

Abstract
Fulranumab, a human IgG2 monoclonal antibody that neutralizes nerve growth factor (NGF), is currently in development for the treatment of pain. Our initial immunogenicity test method was found to be prone to NGF interference, leading to a high apparent incidence of anti-drug antibody (ADA) in phase 1 studies. The ADA immunoassay comprised a homogeneous bridging electrochemiluminescence (ECL) format with biotin and ruthenium-labeled fulranumab bound together (“bridged”) by ADA in test samples for detection. In this assay, NGF produced a false-positive signal due to its ability to bridge fulranumab molecules. Thus, we developed a specificity assay to eliminate the NGF false-positive results. We encountered the challenge of eliminating drug interference as well as drug target interference, and discovered that the acid-dissociation-based pretreatment of samples used for mitigating drug interference dramatically increased drug target interference. Several strategies were investigated to eliminate the NGF interference; yet only one strategy specifically removed NGF and produced true fulranumab-specific ADA results by using competitive inhibition with fulranumab and utilizing an alternative NGF binding antibody to eliminate NGF interference. Using this new method, we confirmed that the high apparent anti-fulranumab antibody incidence (>60%) in clinical study samples was in fact due to fulranumab-bound NGF released during the acid-dissociation step of the ADA testing method. We conclude that our revised method accurately identifies anti-fulranumab antibodies by incorporating steps to eliminate fulranumab and NGF interference. We advise that acid-dissociation pretreatment must not be universally applied to improve ADA assays without investigating its bioanalytical risks versus benefits.
KEY WORDS: anti-drug antibody, assay specificity, drug target interference, immunogenicity, nerve growth factor
INTRODUCTION
Nerve growth factor (NGF) is thought to play a significant role in pain sensation. A secreted factor that controls the sensitivity of primary sensory neurons, NGF is expressed in peripheral tissues in conjunction with pain and is specifically upregulated in injury states both in animal models and in human conditions (1–5). Fulranumab (drug) is a fully human IgG2 monoclonal antibody that specifically neutralizes the biologic actions of NGF, and thus may be of therapeutic benefit in pain states where the mechanism of action involves NGF. It is also anticipated that this drug will provide a new treatment option for chronic pain without sedative effect, a common complaint with many current pain medications. Currently, fulranumab is in clinical phase 2 trials to treat patients with chronic pain.
The administration of therapeutic biological drugs can induce immune responses in subjects. This immune response, usually comprised of anti-drug antibodies (ADA), can produce a range of effects from benign and asymptomatic to altered pharmacokinetics (for example, drug neutralization, abnormal biodistribution, or enhanced drug clearance rates, potentially resulting in altered efficacy) and/or pharmacodynamics and adverse clinical sequelae (6,7). Thus ADA assessment is a critical component for the development of a therapeutic biological drug, and well-designed and specific ADA immunoassays are crucial for appropriately monitoring the drug’s immunogenicity profile. Bridging immunoassay platforms, including enzyme-linked immunosorbent assay (ELISA) and eletrochemiluminescent immunoassays (ECLIA) are often used to detect antibodies directed against therapeutic monoclonal antibodies. Recent publications and regulatory agency guidance documents point to the need for specific immunogenicity assays and a tiered testing scheme to support clinical immunogenicity investigations (8–11). The strategy for measuring ADA involves testing all clinical samples initially with a screening assay that sensitively detects ADA, minimizing the possibility of false-negative results. To that end, the cut point for a screening assay is designed to detect “potentially ADA positive” samples by purposely including a statistical chance of false-positive results. Therefore, a subsequent confirmation assay is a critical component of the tiered ADA testing scheme for eliminating false-positives identified in the screening assay and identifying true ADA-positive samples.
ADA methods can be susceptible to interferents present in the test matrix. The most important interferent in an ADA detection assay is the drug itself, which can lead to false-negative assay results. Pretreatment of samples with acid is widely used to mitigate the interference due to the circulating therapeutic, allowing ADA detection in the immunoassay (12–14). On the other hand, it is underappreciated that the drug’s target can also be an interferent, particularly in bridging immunoassays. With the advent of therapeutic proteins, such as monoclonal antibodies that can form extended complexes with soluble ligands (including soluble forms of some cell-surface receptors) (15) and remain in circulation for long periods of time, the potential of drug target to influence the measurement of ADA can be seen to vary with disease state, treatment regimens and schedules, or regulation of endogenous proteins (16,17). Drug targets can interfere with measurement of ADA, in ways that result in either false-positive or false-negative antibody detection depending on the assay format as well as the concentration, structure, and chemical and biological properties of the targets. Therefore, it is important to evaluate the interference of ADA detection by the drug’s intended targets.
For the assessment of ADA to fulranumab, we developed a three-tiered testing scheme comprising an initial screening assay followed by specificity confirmation assay and a titration assay. Because NGF exists naturally as a homodimer, we speculated that it could bridge the conjugated fulranumab reagents in the ADA detection assay and cause a false-positive signal. Indeed, we found that the screening immunoassay was prone to false-positive NGF interference. To eliminate this interference, the specificity confirmation method initially included the use of Melon™ Gel Resin (Thermo Scientific, Rockford, IL, USA), a proprietary reagent that somewhat selectively removed non-immunoglobulin serum proteins including NGF. However, the inadequacy of this specificity method was suspected after examining the immunogenicity results from a phase 1a healthy volunteer study (with 3% ADA incidence) versus a phase 1b osteoarthritic patient study (with 72% ADA incidence). We doubted that healthy subjects and osteoarthritis subjects would inherently differ to such an extent in their relative ability to induce immune responses against fulranumab. We speculated that NGF bound to fulranumab may accumulate to supra-normal levels in osteoarthritis subjects. When processing such samples for ADA detection (i.e., acid pretreatment for dissociation of ADA and fulranumab), the NGF-fulranumab complex potentially also dissociated to release free NGF that could interfere in the ADA assays to produce false-positive results. Since Melon™ Gel Resin treatment probably reduced “free” unbound NGF levels in samples without removing fulranumab “bound” NGF, the fulranumab “bound” NGF would likely cause false-positive ADA result in the specificity assay. Hence, the elevated NGF levels in clinical samples could have resulted in a significant over-reporting of the immunogenicity incidence in phase 1b osteoarthritis study, whereas this may not have been an issue in the phase 1a healthy volunteer study. Although one may consider ADA over reporting to be preferable to false-negatives, it is equally confounding and should be avoided because it can obscure or lead to inaccurate conclusions when attempting to correlate clinical outcomes against ADA incidence.
Thus, we initiated the development of a new fulranumab ADA specificity assay to confirm positive signals attributed to ADA while eliminating false-positive signals from NGF. We evaluated four different confirmatory assay strategies intended to eliminate false-positive signal. In this article, we demonstrate that only one approach correctly identified the positive ADA samples and eliminated the false-positive samples. In this approach, the majority of NGF was specifically removed using biotinylated anti-NGF antibody and streptavidin-coated beads, and the remaining NGF signal was blocked by a NGF-blocking protein (anti-NGF TrkA peptide fused to human Fc). Only after elimination of NGF signal, it was possible to perform a meaningful competitive inhibition of the samples with unlabeled fulranumab. These findings highlight the importance of a careful examination of drug target interference in ADA methods and the development of approaches to avoid any false reporting of ADA results.
MATERIALS AND METHODS
Equipment
The following instruments and reagents were used: Meso Scale Discovery (MSD) Sector Imager 6000 instruments, MSD Read Buffer, MSD Streptavidin Bind Plates (MSD, Gaithersburg, MD, USA), ELX-405 Plate Washers (BioTek Instruments, Winooski, VT, USA), Spectrophotometric Plate Reader (Molecular Devices, Sunnyvale, California, USA), Thompson 96-well vacuum filtration system, Millipore 96-well filter plates (Catalog No MSFBN6B10 or equivalent), Melon™ Gel IgG spin purification kit (Product No 45206), Streptavidin-coated magnetic beads (Cat# 112.06D, Invitrogen, Grand Island, NY, USA).
