

Abstract
Current treatments for seizure emergencies, such as status epilepticus, include intravenous or rectal administration of benzodiazepines. While intranasal delivery of these drugs is desirable, the small volume of the nasal cavity and low drug solubility pose significant difficulties. Here, we prepared supersaturated diazepam solutions under physiological conditions and without precipitation, using a prodrug/enzyme system. Avizafone, a peptide prodrug of diazepam, was delivered with—Aspergillus oryzae (A.O.) protease, an enzyme identified from a pool of hydrolytic enzymes in assay buffer, pH 7.4 at 32°C. This enzyme converted avizafone to diazepam at supersaturated concentrations. In vitro permeability studies were performed at various prodrug/enzyme ratios using Madin-Darby canine kidney II-wild type (MDCKII-wt) monolayers, a representative model of the nasal epithelium. Monolayer integrity was examined using TEER measurement and the lucifer yellow permeability assay. Prodrug/drug concentrations were measured using HPLC. Enzyme kinetics with avizafone-protease mixtures revealed KM = 1,501 ± 232 μM and Vmax = 1,369 ± 94 μM/s. Prodrug-protease mixtures, when co-delivered apically onto MDCKII-wt monolayers, showed 2–17.6-fold greater diazepam flux (S = 1.3–15.3) compared to near-saturated diazepam (S = 0.7). Data for prodrug conversion upstream (apical side) and drug permeability downstream (basolateral side) fitted reasonably well to a previously developed in vitro two compartment pharmacokinetic model. Avizafone-protease mixtures resulted in supersaturated diazepam in less than 5 min, with the rate and extent of supersaturation determined by the prodrug/enzyme ratio. Together, these results suggest that an intranasal avizafone-protease system may provide a rapid and alternative means of diazepam delivery.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9596-5) contains supplementary material, which is available to authorized users.
KEY WORDS: avizafone enzyme activation, diazepam delivery, hydrophobic drugs, MDCK monolayers, rapid absorption, seizure emergencies, supersaturation
INTRODUCTION
Seizure emergencies such as status epilepticus (SE) involve frequent epileptic attacks lasting for at least 5 min without regaining consciousness. This condition may cause neuronal damage and hence must be treated without delay. Currently, intravenous (i.v.) benzodiazepenes such as diazepam (DZP), lorazepam, or midazolam, are the first-choice drugs for the treatment of SE (1,2). However, i.v. administration requires skilled labor and hospitalization, which are time-consuming processes, and often cause delay in the treatment. Rectal DZP has also been shown to be effective (3), but this route has poor patient compliance. In this regard, the nasal route is an attractive alternative, as intranasal midazolam has been shown to be reasonably safe with clinical efficacy comparable to rectal DZP (4). However, DZP is preferred over midazolam for intranasal delivery due to its high lipid solubility and longer elimination half life (midazolam, 1.8–6.4 h; DZP, 20–100 h) (5,6).
For the treatment of seizure emergencies, 10 mg of DZP must be delivered into the small volume of the nasal cavity (< 200 μL) resulting in a highly concentrated solution (~50 mg/mL) (7). This is difficult to achieve due to the poor water solubility of DZP (<50 μg/mL). However, rapid delivery of lipophilic molecules such as DZP can be made possible by using supersaturated solutions which, compared to saturated solutions, have higher chemical potentials and therefore provide an increased driving force for drug transport (8). Supersaturation has been obtained by sudden changes in pH (9–11), ionic strength (10,12), and use of solid dispersions (13–17).
Supersaturated solutions are thermodynamically unstable against crystallization, making them difficult to store for practical time durations. Therefore, it is preferable to produce supersaturation at the point of administration. For example, Hou et al. showed that addition of water to a saturated DZP-in-water/glycofurol solution creates supersaturated DZP solutions that are stable long enough to cross synthetic membranes several-times faster than saturated DZP (7). In vitro pharmacokinetic studies showed rapid drug absorption from these supersaturated solutions when administered intranasally; these formulations were intolerable to human subjects, however (6).
Recently, we prepared in situ supersaturated drug solutions by coadministering the prodrug form with an activating enzyme in a model system for the nasal mucosa. Supersaturated phenytoin solutions were prepared using fosphenytoin (prodrug) and alkaline phosphatase (enzyme). These prodrug/enzyme mixtures showed several fold faster absorption, without drug precipitation, through Madin-Darby canine kidney-wild type (MDCKII-wt) monolayers, a nasal epithelium model, compared to saturated phenytoin (18). Phenytoin was used in these studies since its prodrug and alkaline phosphatase enzyme are commercially available, and the enzymatic reaction is well understood. However, phenytoin is an unsuitable target for nasal delivery as an antiepileptic owing to its intermediate lipid solubility, poor permeability and hence high dosage requirement (100 mg of drug). Accordingly, there is a need to develop a prodrug/enzyme-based system for the efficient nasal delivery of highly lipophilic and potent antiepileptics such as DZP.
Avizafone (AVF) is an off-patent prodrug of DZP that is available in Europe. It is injected intramuscularly to treat seizures induced by organophosphorous nerve agents (19,20). When administered systemically, it is converted to DZP by endogenous enzymes. However, the precise activating enzymes for AVF are unknown.
In the present work, we have developed a prodrug/enzyme-based system for DZP using AVF as the prodrug. Screening studies were performed to identify an activating enzyme. In vitro permeation studies were performed on prodrug/enzyme mixtures prepared at various prodrug and enzyme concentrations. This system, when administered intranasally, is expected to cause rapid delivery of DZP for effective treatment of SE and other neurological emergencies.
