

Abstract
The concepts, importance, and implications of bioanalytical method validation has been discussed and debated for a long time. The recent high profile issues related to bioanalytical method validation at both Cetero Houston and former MDS Canada has brought this topic back in the limelight. Hence, a symposium on bioanalytical method validation with the aim of revisiting the building blocks as well as discussing the challenges and implications on the bioanalysis of both small molecules and macromolecules was featured at the PITTCON 2013 Conference and Expo. This symposium was cosponsored by the American Chemical Society (ACS)—Division of Analytical Chemistry and Analysis and Pharmaceutical Quality (APQ) Section of the American Association of Pharmaceutical Scientists (AAPS) and featured leading speakers from the Food & Drug Administration (FDA), academia, and industry. In this symposium, the speakers shared several unique examples, and this session also provided a platform to discuss the need for continuous vigilance of the bioanalytical methods during drug discovery and development. The purpose of this article is to provide a concise report on the materials that were presented.
KEY WORDS: bioanalytical, macromolecules, small molecules, validation
INTRODUCTION
This report provides a summary of the symposium entitled “Bioanalytical Method Validation: Concepts, Expectations and Challenges in Small Molecule and Macromolecule” held at the PITTCON 2013 Conference and Expo on March 17–21, 2013 at Philadelphia, Pennsylvania, USA. This symposium was co-sponsored by the American Chemical Society (ACS)—Division of Analytical Chemistry and Analysis and Pharmaceutical Quality Section (APQ) American Association of Pharmaceutical Scientists (AAPS) and was featured on March 18, 2013.
While the importance of bioanalytical method validation has been stressed since the early days of “modern” drug development in the 1950s, the recent high profile problems related to bioanalytical method validation at both Cetero Houston (1, 2) and MDS Canada (3, 4) demonstrates a clear need for continuous vigilance. In both of these cases, failure to comply with the procedures and practices in their bioanalytical facilities led to a loss of reliability in the pharmacokinetic (PK) data generated leading to disastrous implications for pharmaceutical industries that used their services due to underlying regulatory impact on the safety and efficacy of the drug molecule (5). Hence, the time has come to revisit the building blocks of bioanalytical method validation, discuss challenges of doing a proper validation, as well as a need to understand how regulatory agencies (such as the FDA and EMA) view the importance and impact of these methods. This program featured five eminent speakers from academia, industry, and U.S. FDA and was widely attended by pharmaceutical scientists from academia, industry and regulatory agencies, and students.
The purpose of this program was to serve as a forum to discuss the concepts, expectations, and challenges in the bioanalytical method validation of small molecules and macromolecules. In addition, the program allowed for an opportunity to interact with experts from U.S. FDA and to encourage a two-way dialog on the mutual challenges faced by all in this area.
This report summarizes the numerous examples shared during this presentation to highlight the expectations and challenges from a bioanalytical assay of small and macromolecules.
BUILDING BLOCKS OF BIOANALYTICAL METHOD VALIDATION
While the field of bioanalysis has advanced methodologically from simple colorimetry to modern day techniques like LC MS/MS, NMR etc., the underlying principles and expectations of method validation have not changed. Even though our ability to quantify concentrations now reaches down to picograms and femtograms, the mantra of accuracy, precision, sensitivity, and selectivity is as true today as it was with Karl Fischer in 1935 (6).
Fortunately, as our ability to probe lower and lower concentrations have increased, the availability of regulatory guidance’s as to what is required to validate an assay has also evolved. Both the International Conference on Harmonization (ICH) in its “ICH Q2(R1) (7)” document and the FDA’s “Bioanalytical Method Validation Draft Guidance (8)” contain recommendations as to regulatory expectations. Although each Guidance Document is organized somewhat differently, upon examination the concordance is self-evident. However, if one looks deeper, one sees that these guidance’s are nothing more than a restatement of “plain old” good laboratory practices (GLP), the kind taught today in any college level analytical chemistry course (9). This concordance is due to a simple fact “good technique never goes out of style.” Even so, between the “theory of bioanalytical validation” as taught in college and the “reality of bioanalytical validation” in practice, there is still a need for both reinforcement and rededication of efforts.