Drug Antibodies and Assay Reagents
Fulranumab and affinity-purified anti-fulranumab rabbit polyclonal antibody were produced and prepared by Janssen Research and Development, LLC (Radnor, PA, USA). Fulranumab was labeled with biotin as the capture antibody using EZ-Link Sulfo-NHS-LC biotinylation kits (Pierce, Rockford, IL, USA) with 10:1 biotin to fulranumab challenge ratio according to the manufacturer’s instructions. The ruthenium conjugation of fulranumab was carried out using MSD SULFO-TAG™ NHS-Ester according to the manufacturer’s instructions. Recombinant human NGF (catalog# 256-GF) and biotinylated anti-human NGF monoclonal mouse antibody (catalog# 509802) were purchased from R&D Systems (Minneapolis, MN, USA). Pooled normal human serum was obtained from Bioreclamation Inc (Westbury, NY, USA). NGF blocking protein TrkA peptide fused to human Fc was made by Janssen and Development, LLC (Radnor, PA, USA).
Generation of Anti-Fulranumab Antibodies
All animal experiments were conducted in full compliance with local, national, ethical, and regulatory principles and licensing regulations, per the spirit of Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International’s expectations for animal care and use/ethics committees. In accordance with applicable regulations concerning the ethical use of laboratory animals, ten Balb/c mice (12–14 weeks old) were immunized with fulranumab. Lymphocytes were isolated from the immunized mice and were subsequently fused to FO myeloma cells. Solid-phase ELISA was used to screen hybridoma supernatants for fulranumab binding antibodies. The IgG fraction of the hybridoma culture supernatant was purified by protein G after affinity chromatography. Ten mAbs reactive to the variable region of fulranumab were identified and further characterized.
To generate a polyclonal antibody reagent, two cynomolgus monkeys were hyperimmunized by an initial administration of a 50% emulsion of 1 mg fulranumab per kg body weight in Hunter’s TiterMax (CytRx Corp, Los Angeles, CA, USA) followed every third week by booster injections of a 50% mixture of Imject Alum (Pierce, Rockford, IL, USA) and 0.1 mg fulranumab per kg body weight. Blood was collected from the animals after multiple rounds of boosting. Polyclonal antiserum was purified by protein G followed by drug antibody affinity chromatography.
Bridging ECLIA for Fulranumab ADA Screening Assay
A bridging ECLIA with acid dissociation was used to measure ADA (Fig. 1). This MSD-based ECLIA method was developed, optimized, and validated using hyperimmunized monkey polyclonal ADA controls. The samples were pretreated with acid to disrupt the ADA–drug complex before assessment in the bridging ECLIA. The homogeneous bridging ECLIA method allowed for concurrent solution-phase binding of ADA to biotinylated fulranumab and ruthenylated fulranumab. Immune complexes were then captured on MSD streptavidin plates. In brief, the assay procedure was as follows: samples were diluted to 10% in 300 mM acetic acid in a nonbinding polypropylene deep well plate to enable antibody-drug complex dissociation before analysis. Acidified samples were incubated for 30 to 60 min with shaking at ambient temperature. MSD Streptavdin-coated plates were blocked for 30–60 min at room temperature with 300 μL/well of PBS buffer containing 1% BSA. Twenty-five microliters of the acidified sample was transferred to the blocked MSD plates, then 25 μL of master-mix reagent containing 0.25 μg/mL of biotinylated fulranumab and 0.25 μg/mL of ruthenylated fulranumab with 1% BSA in PBS and 0.3 M of a Tris base solution (1 M, pH 9.5) were added to each well of the MSD plate with acid-treated samples. The MSD plates were incubated for 2 h in the dark with shaking. The MSD plates were washed and 150 μL of 2 × MSD Read T-Buffer was added per well before the plates were read on an MSD Sector® Imager 6000. The resulting response was recorded as electrochemiluminescence (ECL) units, and the normalized values (NV) were calculated by dividing the ECL value of each sample by the ECL value of the pooled normal human negative control (NC) serum. All test and control samples were analyzed in triplicate wells. The screening assay cut point was determined by evaluating at least 50 normal human serum samples.
Fig. 1.
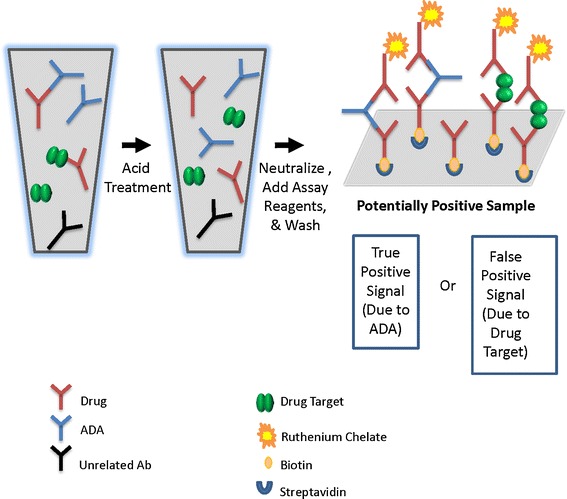
Bridging ECLIA for detection of anti-fulranumab antibody. Acid pretreatment is applied to reduce drug interference in the assay, making it “drug tolerant”
Drug Target Interference
Interference of drug target NGF was evaluated in the fulranumab ADA screening assay by adding increasing levels of recombinant human NGF to the assay in place of the ADA-positive control antibodies. The range of NGF investigated spanned from just below normal reported endogenous levels in healthy subjects to levels higher than projected fulranumab-bound NGF levels in patients administered with fulranumab. This was done with the intention to encompass the potential elevated levels of NGF for the patients in clinical trials. NGF interference was considered positive when levels of NGF in the drug-treated patients yielded a signal above the screening assay cut point in ADA-negative samples.
Evaluation of Signal Inhibition by Fulranumab in the Bridging ECLIA Assay
ADA and NGF both produce positive signals in the screening bridging ECLIA assay (Fig. 1). An experiment was set up to evaluate the ability of added fulranumab to inhibit these signals. Two sets of samples were prepared containing either an anti-fulranumab monkey polyclonal antibody or NGF at 10, 100, and 1,000 ng/mL in normal human serum pool. Each sample was first incubated in assay diluent (1% BSA in PBS) with or without 200 μg/mL of fulranumab for 30 min and then analyzed in the screening assay (refer to previous section of bridging ECLIA for fulranumab ADA screening assay). The NV values were calculated by dividing the ECL response of samples containing NGF or anti-fulranumab polyclonal antibody by the ECL response of the negative human serum pool. The percent inhibition of signal due to fulranumab was calculated as follows: % Inhibition = 100 − 100 × (NV of sample with fulranumab/NV of sample without fulranumab).
Removal of Drug Target Interference with Melon™ Gel Resin Treatment
To test Melon™ Gel Resin treatment to remove drug target interference, samples containing different levels of NGF were treated with Melon™ Gel and tested in the screening assay. Melon™ Gel (Thermo Scientific, Rockford, IL, USA) reagent was filtered, centrifuged, and washed multiple times as per its vendor’s recommendation (18). A volume of 150 μL of the Melon™ Gel mixture was added into each desired well on the 96-well Millipore filter plate. The serum samples containing NGF or ADA at different levels were diluted in the purification buffer provided with the Melon™ Gel Purification kit and added to the wells containing the washed gel. A 96-well polypropylene plate was used to collect the Melon™ Gel-treated samples. After the Melon™ Gel treatment, the flow-through of the samples was analyzed in the screening assay of acid-dissociation bridging ECLIA to evaluate the drug target interference. The signal of positive interference from NGF after Melon™ Gel treatment was compared with the assay screening cut point as well as that of NGF interference without treatment. The monkey anti-fulranumab antibody was also prepared in normal human serum pool and treated with Melon™ Gel. The recovery of ADA signals after the Melon™ Gel treatment was also studied. The percent reduction of signal due to Melon™ Gel treatment was calculated as follows: % Reduction = 100 − 100 × (NV of sample with Melon™Gel treatment/NV of sample without treatment).