MATERIALS AND METHODS
Materials
DZP, tolbutamide (internal standard), enzymes—butyrylcholinesterase (human, 900 U/mL, cat# B4186), dipeptidyl peptidase III (human, 772,727 U/mL, cat# D3571), protease (Aspergillus oryzae, 1474.47 U/mL, cat# P6110), and chemicals used for ‘assay buffer’ preparation were purchased from Sigma. Aminopeptidase N enzyme (human, 45 mU/mL, cat# 164605) was purchased from Millipore. Lucifer yellow, HPLC-grade methanol, acetonitrile, and water, were purchased from Fisher Scientific. Raw materials for AVF synthesis were purchased from Chem-Impex International Inc. (IL, USA). Cell culture reagents (Dulbecco’s modified Eagle’s medium (DMEM), antibiotics, and fetal bovine serum (FBS)) were purchased from Invitrogen. Madin-Darby canine kidney II-wild type cells (MDCKII-wt) cells were generously provided by Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam).
Synthesis and Characterization of Avizafone Dihydrochloride (AVF: Scheme I)
AVF (4) was produced as a dihydrochloride from 5-chloro-2-methyl-aminobenzophenone (1) and (S)-2-(2,6-bis(((benzyloxy)carbonyl)amino)hexanamido)acetic acid (2) employing a two-step procedure (Scheme 1) following a procedure described in a patent (21).
Scheme 1.

Synthesis of avizafone dihydrochloride salt
(S)-Dibenzyl (6-((2-((2-benzoyl-4-chlorophenyl)(methyl)amino)-2-oxoethyl)amino)-6-oxohexane-1,5-diyl)dicarbamate (3). A suspension of dipeptide 2 (3.123 g, 6.62 mmol, finely powdered in a mortar) in anhydrous 1,2-dimethoxyethane (100 mL) was placed under a nitrogen atmosphere and cooled to −20°C (dry ice-acetone bath). To this suspension were added N-methylmorpholine (728 μL, 6.62 mmol) and isobutyl chloroformate (863 μL, 6.62 mmol). The resulting mixture was stirred at −20°C for 1 h. Then the solution was added, in five batches (20 ml portions) through a syringe filter (to remove solids) over a period of 4 h, to a refluxing mixture of 1 (1.627 g, 6.62 mmol) in anhydrous 1,2-dimethoxyethane (100 ml). After refluxing the resulting solution overnight (16 h), the solvent was evaporated under reduced pressure. The resulting residue was dissolved in a small amount of CH2Cl2 and loaded onto an MPLC column containing silica gel (324 g). MPLC separation was performed with EtOAc:hexanes 2:1 (700 mL), then EtOAc:hexanes 3:1 (300 mL), and then EtOAc (1,700 mL). The fractions were collected after the EtOAc elution started. The fractions containing the product were combined and the solvent was evaporated under reduced pressure. Since the residue contained some starting material 2 in addition to the desired product 3, the residue was dissolved in a small amount of CH2Cl2 and filtered through a short (10 cm) column filled with Al2O3 using EtOAc as the eluent (700 ml). After solvent evaporation and drying the residue overnight on high vacuum, compound 3 was obtained in 40% (1.86 g) as orange foam. 1H NMR (400 MHz, CDCl3) δ: 7.28–7.75 (m, 18H, Ar), 5.01–5.05 (m, 4H, 2 CH2O), 4.20 (m, 1H), 3.64–3.71 (m, 1H), 3.84–3.89 (m, 1H), 2.96–3.20 (m, 5H), 1.30–1.87 (m, 6H, 3 CH2). 13C NMR (100 MHz, CDCl3) δ: 193.3, 171.4, 168.5, 156.5, 138.8, 138.4, 136.7, 136.3, 135.8, 134.6, 134.3, 132.4, 132.0, 131.0, 130.1, 130.08, 129.9, 128.9, 128.50, 128.45, 128.1, 128.0, 67.0, 66.6, 54.6, 42.0, 40.3, 37.5, 32.4, 29.4, 22.2, 22.0.
(S)-6-((2-((2-Benzoyl-4-chlorophenyl)(methyl)amino)-2-oxoethyl)amino)-6-oxohexane-1,5-diaminium chloride (4). To a stirring solution of 3 (1.08 g, 1.54 mmol) in dry CH2Cl2 (30 mL) under a nitrogen atmosphere, cooled to −70°C, was added a pre-cooled solution of BCl3 in CH2Cl2 (1.0 M, 50 mL). The mixture was stirred under anhydrous conditions at −70°C for 30 min and then allowed to warm slowly to room temperature overnight. The mixture was evaporated to dryness under reduced pressure, then fresh dry CH2Cl2 (30 mL) was added and the mixture was evaporated again to dryness. This operation was repeated two times with CH2Cl2 and then four times with MeOH (to remove B(OMe)3). The concentrated MeOH solution (10 mL) was then added to anhydrous diethyl ether (750 mL) with vigorous stirring. The solution was left overnight and a fine solid precipitated. The ether solution was decanted with a cannula (double needle transfer under vacuum) and the precipitate was washed with dry ether (3 × 10 mL), dissolved in distilled water (30 mL), shaken with EtOAc (3 × 20 mL) and separated in a separatory funnel. The aqueous solution was lyophilized for 65 h to furnish 58% (385 mg) of compound 4 as a cream-colored solid that was dried overnight in a vacuum desiccator over P2O5. 1H NMR (400 MHz, D2O) δ: 7.40–7.73 (m, 8H, Ar), 3.73–4.20 (m, 3H), 2.95–3.12 (m, 5H), 1.91 (m, 2H, CH2), 1.74 (m, 2H, CH2), 1.48 (m, 2H, CH2). 13C NMR (100 MHz, CDCl3) δ: 197.3, 196.8, 171.6, 170.1, 170.2 169.8, 169.73, 169.70, 139.5, 138.1, 137.1, 136.6, 135.8, 135.6, 134.9, 134.4, 133.2, 132.9, 132.8, 130.8, 130.3, 130.0, 129.5, 129.4, 129.0, 128.8, 128.4, 52.9, 41.8, 41.1, 40.9, 39.0, 37.7, 37.4, 30.23, 30.16, 26.3, 26.2, 21.2, 21.1, 21.0. MS (EI) m/z 431 (M + 1)+. HRMS calculated for C22H26ClN4O3 (M–H)+ 429.1693, found 429.1694. Purity by UPLC 96%. [α22D] = +19.3 ± 0.3 (c 1 in water) (22).