As mentioned before, to validate an analytical method, you must be able to demonstrate the accuracy, precision, sensitivity, and selectivity of the assay over the working range. These elements must also be supported by additional data on the freeze-thaw stability, linearity of the method, robustness, and the ruggedness of the method. Of all of these, robustness and ruggedness are probably the least understood. In short, robustness of a method is a measure of its capacity to remain unaffected by small but deliberate variations in method parameters and provides an indication of its reliability during normal usage (7); while ruggedness is the degree of reproducibility of the test results obtained by the analysis of the same samples under a variety of conditions, such as different laboratories, analysts, instruments, lots of reagents, elapsed assay times, assay temperatures, etc. (10). There is not one measure of robustness that can be found in a textbook or guidance document; it can, however, be considered a reflection of the overall validation and controls used. Controls, not in the sampling sense, but in the sense of a well-documented and implemented series of Standard Operating Procedures (SOPs) that are used. Ruggedness of the method could be determined by analysis of sample lots in different laboratories, by different analysts, and by varying the operational and environmental conditions within the specified parameters of the assay (10).
Ultimately, the foundation of any drug development program must be firmly anchored upon the bioanalytical methods validation. While accuracy, precision, specificity, and sensitivity are the “building blocks” of a good method, but without the “binding” of well written and implemented SOPs as the metaphoric “mortar”, these building blocks, no matter how well stacked are unstable and will bring the entire development program down.
EXPECTATIONS FROM BIOANALYSIS IN CLINICAL PHARMACOLOGY
Drug concentration measurement has traditionally played critical roles in the development of new drugs. Multiple decision-making milestones in new drug development programs rely, at least partially, on the pharmacokinetic (PK) properties and the exposure-response relationships based on pharmacodynamics (PD) endpoints or clinical endpoints. These milestones reflect the go/no-go decision from the preclinical stage through all phases of clinical development, the dose selection for the first-time-in-man study, and the choice of doses for dose-ranging studies and for pivotal studies. Increasingly, drug companies are using exposure-response data and follow learn-and-confirm cycles (11,12) to enhance the productivity and efficiency of the new drug development process.
The PK attributes of a therapeutic moiety at each stage in development may be different and the intended use may dictate the bioanalytical technologies used. At the pre-clinical stages of the drug development, the intended uses may be to characterize the potential PK/PD relationship in an animal model, simulate the PD regimen, and to evaluate the systemic drug exposure and exposure-time course. However, these intended applications may have extensions to them depending on the characteristics of the disease state and the molecule being used. For example, the simulation of PK regimen may have extended applications to calculate safety margin, and determine the safe starting dose in the first in human (FIH) study. In this example, it will be imperative to know the concentrations of the pharmacologically active drug, which is sometimes referred to as the “free” drug.
At the clinical stage of drug development, the requirements are much more population and disease centric. Some of these are listed below:
Provide key PK parameters [e.g., area under the concentration curve (AUC), clearance (CL), volume of distribution (Vd)] in humans and to build the PK/PD model
Understand PK behavior in target populations
Correlate drug exposures to PD or efficacy endpoints
- Model PK/PD profiles to:
- Assess exposure variability and covariate effects
- Simulate PK/PD outcome for a desired regimen at later stages
Assess potential drug-drug interactions
Correlate exposure to safety endpoint to assess the maximum tolerated dose
Based on this understanding of the critical parameters, the analytical methodologies need to fit the intended use. Sometimes, non-validated methods are used for exploratory pilot studies, and scientists should be cautious when interpreting such results. Ultimately, the exposure and response data contribute to the totality of evidence of safety and efficacy submitted for global regulatory reviews, and they are used to support dosing recommendations as well as the clinical pharmacology sections of the product labels. As drug development turns more and more to biologics and macromolecules as therapeutic moieties other assay systems, with their own particularities, will need to be evaluated for their fitness-to-task and our assessment of them.
SCIENTIFIC CHALLENGES IN THE BIOANALYSIS OF SMALL MOLECULES AND MACROMOLECULES
The fundamental goal of bioanalysis is to report an analyte concentration that is an accurate reflection of the concentration in the sample at the time of collection. This is potentially a complex process that requires appropriate collection and storage of the biological sample, analysis by a trained bioanalytical scientist, and then the proper processing and reporting of the data.