Evaluation of Protein G to Selectively Remove IgG in Specificity Assay
To test protein G removal of antibodies from serum samples, monkey anti-fulranumab antibody was added to a normal human serum pool at different levels in 96-well plate. Protein G spin plates were equilibrated by adding 400 μL of binding buffer (Thermo Scientific, product# 45204, Rockford, IL, USA) to each well, and vacuum filtered for 1 min to remove the binding buffer. The protein G plate was then washed by adding 400 μL of assay diluent to each well and centrifuged for 1.5 min at 1,000×g to remove the assay diluent. A volume of 100 μL of serum samples containing ADA or NGF were added to the wells on the protein G plate. The plate was sealed and incubated at room temperature with vigorous (1,100 rpm or greater) shaking on a plate shaker for 20 min. The protein G separation plate was then transferred into the centrifuge and centrifuged for 1.5 min at 1,000×g and the flow-through samples were collected in a collection plate. After immunoglobulin removal by protein G, the flow-through samples were analyzed in the bridging ECLIA assay screening assay and the results were compared with the analysis prior to the removal of immunoglobulins. NGF was also prepared in a normal human serum pool and treated with protein G plate separation. The false-positive signal of NGF as well as the true-positive ADA in the sample flow-through after protein G removal was compared with the signal prior the protein G separation.
Removal of Drug Target with Specific Drug Target Binding Antibodies Immobilized on Beads
To specifically remove NGF, the samples were pretreated with biotinylated anti-target antibody and streptavidin-coated magnetic beads. Normal serum samples were unspiked or spiked with increasing concentrations of NGF up to 1,500 ng/ml. The biotin-anti-NGF solution was prepared by adding 10 μg/mL of biotinylated anti-NGF mouse antibody in assay diluent. For sample pretreatment, 40 μL of samples containing NGF were incubated with 40 μL of the biotin-anti-NGF solution, 40 μL of streptavidin-coated magnetic beads (Dynabeads from Invitrogen, Grand Island, NY, USA) at 10 mg/mL, and 40 μL of assay diluent with NGF blocking protein TrkA peptide fused to human Fc at 40 μg/mL. The plates were sealed and incubated on a plate shaker at a speed that kept beads suspended for 60 min at room temperature. The anti-NGF antibody-immobilized magnetic beads were separated from the solution by applying a magnet externally to the sample vial such that the magnetic beads aggregated on the wall of the wells. Eighty micro-liters of the supernatant were transferred to a set of cluster tubes without disturbing the beads at the bottom of the tubes. The sample supernatant was either analyzed with the screening assay to detect the ADA or further inhibited with 200 μg/mL of fulranumab to calculate the percent inhibition of samples after NGF removal (in order to confirm that a positive response was due to a fulranumab-specific antibody).
Two-Step Specificity Confirmation Assay with Selective NGF Removal and Competitive Inhibition
A two-step specificity confirmation assay was developed with selective NGF blocking and removal followed by competitive inhibition with unlabeled fulranumab. In this assay, two aliquots of each sample were obtained with one aliquot designated as the uninhibited sample and the other one as the inhibited sample. The uninhibited sample was first combined with streptavidin beads, biotinylated anti-NGF MAb solution, and assay diluents with 40 μg/mL of NGF blocking protein at 1:1:1:1 ratio (40 μL each) while the inhibited samples was combined with streptavidin beads, biotin anti-NGF MAb solution, and assay diluents with 200 μg/mL fulranumab at 1:1:1:1 ratio (40 μL each). The NGF blocking protein was used to block any residue NGF left during the NGF removal by biotinylated anti-NGF antibody and streptavidin-coated beads. Both aliquots of samples were incubated for 60 min at 37°C with shaking. A magnetic plate was used to separate the anti-NGF antibody-immobilized magnetic beads from the supernatant. Eighty microliters of the supernatant aliquots were transferred to a new set of cluster tubes. The supernatant of both inhibited and uninhibited samples was then analyzed with the screening assay to detect the ADA. The percent inhibition of signal due to fulranumab was calculated as follows: % Inhibition = 100 − 100 × (NV of inhibited sample/NV of uninhibited sample)
RESULTS
NGF Interference Assessment and Its Inhibition by Fulranumab
Anti-fulranumab binding antibodies in serum samples were measured using a bridging ECL immunoassay employing an acid-dissociation pretreatment step (Fig. 1). A NV value of 1.04 representing the screening assay cut point was determined by calculating the 95th percentile of the distribution of the NV values of naive serum samples. The concentration of affinity-purified anti-fulranumab rabbit polyclonal antibody at the screening cut point was 1 ng/mL and was an indication of the sensitivity of the bridging ECL assay (Fig. 2). The assay reliably detected 10 ng/mL of an anti-fulranumab antibody in the presence of 10 μg/mL of fulranumab (a drug level that is much higher than the anticipated trough levels during treatment). NGF interference in the screening assay was examined by spiking different levels of NGF in undiluted normal human serum. When samples were not processed to remove NGF prior to analysis, NGF interference was observed at 100 pg/mL (above the normal physiological level of <12 pg/mL (20)). Based on the screening assay cut point (1.04 NV), the negative control (ADA-naive and drug-naïve) serum could not tolerate more than 100 pg/mL of NGF prior to producing a false-positive screening result (Fig. 2).
Fig. 2.
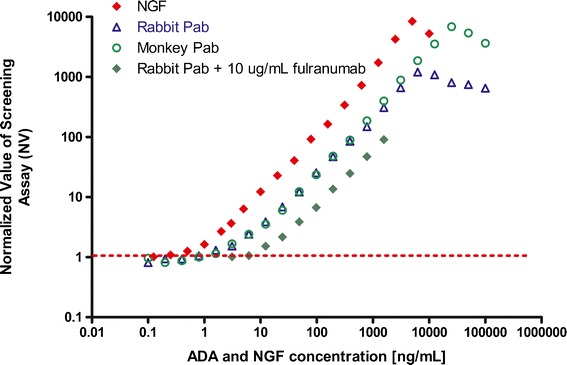
ADA detection sensitivity and NGF detection in the same ECLIA with the acid-dissociation pretreatment step. All samples were prepared in undiluted pooled normal human serum. Serial dilutions of an affinity-purified anti-fulranumab rabbit polyclonal antibody (without or with fulranumab), an affinity-purified anti-fulranumab monkey polyclonal antibody, and NGF alone were tested in the screening assay with acid pretreatment
A confirmatory assay based on the inhibition of positive signal in the screening assay by 200 μg/mL of unmodified fulranumab was evaluated. To that end, the inhibitory effect of fulranumab on the true ADA response as well as the interfering NGF response was examined. This was studied using three concentrations of anti-fulranumab monkey polyclonal antibody (10, 100, and 1,000 ng/mL) and NGF (10, 100, and 1,000 ng/mL). These mock samples were first incubated either with or without 200 μg/mL of fulranumab in assay diluent for 60 min and then tested in the screening assay. Result showed that signal resulting from the anti-fulranumab antibody at 10 ng/mL was completely inhibited (below the screening assay cut point) by 200 μg/mL of fulranumab, and that 100 and 1,000 ng/mL of anti-fulranumab antibody were inhibited by over 90%. A similar inhibition pattern was observed for NGF samples when pre-incubated with 200 μg/mL of fulranumab (data not shown). These results clearly indicated that the false-positive NGF signal in the fulranumab screening ADA assay could not be differentiated from a specific result using competitive inhibition with fulranumab in our confirmatory assay. Therefore, without removing NGF signal prior to analysis, the false-positive signal from NGF would have been misinterpreted as an ADA-positive result, leading to inaccurate reporting of antibody incidence in our clinical studies.
