

Abstract
Although both POPPK and physiologically based pharmacokinetic (PBPK) models can account for age and other covariates within a paediatric population, they generally do not account for real-time growth and maturation of the individuals through the time course of drug exposure; this may be significant in prolonged neonatal studies. The major objective of this study was to introduce age progression into a paediatric PBPK model, to allow for continuous updating of anatomical, physiological and biological processes in each individual subject over time. The Simcyp paediatric PBPK model simulator system parameters were reanalysed to assess the impact of re-defining the individual over the study period. A schedule for re-defining parameters within the Simcyp paediatric simulator, for each subject, over a prolonged study period, was devised to allow seamless prediction of pharmacokinetics (PK). The model was applied to predict concentration-time data from multiday studies on sildenafil and phenytoin performed in neonates. Among PBPK system parameters, CYP3A4 abundance was one of the fastest changing covariates and a 1-h re-sampling schedule was needed for babies below age 3.5 days in order to seamlessly predict PK (<5% change in abundance) with subject maturation. The re-sampling frequency decreased as age increased, reaching biweekly by 6 months of age. The PK of both sildenafil and phenytoin were predicted better at the end of a prolonged study period using the time varying vs fixed PBPK models. Paediatric PBPK models which account for time-varying system parameters during prolonged studies may provide more mechanistic PK predictions in neonates and infants.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-014-9592-9) contains supplementary material, which is available to authorized users.
KEY WORDS: developmental pharmacokinetics, metabolism, paediatrics, time varying
INTRODUCTION
A complex myriad of physiological and biochemical developmental processes can occur over a period of time, such as during normal growth (1,2), pregnancy (3), critical illnesses (4) and disease progression (5–7). Some of these dynamic changes will have an effect on both the pharmacokinetics and pharmacodynamics (PK/PD) of drugs.
Generally, structural PK models determine concentrations using a function (f) as follows: C(t) = f(dose(t), parameters) where the parameters are usually time-invariant values such as rate constants, volume of distribution, amount or activity of an enzyme and so on, that do not change over the simulation interval. In time-variant PK systems, the parameters change as explicit and/or implicit functions of time, C(t) = f(dose(t), parameters(t)). More mechanistic time-variant PK models have the form C(t) = f(dose(t), parameters(g(t),t)), where g is a model parameter growth function (8). The latter is probably more mechanistic, where the parameter can vary explicitly or implicitly as a function of time, an example is the circadian variation in the PK of 5-fluorouracil due to time-variant changes in its metabolism (9). More complicated forms of mechanistic time-variant models include physiologically based pharmacokinetic (PBPK) models where each individual subject’s PK/PD is determined by a complex array of linked covariates that each vary in different ways with time. The later function can be represented as C(t) = f(dose(t), parameters(g(COV(t)),t)), where COV(t) is the time-varying covariates from the baseline covariate defining the PBPK system parameters over time.
The growth/decay of system parameters with time are generally non-linear functions, reaching an asymptote at different points during maturation with a random variability that increases or decreases with time and size. Paediatric population pharmacokinetic studies often account for the effects of body size and the maturation of elimination pathways on specific PK parameters, using a combination of allometric scaling and maturation functions, but these and other included covariates are usually fixed for the time course of the study. This is acceptable for studies with a short duration or where physiological systems have already developed; however, in relation to prolonged drug therapy in neonates, many of the underlying parameters driving PK change drastically during the early stages of life (1,10–12). This area has attracted more researchers in the last few years (13–18) as the limitations of the current available data for the provision of optimal paediatric care have been identified. This, coupled with the increased emphasis on drug research and licencing of medicines in children is driving the need for improved PK/PD models/data in children (14,19–22).