HPLC Method
Concentrations of the prodrug (AVF) and the parent drug (DZP) were obtained by HPLC (Beckman Coulter SYSTEM GOLD: solvent module 126, autosampler 508 and UV detector 166, with 32.0 Karat software). The solvent pump was connected to a Zorbax XDB Eclipse C18 (12.5 × 4.1 mm, 5.0 μm) guard column preceding a Zorbax XDB Eclipse C18 (50 × 2.1 mm, 1.8 μm) analytical column. Chromatographic separation was performed using potassium phosphate (KH2PO4) buffer/acetonitrile (73:27 v/v), pH 2.36 as the mobile phase, at 1 mL/min rate and a run time of 12 min. A 30-μL sample prepared in mobile phase containing 2.5 μg/mL tolbultamide (internal standard) was injected into the column and the chromatogram was obtained at 210 nm. Peak area ratios (drug peak area divided by the area of internal standard from the same injection) were converted to drug concentrations using standard calibration curves (separate for AVF and DZP). The method was validated as per FDA guidelines (23).
Equilibrium Solubility Studies of DZP
DZP (5 mg) was placed in a 4 mL screw-cap glass vial (n = 3) each containing 2 mL assay buffer, pH 7.4 (122 mM NaCl, 25 mM NaHCO3, 10 mM glucose, 10 mM HEPES, 3 mM KCl, 1.2 mM MgSO4, 1.4 mM CaCl2, and 0.4 mM K2HPO4). The vials were placed on an orbital shaker (Shellab, Cornelius, Oregon) at 25, 32 and 37°C for 48 h. Drug suspensions were centrifuged at 13,000 g for 20 min and the supernatant was transferred to a fresh glass vial after filtering through a 0.2-μm membrane. The samples were then analyzed using HPLC.
Preparation of Supersaturated Solutions
Supersaturated solutions of DZP were prepared by incubating the prodrug, AVF, at equivalent molar concentrations, with a small amount of enzyme, in assay buffer pH 7.4. The “supersaturation potential i.e. AVF molar equivalent to supersaturated DZP”, S, was defined as
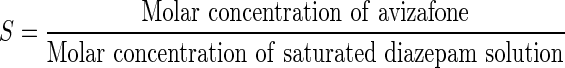 |
Enzyme Screening
To identify the enzyme for activation of AVF, various commercially available esterases/proteases/peptidases (butyrylcholinestease, dipeptidyl peptidase III, aminopeptidase N, protease) were screened. Enzymes at different concentrations (0.25–2.00 U/mL) were incubated with AVF in assay buffer, pH 7.4 in a transparent 96-well plate (Corning, USA) which was placed in an orbital shaker for 10 min at 32°C. At times 0 and 10 min, sample absorbance was noted at 240 nm using a microplate reader (Synergy HT, Biotek Instruments, USA). Enzyme, AVF, DZP, DZP+ enzyme, and blank assay buffer were used as controls. These experiments were performed in duplicate.
Enzyme Kinetics
To evaluate the effect of enzyme concentration on reaction kinetics, the best performing enzyme from the results of screening studies was incubated with AVF (130 μM, S = 1) at different enzyme concentrations (0.125–4 U/mL), in a 1 mL quartz cuvette containing assay buffer, pH 7.4 at 32°C. Absorbance was measured from 0 to 30 min at 316 nm (Cary 100 Bio UV-Vis spectrophotometer with CaryUV software, v.3.0). Enzyme, AVF, DZP, DZP + enzyme, and blank assay buffer were used as controls. These experiments were performed in duplicate.
To evaluate the effect of substrate concentration, 0.25 U/mL enzyme was incubated with various concentrations of AVF (69–3,601 μM, S = 0.5–27.6) in pre-warmed assay buffer, pH 7.4 (1 mL volume). Aliquots (100 μL) were withdrawn and placed immediately in clean glass vials (one for each time point—time 0 and 5 min) shaking at 32°C. At each time point, one vial was withdrawn, to which 900 μL methanol was added to serve as a reaction quencher. Samples were analyzed for AVF and DZP concentrations using HPLC. Blank buffer, enzyme, DZP, DZP+ enzyme, and AVF (no enzyme) were used as controls. The results were an average of three independent experiments. The averaged data was fitted to the Michaelis-Menten model to estimate the kinetic parameters using GraphPad Prism software (version 5.0).
Cell Culture
MDCKII-wt cells were cultured in DMEM media with 10% FBS and antibiotics (100 mg/ml streptomycin, 100 U/ml penicillin and 250 ng/ml amphotericin B) in T-25 flasks at 37°C, 5% CO2 atm. Confluent cells were trypsinized and seeded at 0.5 × 105 cells/mL in a 12-well Transwell plate (0.4 μm pore size, polyester, Corning). Medium was replaced every second day until a cell monolayer was observed (in 4–5 days). All MDCKII-wt cells utilized were between passages 10 and 20.
Membrane Permeability Studies
Permeability studies were performed according to the procedure published previously for prodrug/enzyme/drug systems (18). Briefly, prodrug (AVF) and enzyme at appropriate concentrations were spiked into the apical side (200 μL) of MDCKII-wt monolayers cultured in Transwells, with drug-free assay buffer (1,200 μL) placed in the basal chamber at 32°C in an orbital shaker (60 rpm). At various time points, aliquots were withdrawn from the apical side (25 μL, quenched with 225 μL methanol) and the basal side (200 μL) (with buffer replacement) and analyzed for drug and prodrug concentrations using HPLC. AVF, DZP, enzyme, DZP+ enzyme, blank buffer, untreated cells and blank filters were used as controls. Monolayer integrity was examined before and after the experiments by transepithelial electrical resistance (TEER) measurements. Percent TEER was obtained by normalizing the TEER value of treated cells by the value of untreated cells. Intactness of monolayers was also evaluated using lucifer yellow (100 μM) as a paracellular marker. Only monolayers with a TEER value ≥ 60 Ω cm2 and lucifer yellow permeability < 30 nm/s were used in the experiments. Permeability experiments were performed at various substrate and enzyme concentrations, and the obtained conversion-absorption curves were analyzed in accordance with in vitro pharmacokinetic models developed previously, using Matlab software (18). Results were an average of two independent experiments in duplicate.
