

Abstract
Preventing unfavorable graft-versus-host disease (GVHD) without inducing broad suppression of the immune system presents a major challenge of allogeneic hematopoietic stem cell transplantation. We developed a novel strategy to ameliorate GVHD while preserving graft-versus-tumor (GVT) activity by small molecule-based inhibition of the NF-κB family member c-Rel. Underlying mechanisms included reduced alloactivation, defective gut homing, and impaired negative feedback on IL-2 production resulting in optimal IL-2 levels, which, in the absence of competition by effector T-cells, translated into expansion of regulatory T-cells. c-Rel activity was dispensable for antigen-specific T-cell receptor activation, allowing c-Rel-deficient T-cells to display normal GVT activity. In addition, inhibition of c-Rel activity reduced alloactivation without compromising antigen-specific cytotoxicity of human T-cells. Finally, we were able to demonstrate feasibility and efficacy of systemic c-Rel inhibitor administration. Our findings validate c-Rel as a promising target for immunomodulatory therapy and demonstrate feasibility and efficacy of pharmaceutical inhibition of c-Rel activity.
Keywords: NF-κB, c-Rel, small molecule inhibitor, anti-tumor activity, alloactivation
INTRODUCTION
Allo-HSCT represents a potent therapy of malignant and non-malignant hematologic diseases (1). In the care of patients with malignant diseases it was initially developed to follow high dose chemo/radiation therapy in order to rescue from therapy-related bone marrow failure, however the emphasis has now shifted towards allo-HSCT as a strategy to facilitate GVT activity (2). GVHD remains a major complication of allo-HSCT resulting in significant morbidity and mortality (3, 4), and strategies to suppress GVHD are often associated with broad suppression of the immune system leading to immune deficiency and compromised anti-tumor activity (5). Molecular pathways that have been targeted in preclinical studies to accomplish separation of GVHD from GVT activity include the mammalian target of rapamycin (mTOR), histone deacytelase (HDAC), and NF-κB (via proteasome inhibition) (6). The NF-κB family member c-Rel is a transcription factor that regulates lymphocyte survival and proliferation following antigen receptor triggering and plays dominant roles in inflammation, auto and alloimmunity (7–10). c-Rel regulates expression of many inflammatory cytokines and is expressed in T and B-cells as well as monocytes/macrophages and dendritic cells (11). Immune defects secondary to c-Rel deficiency have been attributed to impaired activation of lymphocytes, in particular T-cells. In T-cells, the main target gene of c-Rel is interleukin 2 (IL-2), a cytokine required for normal T-cell proliferation and differentiation. Previous reports demonstrated that c-Rel-deficient T-cells showed reduced Th1 but normal Th2 responses (12, 13) and are compromised in the generation of anti-inflammatory natural Tregs (14–18) and pro-inflammatory Th17 cells (18, 19). Given the limitations of existing immunosuppression-based therapies for GVHD and the pivotal roles of c-Rel in T-cell proliferation and function, we employed strategies targeting the c-Rel pathway, including small molecule-based inhibition of c-Rel activity (20, 21), to modulate T-cell responses in the context of GVHD and malignant diseases.
RESULTS
c-Rel Expression Is Upregulated During Allo-HSCT
We first analyzed the biological significance of c-Rel for hematopoietic reconstitution and T-cell activation after allo-HSCT. We performed studies assessing c-Rel expression in T-cells in the setting of radiation-induced injury as well as GVHD (Fig. 1A and Supplementary Fig. S1). After irradiation and especially during GVHD, IL-2, CD25 (IL-2Rα), and c-Rel were upregulated, consistent with lymphocyte activation. c-Rel expression was analyzed by both intracellular and intranuclear staining (22), and the results were closely correlated (Fig. 1B and C). Hematopoietic stem cell functions, including the development of lymphoid and myeloid lineages, are not known to be dependent on c-Rel activity. To confirm this, we performed an allo-HSCT utilizing c-Rel−/− donor bone marrow (BM) in a MHC-disparate allo-HSCT model. c-Rel−/− BM engrafted and reconstituted recipients as efficiently as wild-type (WT) BM (Fig. 1D). We also sought to determine if c-Rel activity in cells other than donor T-cells played a role for GVHD development by using different combinations of c-Rel−/− BM and T-cells as donor source (Fig. 1E) as well as utilizing c-Rel−/− mice as recipients (Fig. 1F). c-Rel deficiency in donor BM or in the recipient did not impact on survival or GVHD scores.
Figure 1.

c-Rel expression in donor T-cells is increased after allo-HSCT. A, Sublethally or non-irradiated BALB/c recipients were transplanted with C57BL/6 T-cell depleted (TCD) bone marrow (BM) cells with or without 1 × 106 C57BL/6 WT T-cells. Levels of IL-2, CD25, and c-Rel are shown after gating on either donor or residual recipient T-cell populations on day 7. Values represent mean ± SEM (n = 5). XRT, irradiation; *, P < 0.05; **, P < 0.01. B and C, Splenocytes were stimulated with PMA/Ionomycin for 5 hours and analyzed for intracellular and intranuclear c-Rel expression by flow cytometry. B, Intracellular c-Rel staining was performed following fixation and permeabilization (eBioscience Kit). C, Cytoplasm was removed from c-Rel stained cells by 0.03% saponin with nuclear isolation media (NP-40, nonyl phenoxypolyethoxylethanol) to confirm nuclear localization of c-Rel. D, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 WT or c-Rel−/− TCD BM cells. Thymus, BM, and spleen were analyzed on day 28 after HSCT. Values represent mean ± SEM (n = 4). Data shown in A–D are representative of two independent experiments. NK, Natural Killer; MDSCs, Myeloid-Derived Suppressor Cells; DCs, Dendritic Cells. E, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 WT or c-Rel−/− TCD BM cells with or without 1 × 106 C57BL/6 WT or c-Rel−/− T-cells. Survival curve is shown. F, Lethally irradiated WT C57/BL6 or c-Rel−/− recipients were transplanted with B10.BR TCD BM cells with or without 2 × 106 B10.BR T-cells. Survival curve is shown. Data in E and F are combined from three independent experiments and values represent mean ± SEM (n = 21). ***, P < 0.001.
Recipients of c-Rel-deficient T-cells Exhibit Increased IL-2 Levels Associated With Expansion of Tregs
A recent study found evidence for a role of c-Rel in donor T-cells during acute GVHD in mice (10). We were able to confirm these observations by utilizing c-Rel−/− donor T-cells in mouse models of MHC-disparate as well as a more clinically relevant minor antigen mismatched allo-HSCT, which in both cases resulted in significant amelioration of GVHD as determined by survival, weight loss, histopathology of GVHD target organs, and clinical GVHD scores (Supplementary Fig. S2 and data not shown). Moreover, we evaluated the profiles of donor T-cells in the spleens of recipient mice on day 7 revealing that the effector/naïve (TE/TN) ratios were decreased for both CD4+ and CD8+ T-cells, and we observed lower CD8+ TE/Tregs ratios in mice receiving c-Rel−/− T-cells compared to recipients of WT T-cells (Fig. 2A). We also found increased thymic cellularity on day 14 in recipients of c-Rel−/− T-cells compared to WT T-cells, indicating reduced thymic GVHD (Fig. 2B). Furthermore, c-Rel−/− T-cells expressed significantly lower levels of the intestinal homing marker LPAM-1 on TE cells (Fig. 2C), consistent with the findings that recipients of c-Rel−/− T-cells had much lower numbers of infiltrating donor T-cells in mesenteric lymph nodes (mLN) and small intestines (SI) (Fig. 2D and E). Importantly, the absolute numbers of Tregs infiltrating the SI showed no difference between recipients of WT and c-Rel−/− T-cells (Fig. 2F), reinforcing that the net outcome of the T-cell balance is shifted towards T-cell suppression with significantly lower CD8+ TE/Tregs ratios in the SI of recipients of Rel−/− T-cells (Fig. 2E).