Studying time-based changes in physiology and biochemistry on a mechanistic level through the time course of a prolonged PK/PD study is a complex problem. This is not only because of the non-linearity of the relationship between the data and the PK/PD parameter of interest but also the complexity of the structural model and the interaction between the model parameters and system components such as covariates, the inter- and intra-individual variability, inter-occasional variability and their overall interactions with the study design. Over the last few years, there has been an increasing interest in understanding the covariate–parameter relationship with respect to time over the duration of study. Examples of where this approach has been adopted and applied in POPPK/PD analyses can be seen in the literature for paediatric (23) and adult (24–26) populations. However, whilst many paediatric PK/PD studies have given careful consideration to time-varying covariate effects and implemented it in the population model explicitly as the current age (27), or implicitly as the current weight, these covariates are usually fixed for each individual for the time course of the study (28). Some have demonstrated that model prediction cannot rely on the assumption that covariate–parameter interactions remain constant in different paediatric populations (29–31).
Physiologically, the covariate–parameter relationship depends on age at each point of time and therefore, inclusion of all quantified covariates in a time-dependent manner is potentially important especially for predicting PK during long-term studies performed in neonates. Paediatric PBPK models are inherently suited for this task as the whole physiology can be updated during the time course of a simulation. .
A number of paediatric PBPK models have been reported in the literature and used to predict PK parameters (1,14) and drug–drug interactions (14,32) from birth onwards. Whilst many of these models can predict drug PK over a prolonged time period, to our knowledge, none of them can simultaneously account for changes in the whole physiological parameters over the time course of a simulation. The development of physiological and biochemical processes occurs rapidly in the first few months of life (1,13,33,34); consequently, the pharmacokinetics of drugs may change rapidly during this time. As a result of these changes, particularly in neonates, it is necessary to account for the underlying physiological changes during a prolonged simulation. For instance, a 1-day-old neonate starting on a 28-day course of drug therapy will be 1 month old at the end of the treatment and this should be reflected in the underlying PBPK model. Interestingly, Sharpe et al. showed that twofold increase in clearance of levetiracetam after intravenous administration occurs during the first week of life based on a 7-day study of the pharmacokinetics of the drug in neonates (35).
Aim
The aims of this study are:
To develop a time-varying paediatric PBPK model to predict the PK profile considering the underlying systems parameter that affect PK in paediatrics as ageing occurs during the time course of a study. The paediatric subjects will grow at a ‘normal’ physiological rate during the time course of a clinical trial simulation.
To compare the output of the time-varying paediatric PBPK model against a conventional ‘fixed’ model using existing published prolonged neonatal and infant PK studies to investigate how the PK profiles are affected when the developmental processes are incorporated in the model.
METHODS
Time-Varying Paediatric PBPK Model Building
To determine how necessary it is to frequently update physiological and biochemical parameters within a paediatric PBPK model such that growth and maturation of all system parameters occurred in a seamless manner, it was necessary to map the extent to which key parameters were changing within a given age–time frame. This will determine how often the re-definition of the most sensitive system parameters has to take place. For example, for a 2-day-old neonate given a drug over a 2-week period that is eliminated mainly by hepatic drug metabolism, the expression of the relevant enzymes may increase 10-fold over this period and in this case, it may be necessary to re-define subjects very frequently. The re-defining of a subject within a paediatric PBPK model may increase the predictive accuracy, in particular, during the neonatal and infant period. However, it will also increase the complexity of the model and the extent of the calculations resulting in higher computation burden and longer simulation/evaluation times. Thus, one of the initial decisions in designing the model was to decide on an optimal re-sampling strategy.
Since the overall plan in this paper is to develop default settings for re-defining paediatric PBPK subjects based on the most rapidly changing physiological parameters that may impact PK or PD parameters or profiles, previously reported equations for longitudinal changes in paediatric PBPK model parameters were used (1). For determination of the percentage change in the model parameters with age, a list of all age-related parameters within the Simcyp paediatric PBPK model that may influence drug pharmacokinetics has been considered; this did not include detailed absorption parameters as these are not currently in the model. Equations for many of these parameters are reported elsewhere (1,36). Where a parameter is defined by BSA or WT or HT, age-related equations were substituted into these equations prior to determining the rate of change with age. The equations were entered into Microsoft Excel and used to predict the intended parameter with age. The second stage was then to investigate the percentage change in the parameter with time. The arbitrary decision was taken to allow a maximum of 5% change in prior value before re-defining a subject. The complete analysis of all age-related paediatric PBPK parameters showed the percentage change in a parameter with age using different frequencies of sampling. To determine the minimum necessary re-sampling interval, the values from the fastest changing system parameter were used when evaluating the time-based changing paediatric PBPK model against the conventional paediatric PBPK model.