Statistics
Statistical analysis was performed using GraphPad Prism software version 5.0. For multiple comparisons, one-way ANOVA with Dunnett’s post-test was used. p < 0.05 represented a significant difference.
RESULTS
Analytical Method
Figure 1 represents a typical HPLC chromatogram showing highly resolved peaks for AVF, DZP, and tolbutamide (TLB, internal standard). The developed HPLC method was accurate, precise and sensitive for both AVF (prodrug) and DZP (drug) with a 30 ng/mL limit of detection. To the best of our knowledge, this is the first time an HPLC method has been developed for the co-analysis of AVF and DZP. In addition to the peaks for AVF, DZP, and tolbutamide, a fourth peak was observed in the chromatogram (~2 min, Fig. 1). LC-MS data for this mixture revealed that this peak represents open-ring DZP (chemically 2-(N-methylamino)-5-chlorobenzophenone, MW 302.50), formed due to acidic hydrolysis of DZP (Supplemental 1). Susceptibility of DZP to acid degradation has been reported previously (24). The validation parameters of the developed HPLC method are given in Table I.
Fig. 1.

Typical HPLC chromatogram for AVF, DZP, and the internal standard (tolbutamide, TLB). The samples were analyzed using 73/27 KH2PO4 buffer/acetonitrile, pH 2.36 at 210 nm wavelength. The peak at 1.79 min represents open-ring DZP
Table I.
HPLC Validation Parameters for AVF and DZP
| Parameters | AVF | DZP |
|---|---|---|
| Linearity (R2) | 0.9993 | 0.9995 |
| Accuracy | 100 (2.01) | 100 (1.96) |
| Precision (repeatability), n = 9 | 2.01 | 1.96 |
| Range (μg/mL) | 0.25–8 | 0.125–8 |
| LOD (S/N 2) (μg/mL) | 0.03 | 0.03 |
| LOD (S/N 10) (μg/mL) | 0.25 | 0.125 |
| Aymmetry factor (As) | 2.01 | 1.2 |
| RT (min) | 0.9 | 7.9 |
Equilibrium Solubility
Saturation solubility of DZP (at thermodynamic equilibrium) was observed to be 130 ± 11 μM at pH 7.4 and 32°C, and this was not significantly different from its solubility at other temperatures (122 ± 2 μM at 25°C and 136 ± 5 μM at 37°C).
Enzyme Screening and Enzyme Kinetics
AVF’s (Scheme 1) lysine moiety, attached to DZP via an aminopeptide bond, makes several enzyme classes potential candidates for prodrug conversion, including proteases, peptidases and esterases. Accordingly, from a pool of commercially available enzymes, four enzymes were selected—dipeptidyl peptidase III, aminopeptidase N, a protease from A. oryzae, and butyrylcholinesterase.
As seen in Fig. 2a, UV absorbance of DZP is significantly greater than that of AVF, specifically in the 220–250 nm and 305–320 nm regions. Thus, one might expect to see a net gain in absorbance in these specific UV-Vis regions if prodrug conversion is occurring in systems consisting of AVF spiked with the activating enzyme. This relative increase in absorbance would be due to the appearance of diazepam accompanied by the disappearance of AVF.
Fig. 2.
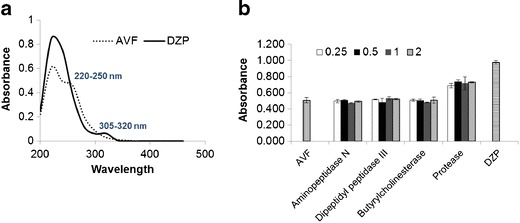
a UV-Vis spectra of AVF and DZP using a microplate reader. b Absorbance of AVF-enzyme mixtures (at 240 nm) prepared using AVF (129.9 μM, S = 1) with different enzymes at 0.25–2 U/mL after 10 min of mixing. Data is reported as sample absorbance minus absorbance of enzyme and blank buffer. Enzyme-free AVF and DZP were used as controls. Mean ± SD. n = 2
When AVF was incubated (in a microplate) with different enzymes, an increase in absorbance (at 240 nm) was observed with A.O. protease after 10 min (Fig. 2b), irrespective of enzyme concentration. This result indicates that A.O. protease causes activation and conversion of AVF. There was no change in absorbance with time of AVF only (without enzyme), DZP only, or AVF with any other enzyme.
In order to accurately examine the effect of A.O. protease concentration on AVF/A.O. protease reaction kinetics, absorbance measurements were performed in a cuvette rather than in a microplate. This change of assay did not influence the spectral characteristics of AVF and DZP (Figs. 2a and 3a). When AVF (130 μM, S = 1) was incubated with A.O. protease at various concentrations at 316 nm (since at 240 nm absorbance > 1), the slope of the absorbance curve (rate of prodrug conversion) was observed to increase with enzyme concentration (Fig. 3b).
Fig. 3.