Figure 2.

c-Rel-deficiency in donor T-cells results in reduced alloactivation and gut homing. A–E, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with or without 2 × 106 C57BL/6 WT or c-Rel−/− T-cells. Data are representative of three independent experiments. Mean values and SEM are presented (n = 5). *, P < 0.05; **, P < 0.01; ***, P < 0.001. TN, naïve T-cells; TE, effector T-cells; mLN, mesenteric lymph nodes; SI, small intestines. A and C, Donor gated T-cell profiles in recipient spleens on day 7 after HSCT are shown. B, Thymic cellularity and numbers of CD4+CD8+ double positive (DP) cells on day 14 are shown. D, Donor gated T-cell profiles in recipient mLN on day 7. E, Donor gated T-cell profiles in recipient SI on day 7. F, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 WT TCD BM cells with 1 × 106 C57BL/6 WT or c-Rel−/− T-cells. Recipient SI were harvested on day 14 and histologic examination was performed. For immunohistochemistry, SI are fixed with 4% paraformaldehyde, embedded in paraffin, and stained with anti-mouse FoxP3 antibody. Three slides sections from each mouse were stained with the antibody and blinded quantitative histologic analysis was performed in 5 mm length of longitudinally sectioned SI tissue. Mean values and SEM are presented (n = 4).
Unlike in vitro (7), we found that a greater percentage of c-Rel−/− donor T-cells produced IL-2 compared to WT T-cells on day 7 after allo-HSCT, resulting in IL-2 serum concentrations above the Kd (meaning that more than 50% of IL-2 receptors will be occupied) in recipients of c-Rel−/− T-cells and in significantly lower levels in recipients of WT T-cells (Fig. 3A and B). STAT5 is a negative regulator of IL-2 production (23, 24). When we analyzed STAT5 phosphorylation (pSTAT5) in c-Rel−/− and WT T-cells we found that splenic c-Rel−/− T-cells harvested 96 hours after allo-HSCT exhibited impaired IL-2-mediated induction of pSTAT5 (Fig. 3C). This lower responsiveness to IL-2 is consistent with decreased expression of CD25 by c-Rel−/− TE cells (data not shown) and suggests decreased negative feedback on IL-2 production resulting in a relative increase in IL-2 secretion compared to WT T-cells. Furthermore, consistent with a previous report (10), we observed a dramatic increase in the numbers of donor-derived Tregs in recipients of c-Rel−/− T-cells starting after day 7 post HSCT (Fig. 3D), whereas host Tregs were hardly detectable. Expression of CD25 on those Tregs was higher than that of WT Tregs (Fig. 3E). These data suggest that competition for IL-2 between TE and Tregs results in promotion of alloactivation in recipients of WT T-cells, and in expansion of Tregs in recipients of c-Rel-deficient T-cells (Fig. 3F). Neuropilin-1 (Nrp-1) was recently identified as a marker for natural Tregs (25, 26). As expected, the vast majority of c-Rel-deficient Tregs in our experiments were Nrp-1− induced Tregs (data not shown). To further investigate the potential role of Tregs in the amelioration of GVHD, we performed in vivo depletion of Tregs using FoxP3-DTR transgenic mice that express the diphtheria toxin receptor (DTR) under control of the FoxP3 promoter (27) as donor T-cell source. Elimination of Tregs in vivo by DT administration on day 13 worsened the survival of recipients of c-Rel-deficient T-cells (c-Rel deficiency was achieved by pre-treatment of donor T-cells with a small molecule inhibitor compound, see below for more details), suggesting that donor Tregs indeed contributed to the amelioration of GVHD (Fig. 3G and Supplementary Fig. S3). Depletion of donor Tregs in recipients of T-cells with normal c-Rel activity did not affect GVHD severity, indicating that in contrast to animals receiving c-Rel-deficient T-cells, the Treg numbers in animals receiving WT T-cells was too low during early GVHD to be biologically significant. Importantly, Tregs from c-Rel−/− mice are functional (28, 29) and could reduce GVHD in our model (Fig. 3H).
Figure 3.

Increased IL-2 production in recipients of c-Rel-deficient T-cells is associated with promoted expansion of regulatory T-cells (Tregs). A–E, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with 1 × 106 C57BL/6 WT or c-Rel−/− T-cells. One of three independent experiments is presented. Values represent mean ± SEM (n = 5). *, P < 0.05; **, P < 0.01. A, Serum levels of IL-2 on day 7 after HSCT are shown. Dotted line indicates signaling threshold of IL-2. B, Secretion of IL-2 from donor CD4+ cells in spleen on day 7. C, Phospho-STAT5 (pSTAT5) levels in IL-2 stimulated splenocytes from WT or c-Rel−/− T-cells transplanted recipients after cytokine stripping with glycine. Expression levels of pSTAT5 in CD25+ donor T-cells are shown. D, Time course analyses of absolute numbers of donor CD4+CD25+FoxP3+ cells are shown. E, Expression level of CD25 in donor Tregs in spleen on day 14. F, Schematic diagram of proposed c-Rel/IL-2 interaction pathways during GVHD. G, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with 1 × 106 C57BL/6 WT or FoxP3-DTR T-cells. T-cells were treated with c-Rel inhibitor or control vehicle for 24 hours before transplantation. Diphtheria toxin (DT) was administered on day 13 after HSCT to eliminate donor Tregs in recipients transplanted with FoxP3-DTR T-cells (n = 5–8). H, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells and 0.5 × 106 C57BL/6 WT T-cells with or without 0.5 × 106 C57BL/6 Tregs or c-Rel−/− Tregs. Control recipients were transplanted with C57BL/6 TCD BM only. Survival curve is shown. Data are combined from two independent experiments (n = 16–18). *, P < 0.05.