Model Performance
Simulations using the paediatric PBPK Model were performed when the system parameters were fixed and also when changing over time. A literature search was undertaken in PubMed to identify longer duration clinical PK studies performed over days or week involving neonates and infants. Only a small number of studies were identified and from these, it was only possible to simulate the concentration profiles of a limited number of subjects for sildenafil and phenytoin. In addition, a case study was developed based on the standard UK treatment regimen for the treatment of neonatal apnoea with theophylline.
Theophylline
A simulation exercise for theophylline plasma concentration after multiple doses was carried out to show that the impact of development had to be included in order to achieve concentrations within the therapeutic range. The British National Formulary for Children standard dosing regimen for the treatment of neonatal apnoea is 4.8 mg/kg iv theophylline followed by 2 mg/kg iv every 12 h (37). This dosage regimen was used in Simcyp paediatric to simulate theophylline PK in a virtual full-term newborn population (<1 day old) with and without the time-based changing physiology activated. The results were output against this therapeutic window for theophylline in neonatal apnoea of 8–12 μg/ml.
Sildenafil
The time-varying changes in the model were applied to sildenafil which is predominantly metabolised by CYP3A4 with a minor role for CYP2C9 to simulate a long-term clinical study where the drug had been given for many days to newborn babies (38). Briefly, 36 term neonates with persistent pulmonary hypertension or hypoxemia were administered intravenous sildenafil as a loading and maintenance dose for up to 7 days starting within 72 h of birth. The dosing regimen consisted of a loading infusion of fixed duration ranging from 5 min to 3 h among treatment groups, followed by a continuous maintenance infusion of variable duration, ranging from 2.6 to 168 h for individual subjects. Mean duration of maintenance infusion for all treatment groups was 77 h, and, except for one subject, all subjects received the maintenance infusion for at least the minimum duration of 48 h specified in the protocol. One group of four subjects received the maintenance infusion only. Plasma concentrations were collected during and after the infusion.
The original study reported only mean loading dose and mean maintenance dose and did not report individual demographics with respect to the weight and dose administered and infusion duration, making it difficult to replicate the exact dosing regimen in each individual. Thus, we have replicated observations from three representative individuals from three groups (one subject in each) for whom enough information is available. For individual 1 (group 1), the mean value for the loading dose was 0.47 mg/h for 0.06 h and the maintenance dose was 0.01 mg/h for 62.9 h, for individual 2 the mean loading dose was 0.18 mg/h for 0.5 h and the maintenance dose was 0.02 mg/h for 108 h and for individual 3 (group 7), no loading dose was given but the maintenance dose was 0.03 mg/h for 163 h. We used this dosing regimen and the default demographic data from the Simcyp simulator to run the simulation in the neonatal age range 0 to 0.001 years, 100 subjects and proportion of females was 0.5. The simulation was run over 8 days to cover the dosing and plasma sampling period. The results from the paediatric PBPK simulation with and without time-based changing physiology were compared to clinical data.