a UV-Vis spectra of AVF and DZP measured in a quartz cuvette using a UV spectrophotometer. b Absorbance at 316 nm, 32°C with time for AVF/A.O. protease mixtures prepared using AVF (S = 1) with A.O. protease at different concentrations (0.125, 0.5, and 4 U/mL). Data is reported as sample absorbance minus absorbance from the A.O. protease alone. Blank buffer, enzyme-free AVF, and DZP (S = 1) were used as negative controls. Mean ± SD. n = 2
UV absorbance was an appropriate method for high-throughput enzyme screening and identification of the activating enzyme. However, this method has the following shortcomings: (1) its inability to distinguish completely between different species (AVF and DZP), (2) its limitation to subsaturated or saturated solutions due to interference from drug precipitates that could possibly be formed at supersaturated concentrations.
To more accurately examine enzyme kinetics at higher saturation levels, HPLC was utilized. AVF and DZP showed unique retention times and therefore could be differentiated using this method (Fig. 1). The possibility of precipitation of supersaturated samples was eliminated by using methanol as the reaction quencher before HPLC analysis, since methanol is a good solvent for diazepam. An example of reaction progress monitored using HPLC with AVF (1,042 μM) and cenz (enzyme concentration) = 0.25 U/mL, is shown in Fig. 4a, in which AVF conversion is accompanied by DZP formation. Complete mass balance was obtained, indicating accuracy of this method in analyzing reactions containing supersaturated drug levels.
Fig. 4.

a Reaction kinetics of AVF-protease mixture prepared using 0.25 U/mL protease and 1041.6 μM AVF at 32°C in a shaker. b Prodrug conversion rate (at 5 min) as a function of its concentration using 0.25 U/mL A.O. protease. Symbols represent the experimental data and the regression line is data fitted to Michaelis-Menten equation (Eq. 1). Mean ± SD. n = 3
When A.O. protease (0.25 U/ml) was incubated with AVF at various concentrations (69–3,601 μM, S = 0.5–27.7), prodrug conversion rate (at 5 min) increased with increasing initial prodrug concentration (cp), followed by saturation (symbols, Fig. 4b). The concentration-rate profile fitted well to Michaelis-Menten equation (solid line, Fig. 4b).
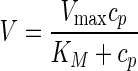 |
1 |
with KM = 1,501 ± 232 μM (s.e.m) and Vmax = 1,369 ± 94 μM/s.
Membrane Permeability Studies
In vitro permeability of AVF and DZP was examined using MDCKII-wt cell monolayers. To begin, DZP (at S = 0.7) or AVF (116–1,993 μM, S = 0.9–15.3, without enzyme) was introduced in the apical side of the monolayer with collection and analysis of both prodrug and parent drug on the basal side at various time points. Taking into account that the drug distributes into both the apical and basal sides, the data was fitted to Eq. (2) (18).
 |
2 |
where x = drug (d) or prodrug (p), cbx= concentration (μg/mL) on the basal side, Va and Vb = apical and basal side volumes, respectively, and CLx = the membrane’s clearance (permeability-area product) to x. As shown in Fig. 5a, DZP accumulated in the basal compartment as per Eq. (2), with CLd = 0.097 ± 0.011 mL/h and Papp = 2.2 × 10−5 cm/s.
Fig. 5.
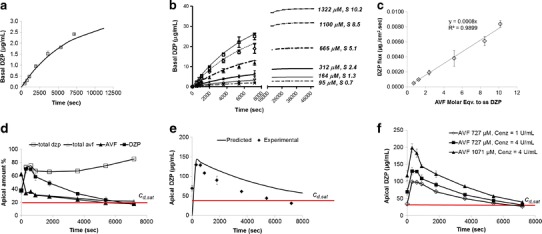
a Permeability of DZP across MDCKII-wt monolayer at near saturation solubility (85.7 μM, S = 0.7) (symbols) with flux 0.00045 ± 0.00007 μg/cm2 s. The curve represents the data fitted to Eq. (2). b Accumulation rate (on the basal side of monolayer) of DZP (symbols) produced from avizafone-protease mixtures prepared with different initial prodrug concentrations (μM). S represents the AVF molar equivalent of supersaturated (ss) DZP. Curves represent the data fitted to Eq. (3). c DZP flux at different ‘S’ values obtained from data (symbols) in b. d Concentration-time profile for the AVF- A.O. protease reaction (AVF at S = 5.6, c enz = 4 U/mL) on the apical side of MDCKII-wt membrane. ‘Total’ amount includes the amount permeating into the basal side. e % of DZP produced from prodrug/enzyme mixture (AVF at S = 5.6, c enz = 4 U/mL) in apical compartment (symbols) compared to predicted values (solid line) obtained using Eq. (4). f Concentration-time profile for DZP produced as a result of prodrug/enzyme mixture introduced onto the apical side prepared at various prodrug/enzyme ratios. In Fig. 5d–f, the horizontal (red) line represents DZP saturation level (S = 1, c d,sat). These experiments were performed in assay buffer, pH 7.4 at 32°C using 12-well Transwell plates. Mean ± SD. n = 4
When MDCKII-wt monolayers were treated apically with AVF at 116 μM, S = 0.9 (no enzyme), a negligible amount of prodrug accumulated in the basal side after 2 h. Although AVF flux increased with increasing prodrug concentration (Supplemental 2), only 10% of the prodrug (at most) permeated into the basal side over 2 h; this poor permeation (apparent permeability, 1–1.5 × 10−6 cm/s) is due to the hydrophilic nature of the molecule. Further, AVF flux saturated at high prodrug concentrations (≥1,130 μM) indicating facilitated membrane transport. In addition, apical solutions showed only 80% prodrug after 2 h (data not shown), indicating that some of the conversion of DZP may be occurring by way of endogenous enzymes that are likely present in the MDCKII-wt cell membranes. Due to the extremely slow prodrug permeation and conversion we ignored those processes in further considerations.