T-cells Treated With a Small Molecule c-Rel Inhibitor Compound Cause Less GVHD
Some natural compounds and their synthetic derivatives inhibit Rel or NF-κB via specific interaction with the Cysteine residues critical for binding to the specific κB-DNA sequence (20). For example, dehydroxymethylepoxyquinomicin (DHMEQ) was shown to inhibit NF-κB binding activity, however, the complex structures and poor pharmacokinetics due to reactive sites found in natural products made them unsuitable for further development. To develop direct Rel inhibitors, we established an assay (fluorescence polarization (FP) assay; for more details, see METHODS) for high throughput screening. Screening of a library of 15,000 compounds yielded 20 hits. We conducted structure activity relationship (SAR) studies of these initial hits using EMSA and finally identified the hydrophobic small molecule Pyrimidinetrione and its derivatives as potent and highly specific inhibitors of c-Rel activity, having a 20–200 fold higher inhibitory effect on c-Rel and NF-κB than on other transcription factors such as Oct1 and AP1 (Fig. 4A and data not shown). These compounds bind c-Rel directly and change the conformation of the protein, inhibiting DNA binding and transcriptional activity. We developed several generations of Pyrimidinetrione derivatives to further optimize the inhibitory effect and found that treatment of activated T-cells with one of those compounds (IT-603) resulted in most efficient inhibition of c-Rel activity as well as IL-2 expression in vitro (Fig. 4B–D) without altering cell viability after 24 hours of incubation (Fig. 4E). IT-603 was used for all subsequent experiments involving chemically induced c-Rel inhibition. Basic characteristic of IT-603 are summarized in Table 1.
Figure 4.

Pre-treatment of donor T-cells with a c-Rel antagonist prevents GVHD in allo-HSCT. A, Results of fluorescence polarization (FP) as well as electrophoretic mobility shift assay (EMSA) are shown (for more details, see METHODS). c-Rel inhibitor compound IT-603 at 2-fold dilutions (2000 to 15.625 nM) was mixed with CD28RE-FITC (10, 3.3, 1.1, 0.33, 0.11 nM) in FP buffer for 30 minutes. Data for 10 nM and 0.33 nM are shown. EMSA for c-Rel, Oct-1, AP-1 at different inhibitor concentrations are shown. (B–E) CD5+ positively selected C57BL/6 splenocytes were treated with two different types of c-Rel antagonists for 24 hours and analyzed after anti-CD3/28 stimulation for another 24–48 hours. Data are representative of more than three independent experiments. B, Representative flow cytometric analysis after 24 hours of anti-CD3/28 stimulation and incubation with inhibitor compound IT-603 at four different concentrations. C, Representative flow cytometric analysis of intracellular c-Rel and IL-2 expressions after 24 hours of anti-CD3/28 stimulation and incubation with inhibitor compound IT-603 at four different concentrations. Arrows indicate gradual loss of the inhibitory effect of the antagonist with decreasing concentrations. D, Representative flow cytometric analysis of intracellular c-Rel and IL-2 expressions after 24 and 48 hours of anti-CD3/28 stimulation and incubation with inhibitor compound IT-603 or IT-901. E, Viabilities of the CD5+ T-cells after 24 hour-incubation with inhibitor compounds IT-603, IT-901, or empty vehicle. Percentages of live/dead cells and annexin V positive pro-apoptotic cells are shown. F and G, Lethally irradiated BALB/c recipients received C57BL/6 TCD BM cells with 0.5 × 106 C57BL/6 WT T-cells after 24 hours of pre-treatment with c-Rel inhibitor compound IT-603 or with empty vehicle solution as a control. Data are representative of two independent experiments. F, Various populations of splenocytes on days 4 and 14 are shown and expressed as the ratio to the total numbers of transplanted T-cells (0.5 × 106). CD4+ and CD8+ T-cells are gated on donor-derived cells. TN, naïve T-cells; TE, effector T-cells. Mean values and SEM are presented (n = 5). *, P < 0.05; **, P < 0.01. G, Survival curve and BW changes (n = 5–10). ***, P < 0.001. H, Plasma samples were analyzed at 30 minutes, 1, 2, 4, 6, and 16 hours after intraperitoneal administration (12 mg/kg) of c-Rel inhibitor compound IT-603. To assess the level of IT-603 in blood, samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS; for more details, see METHODS).
Table 1.
Characteristics of c-Rel inhibitor compounds
| M.W. | Structure | IC50, EMSA | Tumor cell growth inhibition in vitroa (IC50) | Optimum conc. for in vitro T-cell treatment | Duration of inhibitory effectb | Plasma half-lifec | |
|---|---|---|---|---|---|---|---|
| Dexamethasone | 392.46 |
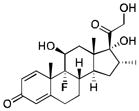
|
N/A | > 20μM | |||
| DHMEQ | 261.23 |
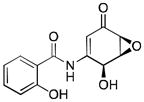
|
40μM | 7μM | |||
| IT-603 | 329.16 |
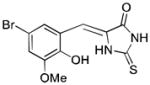
|
3μM | 18μM | 20μM for 24 hrs | < 96 hrs | 2.25 hrs |
Human diffuse large B-cell lymphoma (DLBCL) cell line Ly3 was used.
CD5+ splenocytes were treated with IT-603 for 24 hours and transferred to lethally irradiated recipients. Analysis of c-Rel activity was performed by flow cytometry.
Plasma samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at various time points after 12 mg/kg intraperitoneal injection of IT-603.
DHMEQ, dehydroxymethylepoxyquinomicin; M.W., molecular weight; IUPAC, International Union of Pure and Applied Chemistry; IC50, inhibitory concentration 50; conc., concentration; N/A, not applicable.
We next tested the capacity of IT-603 pre-treated T-cells to induce GVHD. By day 4 after allo-HSCT, T-cells pre-treated with IT-603 expanded less compared to T-cells treated with empty vehicle (DMSO) (Fig. 4F) and even though the inhibitory effect of the compound was temporary and c-Rel activity returned to normal by day 4 after allo-HSCT (data not shown), those T-cells caused significantly reduced GVHD (Fig. 4G). We also observed higher IL-2 secretion from donor CD4+ T-cells on day 4 in this experiment (Supplementary Fig. S4). Of note, we recently developed a DMSO-free lipid-based formulation of IT-603 using the FDA-approved non-ionic surfactant cremophor. Intraperitoneal injection of this formulation was well tolerated and allowed us to study pharmacokinetics (Table 1 and Fig. 4H), in a first step toward development of a c-Rel inhibitor drug.
Treatment of T-cells With a c-Rel Inhibitor Compound Does Not Compromise Anti-tumor Activity
A recent study found evidence indicating that in recipients of c-Rel−/− T-cells GVT activity against A20-TGL mouse lymphoma cells can be intact (10). We confirmed these findings and were, moreover, able to demonstrate that this effect can be sustained with T cell doses as low as 25% of the standard dose (data not shown), and when targeting less immunogenic A20 tumor cells as well as a solid tumor RENCA (renal cell carcinoma, data not shown).