Phenytoin
Phenytoin was administered to 30 (pre-term and term) newborn babies, four of whom were premature (39). Phenytoin therapy was started as an intravenous loading dose of 12 mg/kg phenytoin sodium, given over 15–20 min by syringe driver infusion pump. Twenty-four hours later, the maintenance therapy was started with an intravenous or oral suspension dose of about 8 mg/kg per day, given in divided doses every 12 h for 7–10 days. The duration of the maintenance therapy differed between individuals (7–10 days). The original study included a number of subjects who we had to exclude because they were either pre-term individuals not covered by the current model, did not report the full concentration-time profile or the subjects were not newborn infants. When those individual were excluded, we were left with usable data to perform simulations in just two full-term individuals. These two patients received a loading dose of 12 mg/kg phenytoin given intravenously over 15–20 min and an oral maintenance dose of 8 mg/kg per 24 h given in divided doses every 12 h for 7–10 days. These details were entered into the Simcyp paediatric simulator to match these subjects and their doses as closely as possible. The simulation was run over a 15-day period to cover the dosing and plasma sampling period. The results from the paediatric PBPK simulation with and without time-based changing physiology were compared to clinical data.
RESULTS
Optimising Model Building
The default re-sampling schedule to seamlessly describe the age-related changes in systems parameters was based on the most rapidly changing age-related parameters; these were CYP2A6 and CYP3A4 ontogeny parameters (Fig. 1). Because few drugs are metabolised by CYP2A6 in paediatric clinical practice, the CYP3A4 ontogeny was used to determine the re-sampling time intervals necessary at different ages. The complete summary list of all percentage changes in parameters with time starting at birth is given in the supplementary material (Appendix). For example, there is a 0.02% increase in brain volume from birth to 1 h after birth and a 71.6% increase in CYP3A4 between the first and second hours post birth. The suggested re-sampling frequency based on CYP3A4 is shown in Fig. 2. The graphs of percentage change in parameter with age were calculated using different sampling times. A matrix was constructed, incorporating all parameters, for when it is possible to change to the next sampling level having only <5% change in the parameter. In the case of CYP3A4, the cut off points (days) when the sampling frequency can be reduced to the next level are shown in Fig. 2. Hence, from 0 to 3.5 days, an hourly re-sampling frequency and from between 3.5 and 6.5 days a 6-hourly re-sampling frequency is required and so on. The re-sampling schedule results in all underlying systems parameters being updated to allow for a relatively seamless transition in PK parameters and profiles with age during a long-term simulation.
Fig. 1.
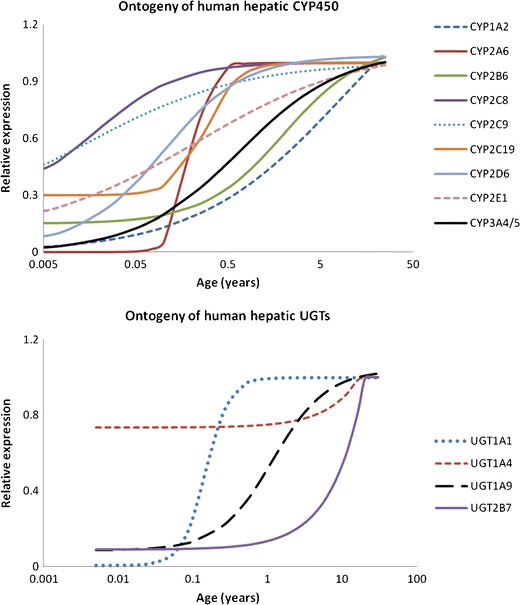
Relative expression of human drug-metabolising enzymes at different ages. Lines represent simulated ontogeny profiles based on ‘best fit’ equations to the original data (see “Discussion”)
Fig. 2.
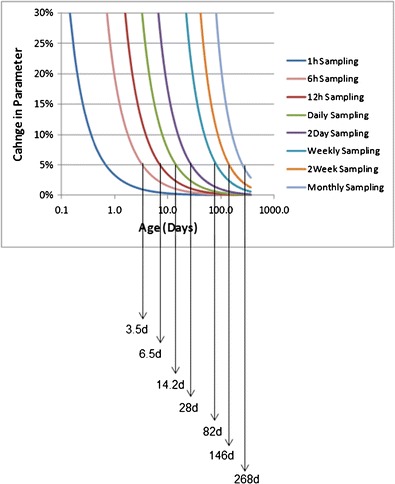
Percent change of CYP3A4 for different sampling periods for different ages and the derived age cut off points for default re-sampling schedule within Simcyp paediatric
Model Performance
A very small number of publications were found involving studies of longer duration in neonates with limited PK information regarding individual demographics and individual pharmacokinetic profiles. In addition, the duration of study for each individual was not reported. The reported mean of population characteristics and study design were used; however, individual rather than population information were used where available. The preliminary results reported below are to illustrate proof of concept for the model and illustrate how time-based changing physiology may improve predictions.