Upon spiking the prodrug with protease (at 4 U/mL) at various prodrug concentrations (95–1,322 μM, S = 0.7–10.2) in the apical compartment, prodrug conversion was followed by drug (DZP) permeation. Figure 5b shows DZP accumulation in the basal side as symbols represented by initial molar concentration of AVF added to the apical side, cap(0), in micromolar units, along with the ratio S = cap(0)/cd,sat, which represents the AVF molar equivalent of supersatured DZP. The obtained permeation data fitted well to Eq. (3) (derived previously in (18)) which predicts drug accumulation on the basal side cbd(t) when both conversion and permeation are occurring (predicted data as solid lines, Fig. 5b).
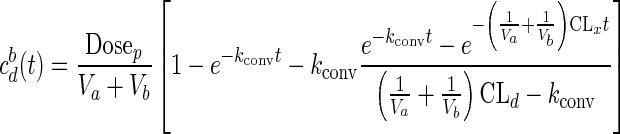 |
3 |
where kconv = (Vmax/KM) = (kcatcenz)/KM, with kcat = 12.7 s− 1, cenz = 108 μM (4 U/mL). Notably, drug accumulation rates (flux) were proportional to S (Fig. 5c) and these were 2- to 17.6-fold greater (at S ≥ 1.3) than the flux obtained with near-saturated DZP (S = 0.7). From the fact that proportionality exists between S and drug accumulation rates, we can conclude that increase in basal drug concentration (below saturated DZP concentration) did not affect the drug permeation rates.
On the apical side, prodrug disappearance corresponded to drug appearance, and there was simultaneous drug disappearance by permeation (AVF at S = 5.6, cenz = 4 U/mL). Complete mass balance was obtained after accounting for AVF and DZP permeating into the basal side (total AVF + total DZP) (Fig. 5d). However, in these prodrug/enzyme mixtures, the prodrug was not completely converted to the parent drug (only 80% conversion) even after 2 h. Figure 5e shows the apical concentration of drug, along with a prediction based on Eq. (4) below and using parameters derived from fits to Eqs. (1) and (4) using data in Fig. 5a–c (18). Equation (4) somewhat overpredicts apical concentrations, but the general trend is reproduced.
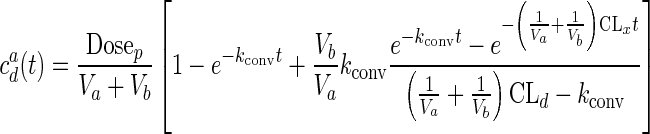 |
4 |
As shown in Fig. 5d–e, supersaturation was achieved as early as 5 min (300 s), after administering AVF and enzyme, as indicated by drug concentrations above the horizontal red line (cd,sat—concentration of saturated DZP). Further, the rate and extent of drug appearance in the apical side could be controlled by the prodrug/enzyme ratio (Fig. 5f).
Monolayer integrity was evaluated with AVF, DZP, enzyme (A.O. protease), AVF + enzyme, DZP + enzyme and blank buffer solutions. As shown in Fig 6, TEER was unaffected by all treatments except prodrug/enzyme mixtures prepared at S = 10.2 (as per ANOVA). However, even with this treatment, the TEER values were above the lowest acceptable limit of 60 Ω/cm2. Therefore, monolayer integrity was not compromised with any treatment employed in our studies.
Fig. 6.

% TEER representing monolayer integrity with various treatments. Numerical value succeeding S represents AVF molar equivalent of supersatured DZP. The monolayers were treated with various samples for 2 h in assay buffer pH 7.4, at 32°C in 12-well Transwells. The data is normalized to TEER value for untreated cells that was considered as 100%. Arrow represents the control group used in one-way ANOVA with Dunnett’s multiple comparison test. Asterisk represents significant difference (p < 0.05) from the control group. AVF avizafone, DZP diazepam , E enzyme (protease) at 4 U/mL, AVFE AVF + protease, DZPE DZP + protease
Control experiments were performed to evaluate the effect of protease (enzyme) concentration on (a) monolayer integrity using TEER and inulin permeability measurements, and (b) DZP permeability. As shown in Supplemental 3, % TEER of the monolayer was unaffected even by the presence of 16 U/mL protease. However, beyond 8 U/mL protease, inulin permeability was greater than 1%, indicating 8 U/mL to be a safe limit. At 4 U/mL which is the protease concentration used in our permeation studies, DZP permeation rate was unaffected by the presence of enzyme (no significant difference in apparent permeability) (Supplemental 4).
DISCUSSION
Epilepsy affects an estimated 3 million people in the USA, making it second only to stroke for debilitating neurological conditions. Contrary to stroke, which primarily affects the elderly, the majority of patients with epilepsy include children and young adults, a population that may require decades of drug therapy. Conditions such as SE are emergencies that require fast delivery of a potent antiepileptic drug such as DZP. Rapid delivery can be obtained by using supersaturated solutions.
Although DZP supersaturation has been obtained previously using water/organic cosolvents (7), with the goal of achieving rapid DZP delivery, this success was limited by tolerance issues or drug precipitation before complete absorption (10,12). With fosphenytoin/alkaline phosphatase as a model system, our group previously prepared supersaturated aqueous solutions of prodrug/enzyme mixtures at the point of administration, and demonstrated enhanced membrane permeation of the product drug, in this case phenytoin, compared to saturated drug solution, without precipitation (18). However, phenytoin is not a suitable candidate for intranasal delivery due to its high dose requirement. The present effort has demonstrated that prodrug/enzyme coadministration also enables the rapid membrane permeation of DZP, which is sufficiently potent that it can be administered intranasally.
AVF is an off-patent DZP prodrug used by the French military to reverse seizures triggered by nerve agents encountered on the battlefield (19,20). However it is not commercially available, and thus it was synthesized by our group. In a preliminary study in dogs, our group demonstrated that, when administered intranasally, the fraction of the AVF absorbed and converted to DZP was only ~30–45% of the total dose, which rendered AVF unacceptable for further development in that particular form (Cloyd and Patterson, personal communication). It was concluded that the highly water soluble AVF does not efficiently cross the nasal mucosa, as was predicted in the current study with our AVF MDCKII-wt permeability results, and that successful intranasal therapy would require coadministration with a suitable converting enzyme. Among the selected panel of enzymes, A.O. protease was shown to cleave the aminopeptide bond in AVF to form DZP, with a conversion rate dependent on enzyme concentration (Fig. 2b).