We then studied how the acid treatment steps in the screening assay affected the inhibition pattern of fulranumab on NGF signals, and tested the hypothesis that acid pretreatment of samples released NGF from its bound state (fulranuma–NGF complex) and caused false-positive signal at low levels. Several sets of samples were prepared with fulranumab at 0, 5, and 100 μg/mL in serum and combined with different levels (i.e., up to 2,000 ng/mL) of NGF in normal human sera. The samples were incubated at room temperature for 2 h to ensure complex formation between NGF and fulranumab. Then the samples were tested in the bridging ECLIA with or without acid pretreatment. In the presence of 100 μg/mL (667 nmol) of fulranumab, concentrations of NGF up to 1,000 ng/mL (76 nmol) did not cause false-positive results when acid dissociation was not employed. On the other hand, 10 ng/mL (0.76 nmol) of NGF pre-incubated with 100 μg/mL (667 nmol) of fulranumab caused false-positive signal when pretreated with acid (Fig. 3). These results demonstrated that fulranumab-bound NGF did not interfere with ADA detection when acid dissociation was not employed. However, when acid dissociation was employed to eliminate fulranumab interference, the low pH conditions released fulranumab-bound NGF, which produced false-positive results. Therefore, acid pretreatment of samples not only dissociated the fulranumab-ADA complex but also the fulranumab-NGF complex, and the resulting “free” ADA or NGF can produce positive results in the assay. Because acid-dissociation pretreatment was extremely valuable to improve the ability of this assay to detect ADA in the presence of drug, it was critical to develop a strategy to remove the impact of NGF from samples before acid-dissociation.
Fig. 3.
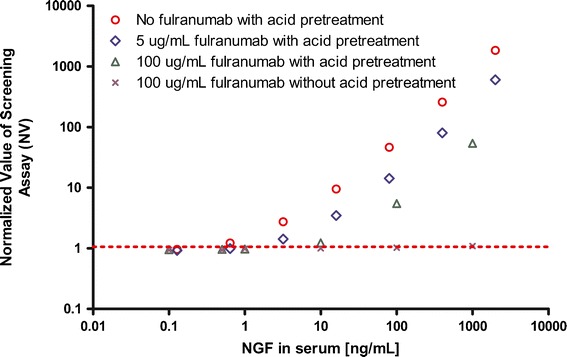
In the ECLIA with acid pretreatment NGF causes a false-positive ADA dose–response curve that is partially and dose dependently suppressed by fulranumab. Increasing concentrations of NGF up to 2,000 ng/mL were spiked into normal human serum. Fulranumab was added to NGF-spiked samples at 5 and 100 μg/mL and incubated for 60 min at room temperature. The NGF samples with or without fulranumab were tested in the screening assay including or excluding an acid-dissociation pretreatment step. The result showed that NGF up to 1,000 ng/mL remained negative in the presence of 100 μg/mL fulranumab in the screening assay without the acid-dissociation pretreatment step
Drug Target Removal by Melon™ Gel Treatment
Melon™ Gel is a commercially available resin that binds non-antibody proteins in serum, such as albumin and transferrin, allowing the antibody to flow through in a mild buffer suitable for downstream applications. Melon™ Gel separation was intended to remove NGF from serum samples as an approach to develop the confirmation assay. Samples containing different levels of NGF and ADA were pre-treated with Melon™ Gel, after which they were analyzed in the screening assay. The results showed over 80% reduction of false high assay signal when samples containing NGF at 25, 50, 100, and 200 ng/mL were pretreated with Melon™ Gel. However, those NGF-containing samples remained positive (above the assay cut point of 1.04 NV) in the screening assay even after the Melon™ Gel treatment. On the other hand, Melon™ Gel pretreatment of samples containing ADA (polyclonal rabbit anti-fulranmab and polyclonal monkey anti-fulranumab) showed less than 50% reduction in ADA-positive signal (data not shown). Therefore, it might be possible to use Melon™ Gel pretreatment to develop a confirmation assay based on the fact that the NGF signal was more effectively inhibited than that of true-positive ADA. Indeed, this was the approach taken when the original confirmation assay was developed utilizing Melon™ gel pretreatment for phase 1 study samples, and the specificity cut-off for an antibody response was <75% inhibition after Melon™ gel treatment. However, we later found that Melon™ gel bound non-specifically to many proteins, including antibodies, even though it removed a greater proportion of NGF signal than ADA signal. Furthermore, the Melon™ gel did not remove NGF/fulranumab complexes as effectively as free NGF. Acid treatment after Melon™ gel separation broke apart the NGF/fulranumab complex and caused significant positive signals from the “freed” NGF as demonstrated in the mock sample study in the later section.
Evaluation of Protein G Plates to Selectively Remove IgG in Specificity Assay
Protein G is an immunoglobulin-binding protein. We found that protein G filter plates had the capacity to deplete several human donor serum samples of antibodies (data not shown). Therefore, a method was developed to use a protein G plate to effectively remove immunoglobulin G from human serum to confirm the true ADA response. In this method, samples identified as positive in the screening assay were treated with protein G plates to deplete the antibodies before the sample flow-through was reanalyzed in the screening assay. A sample that was positive for binding in the screening assay and negative for binding after protein G treatment would be identified as positive for the presence of anti-drug antibody.
In an effort to evaluate the effectiveness of antibody depletion by protein G plates, ADA-positive samples were prepared by spiking a normal human serum pool with different levels (1, 10, 100, and 1,000 ng/mL) of monkey or rabbit PAb against fulranumab. For comparison, concentrations of NGF were prepared in normal human sera as well. The positive ADA antibodies were removed effectively, reducing ADA signals at all four concentrations below the cut-off. On the other hand, the false-positive NGF signals were not affected by protein G treatment and the NGF samples remained positive for binding after the protein G treatment. These results indicated that protein G treatment could be utilized in the confirmatory assay to distinguish ADA from other non-antibody proteins when they were present individually in samples.
To further evaluate the strategy of using protein G to deplete antibodies in the specificity assay, mock samples were generated with combinations of ADA, drug target, and soluble drug in normal human sera at concentrations intended to mimic expected clinical study samples. It was expected that NGF would accumulate in serum in the form of a complex bound to fulranumab. The mock samples were tested in the confirmatory assay with protein G treatment and the results showed that the positive signal due to 20 ng/mL NGF alone was similar before and after protein G treatment, correctly indicating that the signal was not due to ADA (Fig. 4). However, the signal from the mock sample with a combination of 20 ng/mL of NGF and 200 ng/mL of fulranumab was positive before protein G treatment and negative after protein G treatment. This demonstrated that NGF bounded to the fulranumab antibody drug was also depleted by the protein G plates and could be misidentified as ADA positive. In another mixture, the mock samples containing 20 ng/mL of ADA and 20 ng/mL NGF were positive for binding with and without the protein G pretreatment step (because the signal was reduced by only 40% and was not below the screening assay cut point). Therefore, this mock sample would have been misidentified as ADA negative based on the positive signal from ADA plus NGF before protein G treatment and the positive signal due to NGF alone after immunoglobulin depletion by protein G. These experiments demonstrated that the protein G confirmation method could misidentify the ADA status if real samples had NGF complex with drug or if both ADA and NGF were simultaneously present in the sample.