Theophylline Case Study
The result from this exercise (Fig. 3) shows that by re-defining individuals over the simulation study period, the predicted concentration-time profiles at steady state fall within the therapeutic concentration range. However, in the simulation using the baseline characteristics of individuals, the steady state concentrations are above the maximum recommended therapeutic level.
Fig. 3.
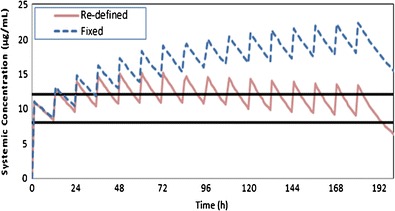
Simulated profiles for theophylline in a virtual full-term newborn population after administration of 4.8 mg/kg iv theophylline followed by 2 mg/kg iv every 12 h. The therapeutic window (8–12 ug/ml) is bound by the two black lines. The dashed line shows the higher level predicted when re-defining individuals in the paediatric-PBPK model was not consider (baseline model) and the solid line shows the achievement of therapeutic level by using the re-defining approach (time-changing physiology paediatric-PBPK model)
Sildenafil Case Study
The result from the sildenafil simulations are given in Fig. 4. The results for subject 1 show a slight improvement in describing the elimination phase of sildenafil in the time-based model. The time-dependent changes in intrinsic clearance via both CYP3A4 and CYP2C9 pathways due to the ontogeny of both enzymes during the course of the simulation can be seen for subject 1 in Fig. 5. The predicted Cmax values were not well recovered using the time-based changing physiology simulation and the reasons for this are unclear. For subjects 2 and 3 in Fig. 4, the overall concentration-time profile, Cmax and elimination phase for sildenafil are much better predicted using the time-based changing physiology-based model compared to the time-fixed model. Note the steady increase in the concentration with time for subjects 2 and 3 in the case of baseline physiology as the elimination and distribution are static. In contrast, the concentration decreases with time in the time-based model as the physiology is continuously growing and the elimination and distribution increase with time.
Fig. 4.
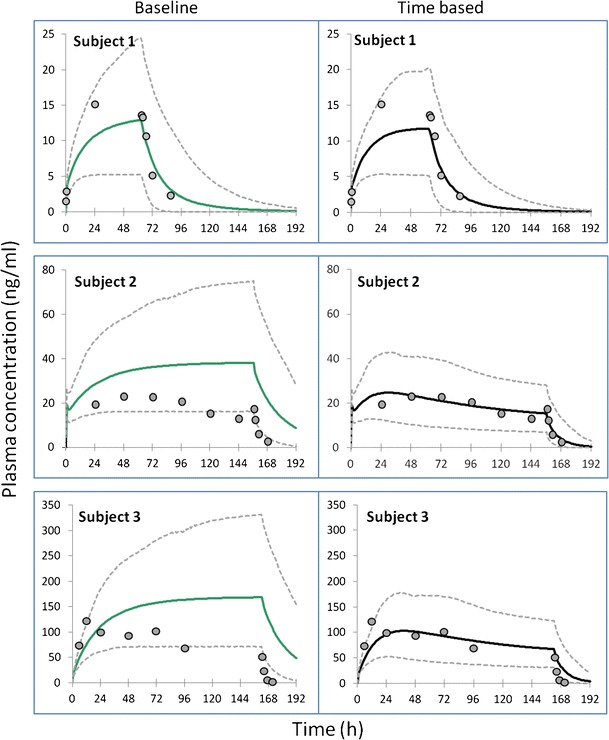
Simulated mean (solid lines) and 95% predictive interval values (dashed lines) of sildenafil plasma concentration over time for three representative subjects using both baseline and time-based changing physiology in the p-PBPK model. Filled circles are the observations from each subject as reported in Mukherjee et al. 2009
Fig. 5.