Even though this enzyme caused rapid prodrug conversion, maximum conversion was only around 75–80% (Figs. 3b and 5d), irrespective of enzyme and prodrug concentrations. Additional studies performed at varying prodrug/enzyme ratios confirmed this result (Supplemental 5). Further confirmation was obtained from TLC studies (data not shown). This finding might be explained by racemization of AVF either during synthesis or during enzymatic reaction, followed by selectivity of the enzyme for one of the isomers. Racemization was qualitatively confirmed by circular dichroism studies, comparing the starting material AVF and unreacted material collected at the AVF elution peak following AVF exposure to enzyme. These samples, while showing equivalent mass spectra, displayed opposing optical activities in circular dichroism studies (Supplemental 6). Moreover, combinations of starting material and unreacted AVF (in reaction mixture) showed CD spectra that qualitatively interpolate between the limiting spectra. These studies likely reflect a putative stereoselectivity of A.O. protease, a common property of enzymes (25–27).
While further studies are needed to clarify this matter, it seems most likely that racemization of AVF occurs during synthesis (NB: polarimetric measurements of AVF exactly matched those specified in the original patent (22)). The unreacted AVF HPLC peak was shown to persist even after dialysis and exposure to fresh enzyme. If A.O. protease is acting as an isomerase, the principle of microscopic reversibility warrants that AVF should flip back and forth between enantiomers, with all AVF ultimately hydrolyzed to diazepam, unless d-AVF is thermodynamically much more stable than l-AVF, which seems quite improbable.
Despite 80% conversion, the AVF-protease mixtures greatly enhanced the rate and degree of drug absorption (up to 18-fold) through MDCKII-wt monolayers, when compared to saturated DZP solution (Fig. 5a–c). Complete mass balance was obtained and drug transport flux increased proportionally to the degree of supersaturation. Microscopic observations during permeation studies revealed no drug crystals in the apical solutions. Together these results demonstrate that the supersaturated state persisted without drug precipitation/crystallization; the observed absence of crystallization was likely due to slow rate of nucleus formation and crystal growth of DZP. The permeation data fitted reasonably well to previously developed in vitro pharmacokinetic models, indicating the applicability of these models to more than one kind of prodrug/enzyme system. However, in the present case, since prodrug conversion was at most 80%, a correction factor was used to compare the model predictions to the data (Fig. 5).
The prodrug/enzyme system studied in this work is capable of not only producing supersaturated solutions quickly (in 5 min or 300 s), but it is also flexible in that the rate and extent of supersaturation can be fine-tuned by adjusting the prodrug/enzyme ratio. This specific feature can be exploited in the development of intranasal delivery systems for DZP, especially for the treatment of seizure emergencies. It should be noted that such a delivery system will require separate formulations of prodrug and enzyme, which will be automatically mixed just prior to nasal administration. Future in vivo studies are needed in order to further strengthen the applicability of the prodrug/enzyme system for human subjects.
CONCLUSIONS
In the current study, the AVF/A.O protease system was shown to be useful for the rapid delivery of DZP across cell monolayers. In this manner, such a system may find application in developing intranasal treatments for neurological conditions such as status epilepticus, where rapid absorption and action are required.
Electronic Supplementary Material
(DOC 244 kb)
ACKNOWLEDGMENTS
We thank the AHC Faculty Research Development Program at the University of Minnesota for research funding (grant#1803-11406-21287-3672675, PI G. Georg). We thank Dr. Subhashree Francis for providing data for Supplemental 1 (LC-MS). We also thank Prof. William Elmquist for providing lab facilities to perform cell studies, and Prof. Karunya Kandimalla for providing the HPLC equipment. Discussions with Profs. James Cloyd, Edward Patterson and Patrick E. Hanna are gratefully acknowledged.
REFERENCES
- 1.Pang T, Hirsch L. Treatment of convulsive and nonconvulsive status epilepticus. Curr Treat Options Neurol. 2005;7(4):247–259. doi: 10.1007/s11940-005-0035-x. [DOI] [PubMed] [Google Scholar]
- 2.Sirven J, Waterhouse E. Management of status epilepticus. Am Fam Physician. 2003;68(3):469–476. [PubMed] [Google Scholar]
- 3.Abou-Khalil B, Wheless J, Rogin J, Wolter KD, Pixton GC, Shukla RB, et al. A double-blind, randomized, placebo-controlled trial of a diazepam auto-injector administered by caregivers to patients with epilepsy who require intermittent intervention for acute repetitive seizures. Epilepsia. 2013;54(11):1968–1976. doi: 10.1111/epi.12373. [DOI] [PubMed] [Google Scholar]
- 4.Holsti M, Dudley N, Schunk J, et al. Intranasal midazolam vs rectal diazepam for the home treatment of acute seizures in pediatric patients with epilepsy. Arch Pediatr Adolesc Med. 2010;164(8):747–753. doi: 10.1001/archpediatrics.2010.130. [DOI] [PubMed] [Google Scholar]