In addition, c-Rel−/− T-cells displayed strong anti-tumor activity against EL4-TGL T-cell lymphoma cells even in the absence of T-cell alloactivation (Fig. 5A). In this syngeneic model, the level of CD25 on c-Rel−/− CD8+ T-cells on day 7 after transplantation was similar to that of WT T-cells (Fig. 5B), in striking contrast to the allo-HSCT setting, where CD25 levels in c-Rel−/− T-cells were significantly decreased (Fig. 5C). We performed a similar experiment using B16-TGL melanoma tumor cells and melanoma-specific syngeneic donor T-cells from Pmel1+/+ mice after incubating those T-cells with the c-Rel inhibitor compound. Pmel1+/+ T-cells retained normal GVT activity under c-Rel-deficient conditions (Fig. 5D), indicating that inhibition of c-Rel activity is not sufficient to prevent antigen-specific TCR activation. To further dissect the role of c-Rel in antigen-specific versus alloactivation of T-cells, we analyzed the expression levels of c-Rel in Pmel1+/+ T-cells stimulated by B16 tumor cells and compared it with that of WT C57BL/6 T-cells stimulated with MHC-mismachted splenocytes (Fig. 5E and F). We found significantly lower levels of c-Rel in Pmel1+/+ T-cells specifically stimulated with target antigen, suggesting that c-Rel activity is less required for antigen-specific T-cell activation. Moreover, T-cell stimulation in the presence of the c-Rel inhibitor compound resulted in reduced c-Rel activity only when allogeneic stimulators were used (Fig. 5E).
Figure 5.

Pre-treatment of donor T-cells with a c-Rel antagonist does not impair GVT activity. A, Lethally irradiated C57BL/6 recipients received C57BL/6 TCD BM cells with 2 × 106 C57BL/6 WT or c-Rel−/− T-cells (syngeneic HSCT). Control mice received BM only. All groups received 1 × 105 luciferase-expressing EL4-TGL tumor cells on day 0. Survival curve is shown (n = 5–8). B, Lethally irradiated C57BL/6 recipients received 5 × 106 C57BL/6 WT or c-Rel−/− T-cells (syngeneic HSCT). All groups received 1 × 106 EL4 tumor cells on day 0. Expression level of CD25 in donor-derived splenic CD8+ T-cells on day 7 is shown. C, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with 2 × 106 C57BL/6 WT or c-Rel−/− T-cells (allo-HSCT). Expression level of CD25 in donor-derived splenic CD8+ T-cells on day 7 is shown. D, Lethally irradiated C57BL/6 recipients received C57BL/6 TCD BM cells with 1 × 106 C57BL/6 Pmel1+/+ T-cells (syngeneic HSCT). T-cells were pre-treated with a c-Rel inhibitor compound IT-603 or with a control vehicle. Control mice received BM only. All groups received 1 × 105 luciferase-expressing B16-TGL tumor cells on day 0. Survival curve is shown (n = 5–8). Data are representative of at least two independent experiments and mean values and SEM are presented (n = 5) in A–D. *, P < 0.05. E, 1 × 106 Pmel1+/+ splenocytes (unmodified, DMSO treated, or IT-603 treated) were cultured in vitro for 32 hours with 2 × 105 B16 tumor cells to induce antigen-specific T-cell stimulation. 1 × 106 C57BL/6 splenocytes (unmodified, DMSO treated, or IT-603 treated) were cultured in vitro for 24 hours with 2 × 105 BALB/c splenocytes to induce allostimulation of T-cells. Unmodified non-stimulated C57BL/6 splenocytes were used for analyzing control c-Rel expression. CD3+ splenocytes were analyzed for c-Rel expression by flow cytometry. Mean values and SEM are presented (n = 4). *, P < 0.05. F, 1 × 106 Pmel1+/+ splenocytes were cultured in vitro for 24 hours with 2 × 105 B16 tumor cells to induce antigen-specific T-cell stimulation. 1 × 106 C57BL/6 splenocytes were cultured in vitro for 24 hours with 2 × 105 BALB/c splenocytes to induce allostimulation of T-cells. Golgi Plug was added at 27 hours of incubation and cells were harvested at 32 hours, followed by flow cytometric analysis with intracellular c-Rel staining (n = 4). G–I, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with 0.5 × 106 C57BL/6 WT T-cells after 24 hours of pre-treatment with a c-Rel inhibitor IT-603. Control mice received C57BL/6 TCD BM cells with 0.5 × 106 C57BL/6 WT T-cells that were pre-treated with empty vehicle control solution. On day 0, HSCT recipients were challenged with 0.25 × 106 luciferase-expressing A20-TGL tumor cells. Data are combined from two independent experiments (n = 3–10). G, Survival curve. *, P < 0.05 versus BM + T + A20-TGL group. H, BW changes. I, The whole-body distribution of tumor cells was monitored using in vivo BLI. Pseudocolor images superimposed on conventional photographs are shown. Data are representative of two independent experiments. J, Lethally irradiated BALB/c recipients were transplanted with C57BL/6 TCD BM cells with 1 × 106 C57BL/6 WT T-cells. c-Rel inhibitor compound IT-603 or control vehicle solution was administered daily from day 10 after allo-HSCT (24 mg/kg, intraperitoneally). Survival curve is shown. *, P < 0.05. K, Human cytomegalovirus (CMV), Wilms tumor 1 (WT1), Epstein-Barr Virus (EBV) specific T-cells are treated with c-Rel inhibitor compound IT-603 for 24 hours in vitro. Viabilities of those T-cells before and after treatment and expressions of c-Rel and IL-2 are shown. Data are representative of more than three independent experiments. L, Human cytomegalovirus (CMV) specific cytotoxic T lymphocytes were treated with c-Rel inhibitor compound IT-603 or control solution for 24 hours in vitro and co-incubated for 8 hours with the following 51Cr-labeled target cells; autologous dendritic cells (DC), autologous DC loaded with CMV peptide (DC/CMV), or HLA mismatched B cells (Allo). Death of target cells was measured by 51Cr release. M, Human peripheral blood mononuclear cells (PBMC) were stimulated on day 0 with either autologous EBV transformed B-cell line (EBV BLCL) or allogeneic HLA mismatched BLCL after co-incubation with the inhibitor compound IT-603 or with control solution (DMSO). PBMC and stimulators were plated at serial dilutions and cultured in the presence of IL-2 for 14 days. IT-603 and DMSO treatment were repeated on day 7 of co-culture. Alloreactivity of human T-cells as well as EBV-specific T-cell activation were analyzed by a 51Cr release cytotoxicity assay on day 15 (see METHODS for more details). Samples were analyzed in multiple replicates in L and M.
Importantly, WT T-cells pre-treated with a c-Rel antagonist mediated GVT activity in several tumor models (Fig. 5D and G–I), indicating that viable and functional T-cells are present. It is therefore highly likely that the diminished capacity of these T-cells to induce GVHD is due to the c-Rel inhibitory mechanism of the small molecule compound and not simply due to a non-specific effect of the in vitro manipulation procedure. However, we did observe that GVT activity of WT T-cells treated with inhibitor compound was slightly less than the GVT activity of c-Rel−/− T-cells, and we found moderate reduction in antigen-specific T-cell activation (but not alloactivation) in vitro in the presence of vehicle alone (Supplementary Fig. S5). As a result of our ongoing efforts to establish less toxic compound formulations we recently succeeded in developing a regimen for systemic administration of the inhibitor compound as GVHD therapy, and we were able to establish in vivo efficacy (Fig. 5J). While administration of empty vehicle was not associated with any signs of toxicity, daily intraperitoneal administration of c-Rel inhibitor solution at a dose of 24 mg/kg for more than two weeks resulted in mild diarrhea and ruffled fur. Importantly, c-Rel deficiency (as a result of c-Rel inhibitor administration or as a result of transplantation of c-Rel−/− BM) did not have a negative impact on hematopoiesis and immune reconstitution (Fig. 1D and data not shown).