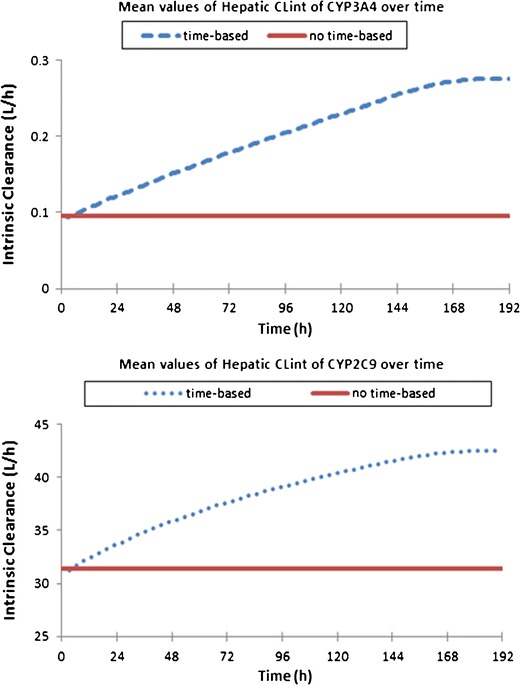
Hepatic intrinsic clearance of sildenafil over time for CYP3A4 (upper panel) and CYP2C9 (lower panel) for representative subject 1 in Fig. 4
Phenytoin Case Study
The results are shown in Fig. 6, although there was some over-prediction of the concentration-time profile, the simulated concentrations using the re-defined subjects were in closer agreement with the actual data. This was especially the case later in the study period when the effects of maturation of underlying processes that may affect phenytoin PK such as CYP2C9 ontogeny would be manifested.
Fig. 6.
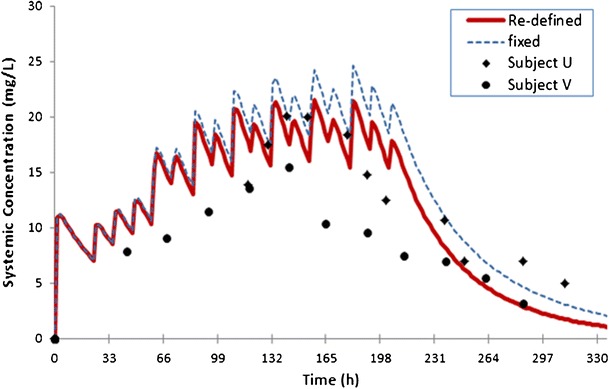
Simulated mean values of plasma concentration of phenytoin over time. Filled circles and diamonds are the observations from individual U and V reported in Loughnan et al., 1977
DISCUSSION
There are a limited numbers of PK studies reported in the literature that were performed in the paediatric age range where a number of covariates were built in the model and allowed to change with age/time during the course of a study. In some cases where covariates were allowed to change with time, the age of the individual subjects was left constant for modelling purposes. Whilst the assumption of a constant covariates–parameter model relationship or fixed age at the start of treatment is probably valid for studies in older children and adults and for those of very short-term treatments in neonates, it is unlikely to be so for studies involving prolonged treatment in the latter group. In general, models for time-varying covariates can provide more valuable information than time–constant covariate as they can provide information leading to a better understanding of the parameter–covariate relationships with respect to time (24,25). Even if the baseline covariate value does not seem important for explaining the inter-individual variability, the time–change in each covariate, which is the aim of this work, can explain the intra-individual or inter-occasion variability terms. In the latter case, it might be of value to screen for a covariate over time at each sampling time, especially in routine care or TDM settings.