Fig. 4.
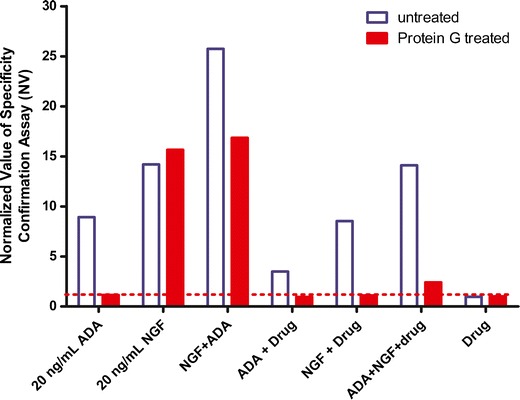
Evaluation of Protein G beads to selectively remove IgG prior to testing ADA in the specificity assay. When test samples contained drug, pretreatment of monkey polyclonal ADA with protein G beads effectively removed IgG and reduced ADA signals below the cut point. The serum sample spiked with 20 ng/mL of NGF and 200 ng/mL of fulranumab showed negative for binding after the treatment with protein G plates
Affinity Capture and Remove Drug Target with Target Specific Binding Antibodies/Proteins
To selectively remove NGF from samples without reducing ADA concentration, a method was developed to treat samples with an alternative monoclonal anti-NGF antibody (from BioLegend, San Diego, CA, USA) immobilized onto magnetic beads and another soluble NGF-blocking protein. The samples of pooled human serum containing increasing concentrations of NGF were tested in the screening assay before and after NGF removal via anti-NGF antibody-coated magnetic beads. Figure 5 showed that without NGF removal the NV ratio increased proportionally with the concentration of NGF so that concentrations of 0.1 ng/mL or greater showed a false-positive signal. However, when NGF was combined with 5 μg/mL of fulranumab, false-positive signals did not occur until NGF reached 2 ng/mL, indicating that fulranumab partially blocked NGF interference in the ADA screening assay. By contrast, NGF concentrations up to 100 ng/mL, either alone or in complexes with fulranumab, were eliminated effectively by treating the samples with anti-NGF antibody-coated beads (Fig. 5). Further, the positive signal of ADA (anti-fulranumab polyclonal rabbit antibody and polyclonal monkey antibody) was not affected by the treatment with anti-NGF beads (data not shown). Therefore, NGF interference was eliminated and ADA detection was preserved when anti-NGF beads were used to remove NGF.
Fig. 5.
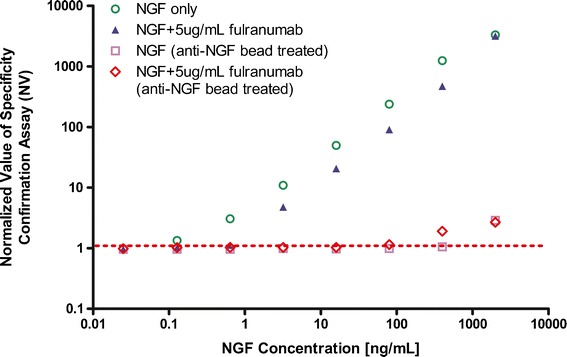
Evaluation of anti-NGF antibody-coated beads to selectively remove NGF. Serum spiked with increasing concentrations of NGF were prepared with or without 5 μg/mL of fulranumab then ADA specificity was tested with and without anti-NGF bead pre-treatment. Up to 100 ng/mL of NGF interference was eliminated and the presence of drug did not influence the outcome
Based on the selective removal of NGF by the anti-NGF beads, a two-step specificity confirmation method was developed. In this method, the samples were treated first with anti-NGF beads as well as NGF blocking protein to remove NGF and then the bead-treated samples were inhibited with fulranumab to confirm the true ADA-positive signal. To establish an appropriate specificity method cut point, 25 individual human serum samples from naïve healthy and diseased individuals were evaluated with the inhibition of fulranumab in the two-step specificity confirmation assay. From the inhibition of those 25 serum samples, the specificity inhibition cut point was determined to be 22.6%. The final immunogenicity testing scheme and interpretation of results for fulranumab clinical studies was illustrated in Fig. 6.
Fig. 6.
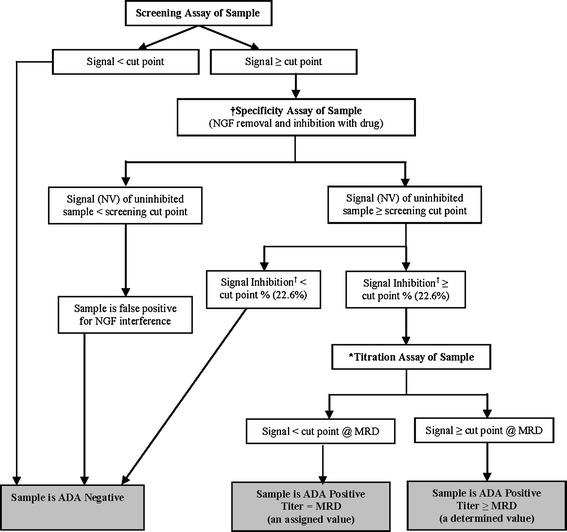
Interpreting the ADA result of a test sample in fulranumab clinical studies (based on the results of three types of assays: Screening, Titration, and Specificity) Dagger sign, the specificity cut point is the minimum percentage inhibition at which, or above, the result indicates a specific immune response. Asterisk, titration assays are performed after the samples are confirmed positive in the specificity assay. Legend: ADA—anti-drug antibody; MRD—minimum required dilution
When treated with fulranumab, patient sera may have different levels of fulranumab, ADA, NGF, and their immune complexes. To mimic this situation, we prepared 11 mock samples containing varying amounts of ADA and/or fulranumab and/or NGF in NHS. Those mock samples were evaluated using the final two-step confirmatory method to identify ADA. For comparison, the mock samples were also tested with the original confirmatory assay, in which Melon™ Gel was used to remove NGF. As shown in Table I, the confirmatory method using Melon™ Gel treatment correctly classified the ADA status of seven out of eleven mock samples, whereas the two-step specificity confirmatory method correctly classified eleven out of eleven mock samples. This demonstrated the superiority of using target specific removal in the confirmatory assay instead of the much less specific Melon™ Gel removal technique.
Table I.
Mock Serum Samples with ADA, NGF, and Fulranumab
| Samples | Melon™ gel confirmation | “Double” confirmation | ADA (+/−) |
|---|---|---|---|
| 20 ng/mL ADA1 + 200 ng/mL NGF | − | + | + |
| 200 ng/mL NGF | − | − | − |
| 20 ng/mL ADA1 (monkey ADA) | + | + | + |
| 20 ng/mL ADA1 + 5 ug/mL fulranumab | + | + | + |
| 20 ng/mL ADA1 + 3 ng/mL NGF | + | + | + |
| 3 ng/mL NGF | − | − | − |
| 5 μg/mL fulranumab + 200 ng/mL NGF | + | − | − |
| 5 μg/mL fulranumab + 3 ng/mL NGF | + | − | − |
| 500 ng/mL ADA1 | + | + | + |
| 500 ng/mL ADA2 (mouse ADA) | − | + | + |
| 2 ng/mL ADA1 | + | + | + |
| No. classified correctly | 7/11 | 11/11 |
Mock serum samples containing different levels of ADA, NGF, and fulranumab were treated with anti-NGF MAbs-coated beads and followed with competitive inhibition of fulranumab in two-step confirmatory assay. Using Melon™ Gel treatment as the confirmatory method, the ADA status for 7 out of 11 mock samples were classified correctly, whereas 11 out of 11 mock samples were classified correctly using the two-step confirmatory method
Clinical Sample Analysis
In a phase 1a single ascending dose clinical study of fulranumab in healthy human volunteers, anti-fulranumab antibodies were detected in 1 out of 34 dosed subjects (3%) using our initial ADA method which utilized Melon™ Gel separation in the specificity confirmation test. In striking contrast, 13 of 18 (72%) dosed patients were positive for anti-fulranumab antibodies in a phase 1b multiple ascending dose clinical study of 24 subjects with osteoarthritis knee pain using the same assay. Realizing that the assay was prone to NGF interference, and that NGF would be increased in patients relative to healthy subjects, we decided to validate the two-step confirmatory method and retest a subset of the phase 1b clinical study samples in order to determine whether the high positive ADA incidence (72%) could be an artifact of NGF interference. Fifteen samples (eight of which were presumed to be ADA positive per the Melon™ Gel confirmatory assay) were selected from the clinical study, de-identified (blinded to the assay operator), and evaluated according to the two-step confirmation method. The retest indicated that all of the samples were in fact ADA negative (Fig. 7).