Our inhibitor compound effectively inhibited c-Rel activity of human T-cells (Fig. 5K). Moreover, in vitro cytotoxicity analysis of human CMV-specific T-cells (30) (Fig. 5L) as well as Wilms tumor 1 (WT1), and Epstein-Barr Virus (EBV) specific T-cells (Fig. 5M and data not shown) demonstrated that inhibition of c-Rel activity did not impair antigen-specific TCR-mediated killing. Alloreactivity of human T-cells was on the other hand dramatically reduced when human peripheral blood mononuclear cells (PBMC) were cultured with HLA-mismatched stimulators in the presence of the inhibitor compound (Fig. 5M). These data reinforce the notion of separation of GVHD from GVT activity through inhibition of c-Rel activity even in human T-cells.
DISCUSSION
The NF-κB/Rel transcription factor family is composed of five members of interacting proteins, c-Rel, p50, p65, p52 and RelB. c-Rel is involved in the pathway downstream of antigen-stimulated canonical NF-κB signal transduction and is crucial for T-cell proliferation and differentiation (7, 8). Studies in mouse models revealed unique roles for c-Rel in the pathophysiology of allergic reactions, autoimmunity, and allogeneic transplantation (10, 11, 31, 32). Our study revealed that deficiency of c-Rel in donor T-cells ameliorated GVHD due to impaired alloactivation and proliferation of TE, decreased homing of donor c-Rel−/− TE to the small intestine, as well as increased Treg-mediated suppression.
Surprisingly, levels of IL-2, one of the key target genes of c-Rel, were increased in recipients of c-Rel−/− T-cells on day 7 after allo-HSCT. We identified a feedback mechanism that resulted in reduced downregulation of IL-2 production by c-Rel−/− T-cells during periods of T-cell activation in the setting of acute GVHD. In addition to the relative increase of IL-2 at a critical time period during early GVHD, we also found that serum levels of Th2 type cytokines, IL-4, IL-5, IL-13 were significantly increased on day 7 after allo-HSCT, and GATA-3 expression of c-Rel−/− T-cells was increased while expression of T-bet was decreased (Supplementary Fig. S6), suggestive of Th2 polarization in recipients of c-Rel−/− T-cells (12, 13). The availability of optimal IL-2 levels in the absence of competition by effector T-cells resulted in expansion of c-Rel-deficient natural Tregs and, more importantly, induction of Tregs. These findings can certainly be considered counterintuitive, given the role of c-Rel for IL-2 as well as Treg generation under physiologic conditions. However, our experiments reveal a differential outcome of the IL-2/STAT5 pathway in wild-type compared with c-Rel deficient T-cells during GVHD. In the setting of GVHD, pSTAT5 in c-Rel deficient donor T-cells is decreased, which over time leads to increased IL-2 levels (due to decreased negative feedback), and renders TE at the same time non-competitive (due to decreased CD25 expression), while allowing Tregs, a T-cell population that expresses CD25 by definition, to outcompete TE. Our findings as well as a recently described similar concept of competition for IL-2 between TE and Tregs (23, 24) indicate that this mechanism may indeed have significant implications for alloactivation and GVHD, and it will be an important line of future research to evaluate the kinetics of TE and Tregs in response to interventions modulating the IL-2 pathway (33). Reported roles of c-Rel in T-cell responses and transplantation immunology in comparison with our findings during GVHD are summarized in Table 2.
Table 2.
Roles of c-Rel in T-cell responses and transplantation immunology
| c-Rel−/− mice (steady state/autoimmune/infection) | Reference # | GVHDa (c-Rel−/− or inhibitor treated donor T-cells) | |
|---|---|---|---|
| Th1 | defective~normal | #12, #13 | decreased |
| Th2 | normal | #12, #13 | increased |
| Th17 | defective | #12, #19 | no change |
| nTreg | defective | #14, #15, #16, #17 | increased |
| iTreg | defective | #17 | increased |
| Graft survival | prolonged | #31, #32 | N/A |
| GVHD survival | prolonged (donor T-cells) | #10 | prolonged |
| IL-2 | defective (in vitro) | #7, #8 | increased |
| TE/TN ratio | decreased (in vitro) | #7, #8 | decreased |
| T-cell activation | defective (in vitro) | #7, #8 | decreased |
| T-cell proliferation | defective (in vitro) | #7, #8 | decreased |
nTreg, natural Treg; iTreg, induced Treg; TN, naïve T cells; TE, effector T cells; N/A, not applicable.
Summary of our findings.
Inhibition of c-Rel activity did not impair GVT activity, as demonstrated by various tumor models in the setting of allogeneic and syngeneic HSCT. This phenomenon was most evident when using c-Rel−/− T-cells as opposed to inhibitor compound treated T-cells, which can be attributed to expected potential limitations of any chemical strategy to inhibit protein function (such as limited duration of the effect, potential off-target effects, and non-specific toxicities). Furthermore, antigen-specific T-cell activation was associated with significantly less c-Rel expression than allostimulation. The redundancy of c-Rel activity for antigen-specific TCR activation provides an underlying molecular mechanism for the observed separation of GVHD from GVT activity: only upon antigen-specific activation (but not alloactivation) c-Rel-deficient T-cells can differentiate into activated effector T-cells that exhibit normal cytotoxicity responses. In addition, we found that in the setting of c-Rel deficiency Treg-mediated suppression of GVHD represents an important additive factor without equally affecting GVT activity. While it is possible that this phenomenon is more pronounced in experimental models where the kinetics of tumor eradication can be much different from real life scenarios, it is important to keep in mind that there is accumulating evidence that Tregs can preferentially impact on GVHD while preserving protective immunity (34, 35). Our findings in the setting of a MHC-matched allo-HSCT indicate that c-Rel activity is also involved in T-cell activation mediated by minor transplantation antigens. This and other important questions regarding the differential requirements for c-Rel activity for TCR triggering will need to be addressed in more depth in future studies, however our current evidence that inhibition of c-Rel activity separates GVHD from GVT activity at the level of TCR signaling has important clinical implications. Administration of donor lymphocyte infusions (DLI) containing high levels of allogeneic cytotoxic T lymphocyte precursors (allo-CTLp) in the early post-transplant period correlates with a high risk of GVHD while infusion of antigen-specific T-cells with low levels of allo-CTLp never causes GVHD. Using limited dilution analysis, we were able to show that pre-treatment of PBMC with our inhibitor compound prevents activation of alloreactive T-cells and results in a 20-fold decrease of allo-CTLp to levels comparable with allo-CTLp levels detected in EBV CTLs that are safely used for adoptive therapy of EBV lymphoproliferative disorders (36).