- 5.Agarwal SK, Kriel RL, Brundage RC, Ivaturi VD, Cloyd JC. A pilot study assessing the bioavailability and pharmacokinetics of diazepam after intranasal and intravenous administration in healthy volunteers. Epilepsy Res. 2013;105(3):362–367. doi: 10.1016/j.eplepsyres.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 6.Ivaturi VD, Riss JR, Kriel RL, Cloyd JC. Pharmacokinetics and tolerability of intranasal diazepam and midazolam in healthy adult volunteers. Acta Neurol Scand. 2009;120(5):353–357. doi: 10.1111/j.1600-0404.2009.01170.x. [DOI] [PubMed] [Google Scholar]
- 7.Hou H, Siegel RA. Enhanced permeation of diazepam through artificial membranes from supersaturated solutions. J Pharm Sci. 2006;95(4):896–905. doi: 10.1002/jps.20600. [DOI] [PubMed] [Google Scholar]
- 8.Ibach H. Nucleation and growth. Physics of surfaces and interfaces. Berlin: Springer; 2006. [Google Scholar]
- 9.Hsieh Y-L, Ilevbare G, Van Eerdenbrugh B, Box K, Sanchez-Felix M, Taylor L. pH-induced precipitation behavior of weakly basic compounds: determination of extent and duration of supersaturation using potentiometric titration and correlation to solid state properties. Pharm Res. 2012;29(10):2738–2753. doi: 10.1007/s11095-012-0759-8. [DOI] [PubMed] [Google Scholar]
- 10.Narayanan J, Liu XY. Protein interactions in undersaturated and supersaturated solutions: a study using light and x-ray scattering. Biophys J. 2003;84(1):523–532. doi: 10.1016/S0006-3495(03)74871-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Sun M, Fan A, Wang Z, Zhao Y. The effect of solute-membrane interaction on solute permeation under supersaturated conditions. Int J Pharm. 2013;441(1–2):389–394. doi: 10.1016/j.ijpharm.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Leichtnam M-L, Rolland H, Wüthrich P, Guy RH. Enhancement of transdermal testosterone delivery by supersaturation. J Pharm Sci. 2006;95(11):2373–2379. doi: 10.1002/jps.20669. [DOI] [PubMed] [Google Scholar]
- 13.Djuris J, Nikolakakis I, Ibric S, Djuric Z, Kachrimanis K. Preparation of carbamazepine Soluplus® solid dispersions by hot-melt extrusion, and prediction of drug-polymer miscibility by thermodynamic model fitting. Eur J Pharm Biopharm. 2013;84(1):228–237. doi: 10.1016/j.ejpb.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 14.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 15.Paradkar A, Ambike AA, Jadhav BK, Mahadik KR. Characterization of curcumin-PVP solid dispersion obtained by spray drying. Int J Pharm. 2004;271(1–2):281–286. doi: 10.1016/j.ijpharm.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 16.Thybo P, Pedersen BL, Hovgaard L, Holm R, Mallertz A. Characterization and physical stability of spray dried solid dispersions of probucol and PVP-K30. Pharm Dev Technol. 2008;13(5):375–386. doi: 10.1080/10837450802244843. [DOI] [PubMed] [Google Scholar]
- 17.Zheng X, Yang R, Tang X, Zheng L. Part I: characterization of solid dispersions of nimodipine prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2007;33(7):791–802. doi: 10.1080/03639040601050213. [DOI] [PubMed] [Google Scholar]
- 18.Kapoor M, Siegel RA. Prodrug/enzyme based acceleration of absorption of hydrophobic drugs: an in vitro study. Mol Pharm. 2013;10(9):3519–3524. doi: 10.1021/mp400272m. [DOI] [PubMed] [Google Scholar]
- 19.Karlsson B, Lindgren B, Waterhouse E, Sandberg M, Sellström A. On the use of diazepam and pro-diazepam (2-benzoyl-4-chloro-N-methyl-N-lysylglycin anilide), as adjunct antidotes in the treatment of organophosphorus intoxication in the guinea-pig. J Pharm Pharmacol. 1990;42(4):247–251. doi: 10.1111/j.2042-7158.1990.tb05401.x. [DOI] [PubMed] [Google Scholar]
- 20.Lallement G, Renault F, Baubichon D, Peoc’h M, Burckhart MF, Galonnier M, et al. Compared efficacy of diazepam or avizafone to prevent soman-induced electroencephalographic disturbances and neuropathology in primates: relationship to plasmatic benzodiazepine pharmacokinetics. Arch Toxicol. 2000;74(8):480–486. doi: 10.1007/s002040000146. [DOI] [PubMed] [Google Scholar]
- 21.Hassall CH, Johnson WH, Krohn A, Smithen CE, Thomas WA, inventors; Phenyl keto derivatives of lysyl glycinamide. AU514778B2, Australia. 1981.
- 22.Hassall CH, Johnson WH, Krohn A, Smithen CE, Thomas WA, inventors; A substituted-phenyl ketone containing a dipeptide residue and a process for the manufacture thereof. GB1517166A, UK. 1974 20 Aug 1974.
- 23.FDA guidelines, Guidance for industry. Q2B Validation of Analytical Procedures: Methodology. November 1996.
- 24.Nudelman NS, de Waisbaum RG. Acid hydrolysis of diazepam. Kinetic study of the reactions of 2-(N-methylamino)-5-chlorobenzophenone, with HCl in MeOH-H2O. J Pharm Sci. 1995;84(8):998–1004. doi: 10.1002/jps.2600840817. [DOI] [PubMed] [Google Scholar]
- 25.Auret BJ, Boyd DR, Breen F, Greene RME, Robinson PM. Stereoselective enzyme-catalysed oxidation-reduction reactions of thioacetals-thioacetal sulphoxides by fungi. J Chem Soc, Perkin Transactions 1. 1981(0):930–3.
- 26.Paul Meloche H, Monti CT, Dekker EE. Enzyme stereoselectivity: the reversible reaction catalyzed by 2-keto-4-hydroxyglutarate aldolase of Escherichia coli. Biochem Biophys Res Commun. 1975;65(3):1033–1039. doi: 10.1016/S0006-291X(75)80489-X. [DOI] [PubMed] [Google Scholar]
- 27.Wildberger P, Brecker L, Nidetzky B. Chiral resolution through stereoselective transglycosylation by sucrose phosphorylase: application to the synthesis of a new biomimetic compatible solute, (R)-2-O-[small alpha]-d-glucopyranosyl glyceric acid amide. Chem Commun. 2013;50(4):436–438. doi: 10.1039/c3cc47249c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 244 kb)