The most reliable bioanalytical data is usually generated using a bioanalytical method that has been validated and applied according to the principles of Good Laboratory Practice (GLP) and Good Clinical Practice (GCP). Method validation is usually connected to bioanalytical assays supporting regulatory aspects of drug development. However, even where data will not be submitted to a regulatory agency, there are strong scientific advantages in using a fully validated method. One underappreciated aspect of validating a bioanalytical method is that the process of validation is an effective tool to identify problematic areas that prevent accurate analyte concentrations from being generated. Problematic areas can include chemical and physical instability of the analyte during collection and storage, interference during analysis by co-eluting molecules, and even the incompatibility of the analyte to detection using LC-MS/MS.
In small molecules, two case studies were presented detailing the development of bioanalytical methods. The first concerned a biomarker assay to measure ex vivo 11β-hydroxysteroid dehydrogenase type 1 (11βHSD-1) activity in human fat samples. The challenge identified in this method was a complex sample collection and manipulation process. The biomarker assay involved excising a gram size piece of fat from a human clinical subject receiving an oral 11βHSD-1 inhibitor, incubating this fat with a substrate of 11βHSD-1, and then calculating 11βHSD-1 activity. Results obtained using an “un-validated assay” were initially very promising, but then found to be impossible to reproduce in subsequent studies. Integrity of the biomarker assay was then addressed from a GLP validation perspective and the validation process highlighted specific areas of errors, including surgical collection of the sample, the stability of radiolabeled substrate, lack of optimization and reproducibility of the assay process, and low sensitivity of the radiometric detector used in the assay. Modifications were made in almost every one of these categories resulting in a rugged and reproducible biomarker assay being developed and applied during clinical testing. This example clearly demonstrates the limitations of using an un-validated method as it adds complexity towards data interpretation. The lessons from this single program were enough to create support for validating important preclinical and clinical biomarker assays to ensure reproducibility, comparability across studies, defined precision and accuracy of results, greater confidence in data, and compatibility to inclusion into a regulatory submission (e.g., supportive exploratory data).
A second example presented which detailed a method developed for the determination of a neuroscience drug in human cerebrospinal fluid (CSF). The challenge in this example was poor solubility of the drug in the CSF biomatrix. CSF is commonly collected for neuroscience drugs during in vivo testing due to the expectation that it is representative of brain concentration. Sample stability issues with the experimental drug in CSF were initially identified during a freeze-thaw experiment performed on assay quality control samples. Freeze-thaw stability is one component of a battery of stability oriented tests performed during GLP method validation. These also include bench-top/short-term stability, refrigerated/frozen stability, long-term stability, stock solution stability, post preparation stability, and incurred sample stability. Poor stability of the CSF drug in sample tubes was attributed to adsorption to the tube walls. This stability concern was then discovered to have larger implications during sample collection based on the use of spinal catheters. Additional experiments indicated that up to 30% of the drug could be lost during sample collection due to adsorption to the catheter tube. Substitution of a different catheter tube material did not alleviate the problem entirely. Additional attempts to reduce adsorption were considered, which included the use of custom tube materials or pretreating the existing catheters to saturate adsorption sites. Eventually, the decision was made to use the existing untreated catheters since the error due to sample loss was within the analytical tolerance required to effectively evaluate the presence of drug in the CSF.
The bioanalytical methods for large molecules generally do not afford adequate understanding of the molecular structure of the measured moiety like small molecules where LC/MS/MS methods are used to assess some degree of certainty on the structural identity of the measured molecule.
Ligand binding assays (LBA) are the most commonly used platforms for the quantification of macromolecules, and the concepts of this method were discussed in this symposium. Basically, the principle behind the LBA assay is a binding reaction of the therapeutic moiety to one or more reagents. Depending on the levels of interaction, in vivo the therapeutic moiety could exist as free (unbound) or complexed (bound to another molecule) or some combination thereof (Fig. 1). Understanding and quantifying the different forms that exist in vivo could be challenging but extremely important to understand the PK/PD relationship and its effect on safety and efficacy.
Fig. 1.