Fig. 7.
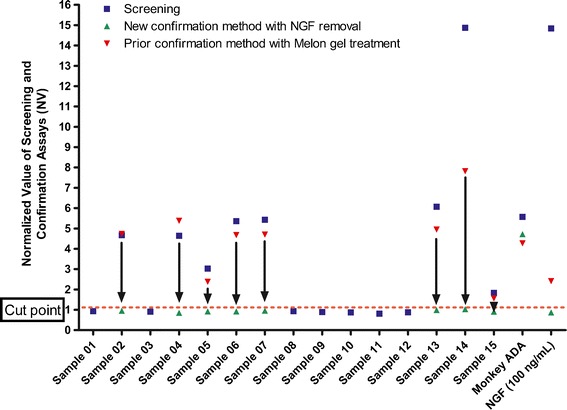
ADA analysis of anonymized clinical samples by the two-step confirmatory method and Melon™ Gel method. Seven samples were negative in both methods. Eight out the 15 samples that were originally identified as ADA positive by the Melon™ Gel method were determined to possess interfering NGF when the double confirmatory method was used
Four phase 2 clinical studies were conducted from which 4,184 samples were obtained from 1,031 pain patients. By applying our tiered ADA testing scheme, we identified large proportions of samples that showed an apparent positive result in the screening assay, most of which were proven to be false-positive results after testing the samples with the specificity confirmation test (Table II). In the clinical trial of patients with lower back pain, subjects with 12-week dosage regimens of fulranumab were compared with those who received placebo. After the initial screening assay, 899 of 1,477 (60.9%) samples were detected as potentially positive for anti-fulranumab antibodies. However, using the two-step confirmatory method, only 19 (1.3%) samples were confirmed to be true ADA-positive samples. In this study, 309 subjects treated with fulranumab were evaluable for immunogenicity assessment (i.e., had post-administration serum samples that were available for analysis). Among these subjects, the ADA incidence was also 1.3% (4/309 subjects). In the trial consisting of patients with knee and hip pain due to osteoarthritis, 380 subjects treated with fulranumab were evaluable for the immunogenicity assessment, of whom only 0.5% (2/380 subjects) were ADA positive. In the chronic knee pain osteoarthritis study, none of the 98 evaluable subjects developed antibodies to fulranumab. And lastly in the clinical study of interstitial cystitis patients, none of the 14 evaluable subjects developed antibodies to fulranumab. Overall, these results from all four clinical studies indicated that the majority of samples detected in the initial screening assay were false-positives due to NGF interference.
Table II.
ADA Detection in Fulranumab Clinical Studies
| Indication (phase 2 study) | Number of samples evaluated for anti-fulranumab antibodies/evaluable patientsa | Screening assay positive (“potentially ADA positive”) samples | Confirmed ADA-positive samples determined using the “double” confirmatory method | Confirmed ADA incidence (ADA-positive patients) determined using the “double” confirmatory method of dosed cohorts |
|---|---|---|---|---|
| Low back pain | 1,477/309 | 899/1,477 (60.9%) | 19/1,477 (1.3%) | 4/309 (1.3%) |
| Knee and hip pain from osteoarthritis | 1,992/380 | 1,106/1,992 (55.5%) | 10/1,992 (0.5%) | 2/380 (0.5%) |
| Chronic knee pain from osteoarthritis | 610/98 | 204/610 (33.4%) | 1/610 (0.2%) | 0/98(0.0%) |
| Interstitial cystitis | 105/14 | 34/105 (32.4%) | 0/105 (0.0%) | 0/14 (0.0%) |
A high rate of apparent ADA detection was found using the screening assay, which was determined to be false-positive results due to NGF. In fact, in all four phase 2 studies fulranumab induced very low or no ADA incidence
aFulranumab-treated subjects with at least one post-administration sample tested for anti-fulranumab antibodies were considered suitable for the assessment of immunogenicity
To support our findings from the ADA methods, the NGF levels were evaluated in false-positive ADA samples from knee/hip pain and chronic knee pain studies. Levels of “total” NGF (i.e., “bound” NGF in complex with fulranumab as well as “free” soluble NGF) were quantified using a validated method (sensitivity of 4.88 pg/mL, data not shown). The relationship between false-positive signal in the ADA screening assay and total NGF levels was studied in samples collected at day 84 following fulranumab administration in the knee/hip pain and chronic keen pain clinical studies.
The false-positive ADA samples in both knee/hip pain and chronic keen pain studies showed a significant positive correlation (P < 0.0001) to total NGF levels in the samples (Fig. 8) with r = 0.839 for knee/hip pain and r = 0.928 for chronic knee pain, respectively. In addition, no significant difference was found between the slopes of linear regression fit for the two studies (0.431 for knee/hip pain and 0.496 for chronic knee pain, respectively). More interestingly, the linear regression fit for both studies had the similar interception of x-axis of log [NGF] concentration at 2.33 for knee/hip pain and 2.29 for chronic keen pain, indicating that approximately 200 pg/mL of NGF, or greater concentrations, caused false-positive signal in the ADA screening assay (NV > 1.04). This result supported our prior findings during the validation of the ADA method that NGF caused a false-positive signal at concentrations exceeding 100 pg/mL in the screening assay. Only seven samples had high false-positive ADA assay signals with no detectable NGF; these false-positive results were probably not caused by NGF interference and were excluded from the correlation. These findings provided additional evidence that the majority of the apparently ADA-positive results from the ADA screening assay were in fact false-positive signals caused by NGF interference.
Fig. 8.

Comparison of false-positive ADA signal with total NGF levels in clinical studies. For both osteoarthritis and chronic knee pain studies, a comparison of false ADA response and total NGF data revealed an increase in false-positive signal (NV) with increased NGF serum concentration. For chronic knee pain study, a number of 59 false-positive samples had the correlation coefficient r = 0.928 with slope of 0.496 and x-axis intercept at 2.29. For osteoarthritis study, a number of 356 false-positive samples had the correlation coefficient r = 0.839 with slope of 0.431 and ×intercept at 2.33
DISCUSSION
We developed a sensitive double-antigen bridging ECLIA with acid-dissociation pretreatment as the screening assay to identify samples that are potentially positive for antibodies against fulranumab. Immune complex dissociation by acid reduces the interference caused by the drug present in the sample (13), and the bivalency of the double antigen bridging format allows for a highly selective and specific detection of most human anti-human antibodies (19). However, due to the fact that drug conjugates are utilized as the capture and detection molecules, this type of assay is susceptible to drug and target interference.