Two recent studies have validated possible uses of a proteasome inhibitor and an IκB kinase inhibitor to target NF-κB (37, 38). These pan-NF-κB inhibitors have broad effects on the downstream signaling pathways and have significant potential to cause serious adverse effects such as increased radiation-induced epithelial damage (39). However, since c-Rel activity is restricted to a small number of hematopoietic lineages, a c-Rel-specific inhibitor will likely have a better safety profile than a pan-NF-κB inhibitor. Indeed, we have evidence that systemic administration of the c-Rel inhibitor compound discovered by us is feasible, safe, and effective. Furthermore, Rel/NF-κB factors are also known for their roles as proto-oncogenes by contributing to tumor growth, survival, drug resistance, and metastasis of lymphoid malignancies, breast, head, and neck cancers (9, 40). We found in a preliminary experiment that intraperitoneal c-Rel inhibitor compound administration displayed antineoplastic activity in a xenograft model of human diffuse large B-cell lymphoma (data not shown).
In conclusion, our data provide for the first time evidence for a differential role of c-Rel for alloactivation versus antigen-specific T-cell activation: while c-Rel activity is critically important for T-cell activation during GVHD, it is dispensable for antigen-specific TCR activation. As a result, inhibition of c-Rel activity reduces the severity of GVHD without compromising anti-tumor activity of T-cells. Our findings validate c-Rel as a highly promising therapeutic target, and we demonstrate biological benefits of inhibition of c-Rel activity in both mouse and human T-cells with a highly specific small molecule compound. Drug development studies are currently underway in an effort to translate this technology from bench to bedside.
METHODS
Mice and Bone Marrow Transplantation
We obtained female C57BL/6 (B6, H-2b), LP/J (H-2b), B10.BR (H-2k), BALB/c (H-2d) from the Jackson Laboratory. B6 mice carrying the c-Rel gene null mutation (c-Rel−/−) were originally generated by inserting the neomycin cassette into the fifth exon of the c-Rel gene (7). c-Rel−/− B6 mice and FoxP3-DTR transgenic B6 mice that express the diphtheria toxin receptor under control of the FoxP3 promoter (27) as well as Pmel1+/+ transgenic B6 mice (41) were maintained at Memorial Sloan-Kettering Cancer Center in accordance with Institutional Animal Care and Use Committee Standards. Mice used for experiments were 6–9 weeks old. Mouse HSCT experiments were performed as previously described (42), with 850 cGy split-dosed lethal irradiation of BALB/c recipients transplanted with bone marrow (5 × 106), T-cell depleted (TCD) with anti-Thy-1.2 and low-TOX-M rabbit complement (Cedarlane Laboratories), or with 1100cGy split-dosed lethal irradiation of B6 or LP recipients transplanted with TCD bone marrow (5 × 106) as well. Donor T-cells were prepared by harvesting donor splenocytes and enriching T-cells by Miltenyi MACS purification of CD5 (routinely >90% purity). For the Treg transfer experiment, highly enriched (routinely >90% purity) CD4+CD25+ T-cells were obtained by positive selection with the Miltenyi MACS magnetic sorting system. In GVT experiments, animals received tumor cells intravenously in a separate injection on day 0.
Small Molecule c-Rel Inhibitor Compounds
Pyrimidinetrione derivatives were previously identified as small molecule c-Rel inhibitor compounds (20, 21). c-Rel inhibitory activity of the compounds was confirmed by fluorescence polarization (FP) as well as electrophoretic mobility shift assay (EMSA) utilizing the DNA-binding property of the c-Rel protein. Detailed methods for these analyses were described previously (7, 20, 43). Unless otherwise indicated, we used IT-603 ((5Z)-5-[(5-bromo-2-hydroxy-3-methoxyphenyl)methylidene]-2-sulfanylideneimidazolidin-4-one, MW = 329.1 g/mol, obtained from ChemDiv) by incubating cells with the compound for 24 hours at a concentration of 20 μM. For high-throughput screening we used a fluorescence polarization (FP) assay that utilizes the DNA-binding property of the c-Rel protein. Specifically, c-Rel binds with high affinity to the CD28 responsive element (CD28RE) in the IL-2 promoter. The differential signals of free CD28RE versus bound CD28-Rel complex was used to screen for compounds that disrupted CD28-Rel interaction.
Assessment of GVHD and GVT; in vivo BLI and cell lines
Mice were monitored daily for survival and weekly for GVHD clinical scores (44). Small intestine, large intestine, liver, and skin samples were evaluated histologically for evidence of GVHD and scored as previously described (45). In GVT experiments, we determined the bioluminescent signal intensity (BLI) of tumor bearing mice twice weekly as described previously (46). We superimposed pseudocolor images showing the whole-body distribution of bioluminescent signal intensity on grayscale photographs and determined total flux (photons s−1) for individual mice. We determined the cause of death (tumor versus GVHD) by necropsy and histopathology as previously described (45). All cell lines used in our experiments were originally obtained from the American Type Culture Collection (ATCC; in 2006). All cells were maintained and propagated according to the recommendations of ATCC and were validated as mycoplasma-negative. All cell lines underwent authentication testing at ATCC; in addition, the histological origin of tumors derived from cancer cell lines in our animal experiments was confirmed by histopathology.
Serum Cytokines Analyses
Blood was collected into microcentrifuge tubes, allowed to clot and centrifuged, and the supernatant was collected. Multiplex ELISA was conducted per manufacturer’s instructions (Millipore). Results were acquired with a Luminex 200 instrument and analyzed with xPONENT software (Luminex Corporation).
Antibodies and Flow Cytometry
All antibodies other than the anti-c-Rel antibody (Santa Cruz Biotechnology) and the anti-CD44 antibody (Biolegend) were obtained from BD Biosciences – Pharmingen. For cell analysis of surface markers, cells were stained for 20 minutes at 4°C in PBS with 0.5% BSA (PBS/BSA) after Fc block, washed, and resuspended in DAPI in PBS/BSA. IL-2 secretion analyses were performed using mouse the IL-2 secretion assay kit per manufacturer’s instructions (Miltenyi Biotech). c-Rel expression was analyzed by intracellular staining after cells were stimulated for 5 hours with Cell Stimulation Cocktail (eBioscience) unless otherwise indicated. Cell surface staining was followed by intracellular staining with the eBioscience kit per manufacturer’s instructions. Dead cells were excluded with LIVE/DEAD Fixable Dead Cell Stain kit (Invitrogen). Intracellular phospho-STAT5 was detected as described (24). Briefly, mice were sacrificed instantaneously and splenocytes were exposed to different concentrations of mouse IL-2 for 10 minutes at 37°C, followed by fixation with 1.6% PFA, and permeabilization with 90% methanol. During this process, membrane-bound IL-2 was stripped from cell surface by a 2-minute incubation with 0.1 M Glycine buffer equilibrated at pH 4.0, followed by a 5-minute wash in RPMI before the exposure to exogenous IL-2. All flow cytometry was performed on an LSR II (BD Biosciences) and analyzed with FlowJo (TreeStar Software).
Pharmacokinetics
Plasma samples were analyzed at 30 minutes, 1, 2, 4, 6, and 16 hours after intraperitoneal administration (12 mg/kg) of c-Rel inhibitor compound IT-603. To assess the level of IT-603 in blood, samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) as previously described (47, 48). Calibration curves were determined for IT-603 to permit conversion of peak areas to the drug amounts against external reference standards. The tandem MS/MS detector (Model ABI/Sciex API 4000, Applied Biosystem, Foster City, California) permitted verification of peak identity as well as a quantitative assessment of the compounds in the samples.