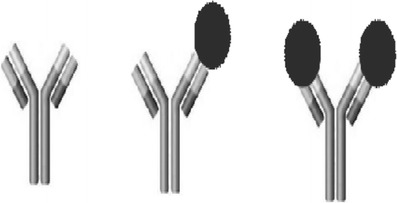
Monoclonal antibodies in study samples can exist in various forms of unbound (free mAb), partially bound (monovalently bound), and fully bound (divalently bound)
For example, in case of LBA for macromolecule bioanalysis, the most critical components are the reagents. Depending on the reagents used in the method, one can ensure what species are being quantified in the system (free, total, complexed). The reagents, primarily antibody against the protein therapeutics of interest, have to be specifically produced for each protein therapeutic agent separately, and in some cases, different reagents are used for preclinical studies versus for clinical studies (13). These assay reagents bind to the particular large molecule at some epitope(s) which represent a small fraction of the large, complex structure; therefore, there could be multiple sets of reagents to choose from during assay method development. Literature examples (13–15) have shown that depending on the reagents used in the immunoassays, the measured concentration may differ. The strong dependence of immunoassays on assay reagents demands thorough characterizations of the assay variables and a rigorous cross-validation plan when assays or reagents are modified. In addition to the critical reagents, the binding affinities, the reference material used in the system, incubation times, and the matrix, all play a role in the accurate and precise measurement of the therapeutic moiety. Multiple LBAs may be needed for the determination of all the parameters necessary to evaluate the PK and PD parameters. Though this is a laborious process, the quality of the results sometimes outweighs the effort.
Furthermore, depending on the molecular epitope(s) engaged with the assay reagents, the measured concentrations may or may not reflect the active (free) drug. For instance, if the protein therapeutics coexists with target ligands in the blood, it can exist as drug-target complex or as un-complexed form. The immunoassay may detect the un-complexed form only representing the free drug concentration if the binding epitopes are the same as those for ligand binding, whereas the immunoassay may detect both forms representing the total drug concentration if the binding epitopes are different from those for ligand binding. Besides the target ligands, other factors may influence the results of immunoassay assays. For example, endogenous immune-reactive materials may interact with the assay reagents leading to higher measured drug concentration, such as quantifiable levels in pre-dose samples. In some cases, the antidrug antibody (ADA) can act as a carrier and prolong the exposure of the protein therapeutics. More often, the presence of ADA is associated with a reduction in drug concentrations and an increase in clearance. When the levels of interfering factors vary over time, a precise characterization of the pharmacokinetic properties is even more challenging. It is thus imperative to deploy a well-designed assay and to rigorously assess the assay performance during validation, during sample analysis (16), and throughout the life-cycle of the method.
The use of fit-for-purpose analytical methods is a pragmatic strategy to establish a meaningful exposure-response relationship for decision-making during development and during regulatory review of the biologics license application (BLA). When a biologic product acts via binding to the target receptor, the free drug concentration would naturally be more useful to describe the exposure-response or receptor occupancy. Therefore, an assay method that utilizes anti-idiotype antibody reagents would be suitable. Romiplostim was a case example where free drug concentrations were used to describe the complex PK-PD relationship involving target-mediated drug disposition and PK-PD interplay (17). The PK-PD characteristics support an individual titration of romiplostim dose based on the clinical response (18) because romiplostim treatment results in an increase in the amount of target receptors and a decrease in its exposure. When a reduction of the circulating target ligand concentration is the treatment objective, target ligand bound to the biological product may prevent a precise determination of free drug concentration. Consequently, the target ligand concentration measurement may be required to establish a meaningful PK-PD relationship. In the case example of omalizumab, total omalizumab concentration, free ligand (IgE) concentration, and total ligand concentrations were measured, whereas the free omalizumab concentration was inferred (or estimated) by using a PK-PD model (19–21). Because a higher free IgE at baseline requires a higher dose of omalizumab, the starting dose for omalizumab was guided by the pre-treatment free IgE level (22)
In summary, the examples shared during the presentation clearly demonstrate:
PK-PD data are key elements for the success in drug development of small molecules and macromolecules, and bioanalytical methods hold the key to obtaining meaningful data.
Using validated bioanalytical methods, early in drug development might have a noted advantage in decision making.
A series of defined experiments should be performed for validation to give a comprehensive picture of a method’s ability to provide accurate results.
Method validation assures an evaluation of accuracy and robustness of an analytical method and gives the best likelihood of reporting an accurate concentration for a biological sample.
Unlike small molecule drug development, biologics development programs need fit-for-purpose bioanalytical methods.
Multiple assays may be necessary to fully understand the PK/PD relationships of biologic therapeutics, and specific reagents are essential to develop LBA ligand binding assays to measure total and free therapeutic protein and it’s corresponding complexed and free target.