Target interference is a common problem in ADA methods for therapeutic antibodies that bind soluble targets such as cytokines. The interference of circulating target on the ADA detection assay depends on the form of the drug target, whether it is “free” or “bound”, and the format of the assay (e.g. with or without acid dissociation). Specifically, if a drug target has two drug-specific epitopes or if a drug target exists as a homodimer or multimer, the “free” target may be able to bridge the assay reagents and cause a false-positive result. We demonstrated that false-positive interference from the NGF homodimer was an important consideration for accurate identification of antibodies to fulranumab. Under experimental conditions, NGF concentrations as low as 100 pg/mL caused false-positive signals in our screening assay. Because endogenous NGF levels are known to be less than 12 pg/mL in healthy humans (20), no impact on the ADA assay is anticipated in healthy donor samples. However, disease and exposure to therapeutic drugs have been associated with increased production of many targets. Furthermore, therapeutic monoclonal antibodies can act as a carrier, allowing targets such as NGF to accumulate in a “bound” state in treated patients.
While it is expected that accumulated NGF is bound to excess fulranumab and unable to interfere in a typical immunoassay not involving acid pretreatment, this “bound” NGF can be released under low pH conditions and cause a false-positive signal in a “drug tolerant” ADA detection method that includes acid-dissociation pretreatment. We observed a significant improvement in drug tolerance with acid pretreatment in the screening assay (20 ng/mL of positive control remained positive in the present of 50,000 ng/mL fulranumab) compared to without acid pretreatment (20 ng/mL of positive control were negative in the presence of 100 ng/mL of fulranumab). However, it appears that the acid step can displace not only ADA, but also target bound to drug. When the target is multimeric, such as the NGF homodimer, it can bridge drug conjugate reagents and cause a false-positive signal in the ADA assay. Prior to this discovery, this phenomenon led to false-positive signals in a majority of the samples resulting in an apparent high incidence of ADA.
According to our tiered testing scheme, we originally developed a competitive inhibition assay to confirm the specificity of the “potentially positive” ADA detected in our screening assay. In this confirmation method, samples were preincubated with or without a high concentration of drug to inhibit the assay signal beyond a predetermined inhibition cut point value. Inhibition beyond the cut point confirms the presence of an anti-drug antibody. In this initial confirmation assay, a fulranumab-based inhibition test could not distinguish between anti-fulranumab antibodies and NGF because signals produced by either could be competitively inhibited by fulranumab.
A variety of strategies have been used to confirm drug-specific reactivity in potentially ADA-positive samples (21). However, past work has largely ignored the effect of drug-bound target interference on these assays. Of note, even when investigators have determined the effect of target in their ADA methods, the focus had been on free target which is typically very low in the blood. What we have discovered and described in this paper is that the acid-dissociation pretreatment of samples (to reduce drug interference) frees up target that has accumulated to high concentrations in a bound state (immune complex with the therapeutic mAb drug). The ability to differentiate positive samples from false-positive samples is critical if one hopes to interpret the influence that ADA may have on patient treatment. We investigated several strategies intended to improve the anti-fulranumab specificity assay. Not presented here was an initial attempt to circumvent this problem by developing a confirmatory assay in a sandwich format. Unlike the bridging format, a sandwich format should not allow target to cause false-positive results. Therefore, we developed a confirmation assay in a sandwich format with biotinylated fulranumab F(ab′)2 capture and ruthenylated rabbit anti-human IgG Fc-based detection. As predicted, the assay was indeed resistant to NGF interference (at concentrations 100 times of physiological levels). Nevertheless, this assay was not pursued because it was approximately 100 times less sensitive for ADA detection than the bridging assay. Hence, we adopted the latter format for both screening and confirmatory assays.
To address NGF interference and to eliminate the false-positive results in the fulranumab ADA confirmation assay, several sample pretreatment options were evaluated for their effectiveness to distinguish true antibody responses from non-antibody interferences. One method tested was sample pretreatment with Melon™ Gel. Use of non-antibody binding gels like Melon™ Gel to reduce non-specific factor was reported to remove soluble drug target as well as other non-antibody proteins (22). Hence, in our initial confirmation assay, we had applied Melon™ Gel pretreatment to reduce the NGF-positive interference in samples and applied it in our phase 1 studies. However, the high apparent ADA incidence in the phase 1b study, and our subsequent investigations with mock samples indicated that the confirmation assay involving Melon™ Gel was ineffective because it frequently failed to distinguish ADA from NGF interference that induced false-positive results.
Another common strategy to confirm ADA-positive samples involves depleting antibodies from samples identified as “potentially positive” in the screening assay and then reanalyzing those depleted samples, wherein a loss of assay signal infers antibody induced signal (21). While this strategy cannot differentiate specific antibody reactivity versus non-specific antibody reactivity, it can be particularly useful when the nonspecific interference with the assay is caused by a non-antibody serum protein, such as NGF. Protein A/G/L-coated plates or beads were reported to be utilized to remove the antibodies from samples to confirm anti-drug antibody response toward therapeutic proteins as well as the presence of neutralizing antibodies in a biological assay (23,24). It is important that the method used to remove immunoglobulin in this strategy is specific for immunoglobulin removal and not removing other non-antibody interferents. Our experiments with protein G plate pretreatment of serum samples demonstrated that protein G plates were able to remove serum immunogloblin effectively up to 1,000 ng/mL of ADA, while the serum NGF signal was not affected by protein G treatment. This initial testing of the specificity of immunoglobin removal by protein G plates indicated that protein G plates could be suitable to distinguish false NGF response from that of true ADA. However, after we tested mock serum samples spiked with combinations of ADA, NGF, and fulranumab with protein G plates, the results became more complicated, especially when NGF-drug complexes were present. One mock sample contained approximately equal molar amounts of NGF (20 ng/mL) and fulranumab (200 ng/mL) spiked into naive human serum to mimic clinical samples that may contain elevated levels of NGF bound with fulranumab. As expected, this sample showed strong positive signals in the ADA screening assay because the acid-dissociation step disrupted the NGF/drug complex and the released NGF to produce a signal in the screening assay (a false-positive ADA result). After this mock sample was treated with the protein G plate to remove all of the immunoglobulin and reanalyzed in the screening assay, the signal was negative. This experiment clearly indicated that the protein G plate effectively removed the fulranumab (a human IgG2 antibody) as well as fulranumab-bound NGF, which led to a depletion of signal in the assay and an erroneous inference of ADA confirmation. This experiment demonstrated that when designing a confirmation assay, it is critical not only to understand possible free drug target levels that could interfere in assays but also the drug and target complexes that can form in circulation after the administration of drug. To understand the levels of free and bound NGF in our study patients, we separately developed and validated quantification methods. Among patients who received fulranumab treatment, the concentration of NGF was below 20 pg/mL prior to treatment. However, after treatment, the total NGF (free NGF plus fulranumab-bound NGF) was observed as high as 20,000 pg/mL, the majority of which was found to be in the fulranumab-bound state (data not shown). Based on our studies with mock samples and the evaluation of NGF concentrations in clinical samples, we concluded that protein G depletion could not reliably confirm ADA specificity for our clinical study samples. This finding indicated the inadequacy of the specificity methods we attempted thus far, leading us to consider selective removal of all NGF (both free and bound NGF) from samples before performing a competitive inhibition-based ADA specificity confirmation assay.
Therefore, our final strategy involved a two-step specificity confirmation assay that first used anti-NGF antibody-coated beads to selectively remove NGF (both free and bound to fulranumab), followed by competitive inhibition with fulranumab. Using this two-step confirmation assay, we could correctly identify the ADA status of the mock samples containing varying amounts of ADA, fulranumab, and NGF in combination. Analysis of samples from four phase 2 clinical studies showed that high false-positive ADA results (>50%) were observed in the screening assay and that most of the positive samples were categorized as NGF interference in the two-step confirmation assay, resulting in a true ADA-positive incidence of <1.3%. False-negative and false-positive data from inaccurate test methods could lead to flawed correlations of ADA with clinical safety, pharmacokinetics, and efficacy results. For example, an apparent “high incidence” of ADA would poorly correlate with the few truly ADA related adverse events, leading to the erroneous conclusion that ADA was inconsequential despite the “high incidence”.