Human T-cell Cytotoxicity and Limited Dilution Assays
In vitro cytotoxicity analyses of human CMV-specific T cells as well as human Wilms tumor 1 (WT1) and human Epstein-Barr Virus (EBV) specific T cells were performed as previously described (30). In brief, CMV specific T cells were generated from PBMC of a healthy donor by repeated in vitro stimulations with an autologous EBV transformed B-cell line (EBV BLCL) loaded with a pool of CMVpp65-derived pentadecapeptides overlapping by 11 amino acids and including the entire sequence of the protein. T cells were tested on day 34 of culture in a standard 51Cr release assay against a panel of targets including autologous dendritic cells without CMVpp65 peptide, autologous dendritic cells loaded with a CMVpp65 peptide, and fully HLA mismatched allogeneic BLCL. The effector target ratio was 25:1. We compared unmodified T cells with control-treated and compound-treated T cells (pre-treatment for 24 hours in media containing IT-603 compound at 20 μM).
The frequency of EBV-specific T cells and alloreactive T cells were measured in the PBMC from a normal EBV seropositive donor by limited dilution analysis (LDA) as previously described (36). Briefly, the aliquots of PBMC were stimulated on day 0 with either autologous EBV BLCL or allogeneic HLA mismatched BLCL after co-incubation with the inhibitor compound IT-603 or with the control solution (DMSO). The stimulated PBMC were plated at serial dilutions and cultured in the presence of IL-2 for 14 days. The CTL precursor frequencies were measured in a 51Cr assay against either autologous BLCL or allogeneic HLA mismatched BLCL.
HLA type of the PBMC donor: A0201 A3601 B 44WRJ B1510 C0501 C04JERF DRB1 0401 DRB1 0301 DQB1 0301 DQB1 0201
HLA type of the HLA mismatched BLCL: A2601 B 0801 C0701 DRB1 1501 DQB1 0602 (homozygous by all alleles)
Statistics
Data are presented as mean ± SEM. Survival data were analyzed with the Mantel-Cox log-rank test. For nonsurvival pointwise analyses, unpaired t test was used for comparisons between two experimental groups, or nonparametric Mann-Whitney U test was used for non-Gaussian distributions, and ANOVA was used for comparisons of more than two groups. All statistical analyses were performed using GraphPad Prism 5 (La Jolla, CA). A P value of less than 0.05 was considered statistically significant.
Supplementary Material
SIGNIFICANCE.
Chemical inhibition of c-Rel diminishes alloactivation while preserving antigen-specific T-cell receptor activation, revealing redundancy of c-Rel in T-cell mediated anti-tumor activity of both mouse and human T-cells. Our study provides a highly innovative immunomodulatory approach that has true potential for drug development and clinical application with broad therapeutic implications including allo-tolerance induction after allo-HSCT, as well as anti-tumor therapies.
Acknowledgments
Grant Support
This work was supported by National Institutes of Health award numbers R01-HL069929 (to M.R.M. van den Brink), R01-AI100288 (to M.R.M. van den Brink), R01-AI080455 (to M.R.M. van den Brink), R01-AI101406 (to M.R.M. van den Brink), and 1K08CA160659-01 (to J.L. Zakrzewski). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Support was also received from the Radiation Effects Research Foundation (RERF-NIAID) (to M.R.M. van den Brink), and The Uehara Memorial Foundation (to Y. Shono), Lymphoma Research Foundation Post-Doctoral Fellowship Research Grant (to Y. Shono), MSKCC Center for Molecular Imaging and Nanotechnology (CMINT), The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center funded by Mr. William H. Goodwin and Mrs. Alice Goodwin, The Lymphoma Foundation, Alex’s Lemonade Stand, The Geoffrey Beene Cancer Research Center at Memorial Sloan-Kettering Cancer Center, and The Peter Solomon Fund.
We appreciate the invaluable help of the Laboratory of Comparative Pathology, the Analytical Pharmacology, and the Molecular Cytology Core Facility (supported by Cancer Support Grant NCI P30-CA008748) of Memorial Sloan-Kettering Cancer Center. T. Merghoub and A.Y. Rudensky (Memorial Sloan-Kettering Cancer Center) provided PMEL1 and FoxP3-DTR mice. We thank E. Velardi, J.A. Dudakov, A.M. Hanash, R.R. Jenq, L.F. Young, A. Ghosh, A.M. Holland, and T. Chinen for helpful discussion.
Footnotes
Disclosure of Potential Conflicts of Interest
HCL is a co-inventor of IT-603 and the CEO of ImmuneTarget, Inc. JLZ is a scientific advisor for ImmuneTarget, Inc. No potential conflicts of interest were disclosed by the other authors.
Author’s Contributions
Conception and design: Y. Shono, J.L. Zakrzewski
Development of methodology: Y. Shono
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Y. Shono, A.Z. Tuckett, J.J. Tsai, O.M. Smith, M.L. West, N.V. Singer, D. Pankov, C.V. Undhad, S. Ouk, H. Liou, E. Doubrovina, R.J. O’Reilly
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Y. Shono, G. Altan-Bonnet, J.E. Oyler, G.F. Murphy, C. Lezcano, C. Liu
Writing, review, and/or revision of the manuscript: Y. Shono, J.L. Zakrzewski, M.R.M. van den Brink
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): Y. Shono
Study supervision: J.L. Zakrzewski, M.R.M. van den Brink
References
- 1.Appelbaum FR. The current status of hematopoietic cell transplantation. Annu Rev Med. 2003;54:491–512. doi: 10.1146/annurev.med.54.101601.152456. [DOI] [PubMed] [Google Scholar]
- 2.RRJ, vdB MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010;10:213–21. doi: 10.1038/nrc2804. [DOI] [PubMed] [Google Scholar]
- 3.JLF, RL, NJC Pathophysiologic mechanisms of acute graft-vs-host disease. Biol Blood Marrow Transplant. 1999;5:347–56. doi: 10.1016/s1083-8791(99)70011-x. [DOI] [PubMed] [Google Scholar]
- 4.Wingard JR, Majhail NS, Brazauskas R, Wang Z, Sobocinski KA, Jacobsohn D, et al. Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2230–9. doi: 10.1200/JCO.2010.33.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MNB, DR, WJM Immunotherapy following hematopoietic stem cell transplantation: potential for synergistic effects. Immunotherapy. 2010;2:399–418. doi: 10.2217/imt.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowler DH. Shared biology of GVHD and GVT effects: potential methods of separation. Critical reviews in oncology/hematology. 2006;57:225–44. doi: 10.1016/j.critrevonc.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. 1999;11:361–71. doi: 10.1093/intimm/11.3.361. [DOI] [PubMed] [Google Scholar]
- 8.Visekruna A, Volkov A, Steinhoff U. A key role for NF-kappaB transcription factor c-Rel in T-lymphocyte-differentiation and effector functions. Clinical & developmental immunology. 2012;2012:239368. doi: 10.1155/2012/239368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fullard N, Wilson CL, Oakley F. Roles of c-Rel signalling in inflammation and disease. The international journal of biochemistry & cell biology. 2012;44:851–60. doi: 10.1016/j.biocel.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y, Wang D, Kaosaard K, Liu C, Fu J, Haarberg K, et al. c-Rel is an essential transcription factor for the development of acute graft-versus-host disease in mice. Eur J Immunol. 2013 doi: 10.1002/eji.201243282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liou HC, Hsia CY. Distinctions between c-Rel and other NF-kappaB proteins in immunity and disease. Bioessays. 2003;25:767–80. doi: 10.1002/bies.10306. [DOI] [PubMed] [Google Scholar]