Thorough characterizations of the assay, rigorous validation, careful assay execution, diligent monitoring of assay performance, and thoughtful presentation of data in regulatory submissions are critical to provide adequate support to the regulatory review and ultimately the product label.
Ultimately, if the goal of any development program is to produce meaningful data that is actionable from a safety and efficacy standpoint, then we must also look at the implications when there is a breakdown of process.
REGULATORY IMPLICATIONS
Taking a broader view, bioanalytical validation is the cornerstone of not only PK and clinical pharmacology, but also in clinically based decision making. If one does not have confidence in the data, and its collection, then how can one make rational dosing decisions based on the collected data? If one is not able to make rational dosing decisions, then how can one either determine with any degree of certainty the safety and efficacy of a drug product, let alone prepare a package insert?
To understand the issues involved and how they may crop up, we need to adopt a holistic approach to analytical validation. Not focusing exclusively on the number of quality control samples or standard curve preparation, per se, but the entire process from method selection, through validation, execution, and final report writing. This broad view is necessary as, like a weak link in a chain, a single point failure here can negate the entire process-no matter how well designed. While there can be many reasons for a failure of analytical technique, some of the more common errors encountered by FDA reviewers include, but are certainly not limited to:
Mislabeled samples
Improper shipping of samples
A flawed extraction
Analytical problems
Calculation issues
Analysis/reporting issues
Most of these issues are readily apparent to those familiar with the actual performance of analytical methods. What is not often seen is how they can become a cascade resulting in project failure and delay to market. An illustrative example of how these issues can combine to undermine a bioanalytical method is presented below, sufficiently obfuscated to disguise the drug involved. This example contains at its center examples of errors 1, 4, 5, and 6 from the previous list.
A Failure of Data/System Integrity
We have all seen data where one sample is grossly elevated relative to either sample before or after, and yet, even though the amount of mass transfer this represents is not possible, how many times does this data escape unchallenged and fit into the calculation of AUC or Cmax? In this instance, a contractor handling the data analysis for a sponsor was not able to calculate half-life for an intravenous (IV) drug in 12 of 16 subjects in one and 11 of 18 subjects in another study. Both studies were single dose studies of what was known to be a one compartment drug. Examination of the raw data revealed that for these 23 subjects, at either 6 or 12 h, there was a massive spike in the plasma concentration in a single sample (4, analytical problems), the computer algorithm calculating half-life could not reconcile this and stopped the calculation for that subject and moved on. AUCs were, however, constructed from this data and showed the existence of what could almost be called a subpopulation for absorption (5, calculation issues)! Instead of conducting a failure-mode analysis of this data, the report from the computer was initialed, signed off, and sent to a report writer, who in absence of any information from the contrary, wrote a report with the suspect data intact and unchallenged. This report was subsequently signed off by the analyst, Director of Analytical Services for the contractor, the Pharma Project Director, and the Vice-President for Scientific Services at the sponsor prior to submission of the NDA (6, analysis/reporting issues). Even though these signature supposedly indicated that they had read and agreed with the data contained within, it is obvious that they had not read it with sufficient detail but had in fact relied on the integrity of the systems due diligence to catch errors.
This is a disturbing example as it shows both that there appears to be both a reliance on the computer to examine the data and a lack of human oversight to actually examine the raw data sets in even a cursory way. The signal that out of 34 subjects, the computer could not calculate a standard parameter for fully three-quarters of the subjects should have elicited at least a raised “eyebrow” and questions. Whether or not there were issues of mislabeling of samples or shipment/stability issues is irrelevant. The fact that everyone went along with the report writer, who is probably the least likely person to raise an issue, shows a failure to understand even the potential of failure and its ramifications.