Although p-PBPK models are more complex and costly to build compared to the top-down approaches, the ability to potentially update the whole physiology during a simulation is an advantage. During the time course of a simulation, not only will liver size be increased in an individual but also all the other covariates such as renal function and expression of albumin and thus protein binding will also change. This would be very challenging to achieve using conventional compartmental PK analysis.
A time-based PBPK model that accounts for physiological growth/decay in its parameters has been developed for the paediatric population and implemented within the Simcyp Simulator (V12 Release 1). To our knowledge, this is the first paediatric PBPK model that accounts for re-defining or re-sampling the simulated subject’s physiology during the course of treatment. Within the paediatric PBPK model all individual characteristics impacting on drug PK will develop as the individuals’ age/grow and will account for intra-individual variability, which stems from changes in system parameters. For example, for a drug metabolised by CYP3A4, a parameter that has a rapid ontogeny post birth, the PK of a drug given during the first months of life is likely to change rapidly due to growth and maturation. The described model also accounts for inter-individual variability of the simulated characteristics of individuals at specific time points e.g. where a parameter such a body surface area changes with age but is different between males and females,
The main advantage of this approach is that it allows more realistic modelling of simulated subjects and as a result, PBPK models may perform better for the prediction of PK parameters and individualisation of dosage regimens in the paediatric population. During growth, changes in body composition, hepatic and renal functions are responsible for alterations in PK/PD parameters and hence, the therapeutic outcomes across different ages, which necessitates dose adjustment in some cases. This is well known in drug development and clinical arenas and a significant body of publications address these issues (1,16,21,40–45). For example, during growth, the volume of distribution and clearance of lipid-soluble drugs increase in young adults compared with infants (46). The benefit from the time-varying PBPK model is not just in the fact that it accounts for all known interacting physiological processes, but that it keeps them updated over time.
The results presented here for sildenafil, phenytoin and theophylline illustrate applications of the age-varying PBPK model, accounting for the changing physiology over the course of the prolonged studies. This approach established the basis of describing the developing disposition kinetics during the treatment course in a dynamic predictive way. Although the results are very preliminary and further validation is required, the examples show proof of concept, the theophylline example showing the importance of considering growth and development when establishing dosage regimens for drugs with a narrow therapeutic index. The sildenafil simulations generally show improvements in the prediction of PK parameters and concentration-time profiles using the time-based physiology approach. However, these examples are limited to data from individuals from the Mukherjee et al. paper (38) as no information was given on individual subject demographics and this limited our ability to simulate the data to just three representative subjects, the limitations are clearly stated in the ‘Methods’ section. The smooth nature of the intrinsic clearance-time curves (Fig. 5) shows that the re-sampling frequency in the time-based changing physiology model is frequent enough to prevent stepwise changes in these intrinsic clearance values.
One of the limitations of the current model is uncertainty around the development and ontogeny of some of the underlying processes governing PK prediction in the paediatric age range and consequently, a need to refine these in existing paediatric PBPK models as data become available. The ontogeny profile for CYP3A4 (Fig. 1) within the Simcyp paediatric simulator is based on in vitro data which is expressed relative to adults, data is taken from a number of sources (47–52) and the ontogeny function derived using non-linear regression. Overall evidence from the literature from both in vitro and in vivo data points to a continuous rather than stepwise change in CYP3A4 expression with age. A recent study to determine a maturation model for midazolam clearance (CYP3A) analysed the allometrically scaled age-related change in midazolam iv clearance using NONMEM, the best fit model, was a continuous sigmoidal-shaped curve (53). Reference sources for the other CYP enzyme ontogeny are published previously (1), the UGT ontogeny profiles are previously unpublished but a list of references is available from Simcyp Limited (support@simcyp.com). An additional limitation in the current re-defined paediatric PBPK population model is that it only considers a healthy paediatric population. Disease progression/regression and the effects these have on drug disposition are not considered. However, as more information becomes available, a model describing both age and disease changes can also be developed. Lack of adequate clinical studies available in the literature, where long-term studies in young children are conducted, limits the possibility of assessing the model performance for a bigger dataset. Such studies may be difficult to achieve due to logistical and ethical issues, consequently; although the concept is interesting, at the current time, it is difficult to extensively validate the model. The use of the model in clinical study design may allow more validation to be performed in future. Additional challenges remain to develop and validate the model for pre-term individuals where post-natal age range varies from hours to months at the start of treatment.