- 12.Reinhard K, Huber M, Wostl C, Hellhund A, Toboldt A, Abass E, et al. c-Rel promotes type 1 and type 17 immune responses during Leishmania major infection. Eur J Immunol. 2011;41:1388–98. doi: 10.1002/eji.201041056. [DOI] [PubMed] [Google Scholar]
- 13.Hilliard BA. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. Journal of Clinical Investigation. 2002;110:843–50. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grigoriadis G, Vasanthakumar A, Banerjee A, Grumont R, Overall S, Gleeson P, et al. c-Rel controls multiple discrete steps in the thymic development of Foxp3+ CD4 regulatory T cells. PLoS One. 2011;6:e26851. doi: 10.1371/journal.pone.0026851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guckel E, Frey S, Zaiss MM, Schett G, Ghosh S, Voll RE. Cell-intrinsic NF-kappaB activation is critical for the development of natural regulatory T cells in mice. PLoS One. 2011;6:e20003. doi: 10.1371/journal.pone.0020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vang KB, Yang J, Pagan AJ, Li LX, Wang J, Green JM, et al. Cutting edge: CD28 and c-Rel-dependent pathways initiate regulatory T cell development. J Immunol. 2010;184:4074–7. doi: 10.4049/jimmunol.0903933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–40. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan Q, Chen YH. Nuclear factor-kappaB in immunity and inflammation: the Treg and Th17 connection. Adv Exp Med Biol. 2012;946:207–21. doi: 10.1007/978-1-4614-0106-3_12. [DOI] [PubMed] [Google Scholar]
- 19.Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J Exp Med. 2011;208:2321–33. doi: 10.1084/jem.20110462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouk S, Liou ML, Liou HC. Direct Rel/NFkB inhibitors: structural bassis for mechanism of action. Future Med Chem. 2009;1:1683–707. doi: 10.4155/fmc.09.96. [DOI] [PubMed] [Google Scholar]
- 21.Liou HC. Methods and compositions for targeting c-Rel. United States Patent Application Publication. 2010
- 22.Doan LL, Tanner MK, Grimes HL. Intranuclear staining of proteins in heterogeneous cell populations and verification of nuclear localization by flow cytometric analysis. Journal of Immunological Methods. 2003;279:193–8. doi: 10.1016/s0022-1759(03)00184-4. [DOI] [PubMed] [Google Scholar]
- 23.Hofer T, Krichevsky O, Altan-Bonnet G. Competition for IL-2 between Regulatory and Effector T Cells to Chisel Immune Responses. Frontiers in immunology. 2012;3:268. doi: 10.3389/fimmu.2012.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feinerman O, Jentsch G, Tkach KE, Coward JW, Hathorn MM, Sneddon MW, et al. Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Molecular systems biology. 2010;6:437. doi: 10.1038/msb.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. 2012;209:1723–42. S1. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209:1713–22. S1–19. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature immunology. 2007;8:191–7. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 28.Deenick EK, Elford AR, Pellegrini M, Hall H, Mak TW, Ohashi PS. c-Rel but not NF-kappaB1 is important for T regulatory cell development. Eur J Immunol. 2010;40:677–81. doi: 10.1002/eji.201040298. [DOI] [PubMed] [Google Scholar]
- 29.Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009;206:3001–14. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trivedi D, Williams RY, O’Reilly RJ, Koehne G. Generation of CMV-specific T lymphocytes using protein-spanning pools of pp65-derived overlapping pentadecapeptides for adoptive immunotherapy. Blood. 2005;105:2793–801. doi: 10.1182/blood-2003-05-1433. [DOI] [PubMed] [Google Scholar]
- 31.Yang H, Thomas D, Boffa DJ, Ding R, Li B, Muthukumar T, et al. Enforced c-REL deficiency prolongs survival of islet allografts1. Transplantation. 2002;74:291–8. doi: 10.1097/00007890-200208150-00002. [DOI] [PubMed] [Google Scholar]
- 32.Finn PW, He H, Ma C, Mueller T, Stone JR, Liou HC, et al. Molecular profiling of the role of the NF-kappaB family of transcription factors during alloimmunity. J Leukoc Biol. 2002;72:1054–62. [PubMed] [Google Scholar]
- 33.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484:529–33. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–50. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 35.Zheng J, Liu Y, Liu Y, Liu M, Xiang Z, Lam KT, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Science translational medicine. 2013;5:168ra9. doi: 10.1126/scitranslmed.3004943. [DOI] [PubMed] [Google Scholar]
- 36.Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–56. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun K, Welniak LA, Panoskaltsis-Mortari A, O’Shaughnessy MJ, Liu H, Barao I, et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8120–5. doi: 10.1073/pnas.0401563101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vodanovic-Jankovic S, Hari P, Jacobs P, Komorowski R, Drobyski WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. 2006;107:827–34. doi: 10.1182/blood-2005-05-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egan LJ. I B-kinase -dependent NF- B activation provides radioprotection to the intestinal epithelium. Proceedings of the National Academy of Sciences. 2004;101:2452–7. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–47. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 41.Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity. 2012;37:339–50. doi: 10.1016/j.immuni.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liou HC, Sha WC, Scott ML, Baltimore D. Sequential induction of NF-kappa B/Rel family proteins during B-cell terminal differentiation. Molecular and cellular biology. 1994;14:5349–59. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Jr, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–9. [PubMed] [Google Scholar]
- 45.Petrovic A, Alpdogan O, Willis LM, Eng JM, Greenberg AS, Kappel BJ, et al. LPAM (alpha 4 beta 7 integrin) is an important homing integrin on alloreactive T cells in the development of intestinal graft-versus-host disease. Blood. 2004;103:1542–7. doi: 10.1182/blood-2003-03-0957. [DOI] [PubMed] [Google Scholar]
- 46.Zakrzewski JL, Kochman AA, Lu SX, Terwey TH, Kim TD, Hubbard VM, et al. Adoptive transfer of T-cell precursors enhances T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Nat Med. 2006;12:1039–47. doi: 10.1038/nm1463. [DOI] [PubMed] [Google Scholar]
- 47.Clarke JL, Pao W, Wu N, Miller VA, Lassman AB. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. Journal of neuro-oncology. 2010;99:283–6. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin W, Tao H, Chen Y, Chen Z, Wu N. Sensitive, accurate and simple liquid chromatography-tandem mass spectrometric method for the quantitation of amphotericin B in human or minipig plasma. Journal of chromatographic science. 2012;50:636–43. doi: 10.1093/chromsci/bms049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.