The underlying issue here is not new. The airline industry is currently facing this issue which they call the “glass cockpit problem (23)”. The advantage of today’s modern aircraft is that they are literally quite capable of flying themselves, except for the taxiing stage where navigating around ground baggage carts requires (at this time!) human observation and judgment. The problem with this approach is that the pilot, the highly skilled expert, is less directly involved in the “physical act” of flying and is more and more becoming a systems manager. Up until recently, flying an aircraft was a very tactile exercise, with pilots pulling and pushing on control yokes and pedals, the physical resistance maintained a situational awareness of their actions and the planes response. Now, through the use of multiple computers and algorithms designed to smooth-out control inputs to present smooth flight, much of the feedback to control inputs is dampened. This lack of physical confirmation can result in the loss of situation awareness at those very moments when quick decisive action is most called for. The result of this loss of awareness has resulted in tragic loss of life when the pilots became confused or distracted by the technology and did not assess the situation properly (24). For the airlines, being appropriately risk adverse, have countered with beefed up training schedule and crew resource management training to ensure that situational awareness is maintained by recreating in simulators the conditions that led to aircraft loss. In bioanalytical validation, the steps we use to prepare new assays and training to maintain the currency of our staff are direct analogs to these steps. Spiked samples, incurred sample reanalysis, and adherence to not only SOPs but norms of what a good analytical method is can minimize but not eliminate these concerns. The price of a quality system is eternal vigilance to performance.
Returning to the example given here, while not as dramatic as the loss of an aircraft, the failure of all levels to provide oversight ultimately cost the sponsor, in this example, in terms of time to market and market share by arriving on the market late. For the contractor, the reputational loss was nearly fatal to the contractor.
From a regulatory point of view, the issue is ultimately one of trust. Regulators trust that sponsors do “due diligence” in both the oversight of their contractors and their internal processes of data analysis and report writing. The finding of this kind of failure that encompasses “potentially” mislabeled samples, analytical issues, calculation issues, and report writing issues is hopefully a rare confluence of problems in a single study. Even so, a single point failure can be just as catastrophic if it is not caught. Of course, once such a failure is found, the regulator is duty bound to both take action and to examine the process deeper to see where the failure lay and take remedial action. We have in the introduction mentioned the examples of MDS Canada and Cetero (Houston). The cost to MDS and Cetero is well known, the cost to the individual sponsors in the diversion of resources to correct these issues and the pall it casted over the industry (reputation wise) is incalculable.
One can never expect the unexpected; however, in the case of bioanalytical validation, one CAN anticipate likely failure modes and single point failures and design systems to detect deviations. Hoping that things will work as designed is not a mitigation strategy. Over reliance on the computer to do quality control is not a strategy either. We do not have to be “luddites” when it comes to the use of technology in data acquisition and analysis, but we do need to exercise real and effective control of the processes.
PROGRAM CONCLUSIONS
Although the principles of bioanalytical validation are well understood and a part of key curricula of any collegiate chemistry program, due diligence in their application are necessary. Thru the presentations presented at this symposium, representatives from academia, regulatory agencies, and the pharmaceutical industry gathered to present their perspectives on both the methods of bioanalysis and the challenges ahead as we move into the future of macromolecules and technological advances in small molecules. Practical examples of real world bioanalytical challenges were presented along with recommendations on how we can move forward while maintaining appropriate levels of control, thus maintaining our trust in drug development.
“It is not enough to do your best; you must know what to do, and then do your best.”
-W. Edwards Deming
Acknowledgments
The authors thank Craig Lunte, Ph. D., the first speaker in this symposium for sharing his views on the building blocks of bioanalytical method validation.
Disclaimer
The views expressed here are the views of the scientists and do not represent the policy of their respective organizations.
References
- 1.FDA finds U.S. drug research firm faked documents. 2011 [Accessed on March 07, 2014]; Available from: http://www.reuters.com/article/2011/07/26/us-fda-cetero-violation-idUSTRE76P7E320110726.
- 2.Cetero Reaches Final Resolution with the Food and Drug Administration 2012 [Accessed on March 07, 2014]; Available from: http://www.prnewswire.com/news-releases/cetero-reaches-final-resolution-with-the-food-and-drug-administration-148658885.html.
- 3.MDS hit by FDA warning on pharmacokinetic data. 2007 [Accessed on March 07, 2014]; Available from: http://www.pharmatimes.com/Article/07-01-15/MDS_hit_by_FDA_warning_on_pharmacokinetic_data.aspx.
- 4.FDA requires new pharmacokinetic data from MDS Pharma clients. 2007 [Accessed on March 07, 2014]; Available from: http://www.pharmamanufacturing.com/blogs/onpharma/fda-requires-new-pharmacokinetic-data-from-mds-pharma-clients/.
- 5.Handbook of LC-MS bioanalysis: best practices, experimental protocols, and regulations 2013.