This study demonstrated that a lack of an appropriately characterized confirmation assay for ADA detection can result in a dramatically inaccurate ADA incidence. Our work underscores the importance of developing ADA methods that are not only capable of resisting drug interference, but also capable of providing accurate ADA results despite the presence of free or bound target.
CONCLUSION
The ability to accurately detect the presence of antibodies toward protein therapeutic drug in patient serum samples depends on a well-designed screening assay and a highly selective confirmation assay. We developed a screening assay with acid pretreatment to detect the ADAs toward the monoclonal human anti-NGF drug fulranumab in all samples. Despite the advantage of improving drug tolerance, the widely implemented acid treatment procedure was found to cause unintended false-positive interferences due to drug target NGF. We demonstrated that the homodimeric drug target NGF interfered with accurate ADA detection and that the acid pretreatment procedure included in the screening assay made target interference worse by releasing additional target from drug-target immune complexes. Drug bound NGF presented the greatest challenge and different strategies were carefully designed and thoroughly investigated to eliminate the interference. After numerous attempts, we developed a novel two-step confirmatory assay with selective removal of drug bound NGF to confirm the true ADA response. Finally, the two-step confirmatory assay provided accurate measurement of ADA responses in four clinical studies. The results showed very low ADA incidence for all the studies. Furthermore, we advise that acid-dissociation pretreatment in ADA detection assay must not be universally applied to improve drug tolerance in ADA assays without cautious examination of its analytical risks versus benefits.
ACKNOWLEDGEMENTS
The authors thank Monica Keen and George Gunn (Janssen R&D LLC) for providing relevant information from the clinical studies, Thomas McIntosh (Janssen R&D LLC) for the information about NGF levels in clinical studies.
Conflict of Interest
All authors are employees of Janssen Research and Development, LLC., a division of Johnson and Johnson Inc. This work was partially presented as a poster presentation at the 2012 AAPS National Biotechnology Conference, May 21–22 in San Diego, CA, USA.
REFERENCES
- 1.Aloe L, Probert L, Kollias G, Bracci-Laudiero L, Spillantini MG, Levi-Montalcini R. The synovium of transgenic arthritic mice expressing human tumor necrosis factor contains a high level of nerve growth factor. Growth Factors. 1993;9:149–155. doi: 10.3109/08977199309010830. [DOI] [PubMed] [Google Scholar]
- 2.Bai G, Kusiak JW. Nerve growth factor up-regulates the N-methyl-d-aspartate receptor subunit 1 promoter in PC12 cells. J Biol Chem. 1997;272:5936–5942. doi: 10.1074/jbc.272.37.23172. [DOI] [PubMed] [Google Scholar]
- 3.Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- 4.Roekaeus A, Jiang K, Spyrou G, Waschek JA. Transcriptional control of the galanin gene. Tissue-specific expression and induction by NGF, protein kinase C, and estrogen. Ann NY Acad Sci. 1998;863:1–13. doi: 10.1111/j.1749-6632.1998.tb10679.x. [DOI] [PubMed] [Google Scholar]
- 5.Winston J, Toma H, Shenoy M, Pasricha PJ. Nerve growth factor regulates VR-1 mRNA levels in cultures of adult dorsal root ganglion neurons. Pain. 2001;89:181–186. doi: 10.1016/S0304-3959(00)00370-5. [DOI] [PubMed] [Google Scholar]
- 6.Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24:1720. doi: 10.1016/S0149-2918(02)80075-3. [DOI] [PubMed] [Google Scholar]
- 7.Koren E, Zuckerman L, Mire-Sluis AR. Immune responses to therapeutic proteins in humans—clinical significance, assessment, and prediction. Curr Pharm Biotechnol. 2002;3:349–360. doi: 10.2174/1389201023378175. [DOI] [PubMed] [Google Scholar]
- 8.US Department of Health and Human Services, FDA (CDER, CBER). Guidance for industry—assay development for immunogenicity testing of therapeutic proteins. 2009. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM192750.pdf. Accessed 1 Aug 2013.
- 9.CHMP. Guideline of immunogenicity assessment of biotechnology derived therapeutic proteins. London: EMEA Guidance Document;2008.
- 10.Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333:1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48:1267–81. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Patton A, Mullenix MC, Swanson SJ, Koren E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J Immunol Methods. 2005;304:189–195. doi: 10.1016/j.jim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 13.Quinn TC, Kline R, Moss MW, Livingston RA, Hutton N. Acid dissociation of immune complexes improves diagnostic utility of p24 antigen detection in perinatally acquired human immunodeficiency virus infection. J Infect Dis. 1993;167:1193–1196. doi: 10.1093/infdis/167.5.1193. [DOI] [PubMed] [Google Scholar]
- 14.Roch AM, Delcros JG, Ripoll JP, Thomas V, Richard J, Quash G. A novel covalent enzyme-linked immunoassay (CELIA) for simultaneously measuring free and immune complex-bound antibodies of defined specificity: I. Application to naturally occurring antipolyamine antibodies in human sera. J Immunol Methods. 1990;4:1–11. doi: 10.1016/0022-1759(90)90312-J. [DOI] [PubMed] [Google Scholar]
- 15.Chen K, Page JG, Schwartz AM, Lee TN, DeWall SL, Sikkema DJ, et al. False-positive immunogenicity responses are caused by CD20+ B cell membrane fragments in an anti-ofatumumab antibody bridging assay. J Immunol Methods. 2013;394:22–31. doi: 10.1016/j.jim.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Doucet J, Avrameas A. A novel method for quantitative measurement of a therapeutic monoclonal antibody in the presence of its target protein using enzymatic digestion. J Pharm Biomed Anal. 2010;52:565–570. doi: 10.1016/j.jpba.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 17.Zhong ZD, Dinnogen S, Hokom M, Ray C, Weinreich D, Swanson SJ, et al. Identification and inhibition of drug target interference in immunogenicity assays. J Immunol Methods. 2010;355:21–28. doi: 10.1016/j.jim.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, NY, USA, 1988;298–299.
- 19.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289:1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Gold SM, Schulz KH, Hartmann S, Mladek M, Lang UE, Hellweg R, et al. Basal serum levels and reactivity of nerve growth factor and brain-derived neurotrophic factor to standardized acute exercise in multiple sclerosis and controls. J Neuroimmunol. 2003;138:99–105. doi: 10.1016/S0165-5728(03)00121-8. [DOI] [PubMed] [Google Scholar]
- 21.Swanson S, Chirmule N. Assessing specificity for immunogenicity assays. Bioanalysis. 2009;1:611–617. doi: 10.4155/bio.09.41. [DOI] [PubMed] [Google Scholar]
- 22.Qin S, Qiu W, Ehrlich JR, Ferdinand AS, Richie JP, O’Leary MP, et al. Development of a “reverse capture” autoantibody microarray for studies of antigen-autoantibody profiling. Proteomics. 2006;6:3199–3209. doi: 10.1002/pmic.200500673. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez S, Barger T, Zhou L, Hale M, Mytych D, Gupta S, et al. Strategy to confirm the presence of anti-erythropoietin neutralizing antibodies in human serum. J Pharm Biomed Anal. 2011;55:1265–1274. doi: 10.1016/j.jpba.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 24.Gupta S, Indelicato SR, Jethwa V, Kawabata T, Kelley M, Mire-Sluis AR, et al. Recommendations for the design, optimization, and qualification of cell-based assay used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321:1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]