]
- 35.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 36.Fellouse FA, Sidhu SS. Making antibodies in bacteria. In Making and using antibodies. A practical handbook. G.C. Howard, and M.R. Kaser, CRC Press, Boca Raton, Florida, 2007, pp 157-177. [Google Scholar]