- 6.Fischer K. Neues verfahren zur maßanalytischen bestimmung des wassergehaltes von flüssigkeiten und festen körpern. Angew Chem. 1935;48(26):394–396. doi: 10.1002/ange.19350482605. [DOI] [Google Scholar]
- 7.ICH Harmonised Tripartite Guideline—validation of analytical procedures: text and methodology Q2(R1). 2005 [Accessed on March 04, 2014]; Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf.
- 8.Draft guidance for industry—bioanalytical method validation. 2013 [Accessed on March 04, 2014]; Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM368107.pdf.
- 9.Christian GD. Anal Chem. Sixth ed. John Wiley & Sons, Inc.
- 10.United States Pharmacopeia Chapter 1225. 2006 [Accessed on March 07, 2014]; Available from: http://www.pharmacopeia.cn/v29240/usp29nf24s0_c1225.html.
- 11.Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61(3):275–291. doi: 10.1016/S0009-9236(97)90160-0. [DOI] [PubMed] [Google Scholar]
- 12.Miller R, Ewy W, Corrigan BW, Ouellet D, Hermann D, Kowalski KG, et al. How modeling and simulation have enhanced decision making in new drug development. J Pharmacokinet Pharmacodyn. 2005;32(2):185–197. doi: 10.1007/s10928-005-0074-7. [DOI] [PubMed] [Google Scholar]
- 13.Blasco H, Lalmanach G, Godat E, Maurel MC, Canepa S, Belghazi M, et al. Evaluation of a peptide ELISA for the detection of rituximab in serum. J Immunol Methods. 2007;325(1–2):127–139. doi: 10.1016/j.jim.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Fischer SK, Yang J, Anand B, Cowan K, Hendricks R, Li J, et al. The assay design used for measurement of therapeutic antibody concentrations can affect pharmacokinetic parameters: case studies. MAbs. 2012;4(5):623–631. doi: 10.4161/mabs.20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall MP, Gegg C, Walker K, Spahr C, Ortiz R, Patel V, et al. Ligand-binding mass spectrometry to study biotransformation of fusion protein drugs and guide immunoassay development: strategic approach and application to peptibodies targeting the thrombopoietin receptor. AAPS J. 2010;12(4):576–585. doi: 10.1208/s12248-010-9218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Booth B. Incurred sample reanalysis. Bioanalysis. 2011;3(9):927–928. doi: 10.4155/bio.11.69. [DOI] [PubMed] [Google Scholar]
- 17.Wang YM, Krzyzanski W, Doshi S, Xiao JJ, Perez-Ruixo JJ, Chow AT. Pharmacodynamics-mediated drug disposition (PDMDD) and precursor pool lifespan model for single dose of romiplostim in healthy subjects. AAPS J. 2010;12(4):729–740. doi: 10.1208/s12248-010-9234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romiplostim (NplateTM) Prescribing Information. 2013 (May); [Accessed on March 03, 2014] Available from: http://pi.amgen.com/united_states/nplate/nplate_pi_hcp_english.pdf.
- 19.Meno-Tetang GM, Lowe PJ. On the prediction of the human response: a recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin Pharmacol Toxicol. 2005;96(3):182–192. doi: 10.1111/j.1742-7843.2005.pto960307.x. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol. 2007;63(5):548–561. doi: 10.1111/j.1365-2125.2006.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe PJ, Renard D. Omalizumab decreases IgE production in patients with allergic (IgE-mediated) asthma; PKPD analysis of a biomarker, total IgE. Br J Clin Pharmacol. 2011;72(2):306–320. doi: 10.1111/j.1365-2125.2011.03962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omalizumab (XolairTM) Prescribing Information. 2010 (July); [Accessed on March 03,2014] Available from: http://www.gene.com/download/pdf/xolair_prescribing.pdf.
- 23.Glass cockpits. The First Aviation Wiki Journal 2011 [Accessed on March 03, 2014]; Available from: http://aviationknowledge.wikidot.com/aviation:glass-cockpits.
- 24.Final AF447 report suggests pilot slavishly followed flight director pitch-up commands AINonline2012 [Accessed on March 03, 2014]; Available from: http://www.ainonline.com/aviation-news/2012-07-08/final-af447-report-suggests-pilot-slavishly-followed-flight-director-pitch-commands.