CONCLUSION
To account for the time-varying changes in physiological and biochemical parameters with age, a higher level of complexity in the PK models are required. PBPK models are highly suited to this task, as they can be used to account for all the underlying physiological changes and their impact on pharmacokinetics to better predict drug disposition and the effects of new dosage regimens. Although such models are still evolving and users must appreciate their limitations, they may help to reduce the number of time- and cost-intensive long-term clinical trials that need to be performed. These models can also be considerably useful in designing clinical studies prospectively and optimising the dosage regimens and thus, their synergistic use with the conventional PK approach can be envisaged in decreasing the time and costs involved in drug development. Together with physicochemical properties of the drug, these models may also help to identify agents with undesirable properties at an early stage.
The application of the time-based physiological changes described in the paper for the paediatric population can be extended to other populations such as cancer patients, pregnancy and disease progression models where physiology and biochemistry are changing with time.
Electronic Supplementary Material
(DOCX 35.3 kb)
Acknowledgments
The authors would like to thank James Kay and Eleanor Savill for their assistance with the preparation of this manuscript.
References
- 1.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 2.Boss GR, Seegmiller JE. Age-related physiological changes and their clinical significance. West J Med. 1981;135(6):434–440. [PMC free article] [PubMed] [Google Scholar]
- 3.Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6):365–396. doi: 10.2165/11597440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Boucher BA, Wood GC, Swanson JM. Pharmacokinetic changes in critical illness. Crit Care Clin. 2006;22(2):255–271. doi: 10.1016/j.ccc.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Joerger M. Covariate pharmacokinetic model building in oncology and its potential clinical relevance. AAPS J. 2012;14(1):119–132. doi: 10.1208/s12248-012-9320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89(2):381–410. doi: 10.1152/physrev.00016.2008. [DOI] [PubMed] [Google Scholar]
- 7.Gough DB, Heys SD, Eremin O. Cancer cachexia: pathophysiological mechanisms. Eur J Surg Oncol. 1996;22(2):192–196. doi: 10.1016/S0748-7983(96)90905-1. [DOI] [PubMed] [Google Scholar]
- 8.Uchizono JA, Lane JR. Empirical pharmacokinetic/pharmacodynamic models. In: Ette EI, Williams PJ, editors. Pharmcometrics: the science of quantitative pharmacology. Hoboken: Wiley; 2007. pp. 529–546. [Google Scholar]
- 9.Bressolle F, Joulia JM, Pinguet F, Ychou M, Astre C, Duffour J, et al. Circadian rhythm of 5-fluorouracil population pharmacokinetics in patients with metastatic colorectal cancer. Cancer Chemother Pharmacol. 1999;44(4):295–302. doi: 10.1007/s002800050980. [DOI] [PubMed] [Google Scholar]
- 10.Johnson TN. Modelling approaches to dose estimation in children. Br J Clin Pharmacol. 2005;59(6):663–669. doi: 10.1111/j.1365-2125.2005.02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdel-Rahman SM, Reed MD, Wells TG, Kearns GL. Considerations in the rational design and conduct of phase I/II paediatric clinical trials: avoiding the problems and pitfalls. Clin Pharmacol Ther. 2007;81(4):483–494. doi: 10.1038/sj.clpt.6100134. [DOI] [PubMed] [Google Scholar]
- 12.Knibbe CA, Danhof M. Individualized dosing regimens in children based on population PKPD modelling: are we ready for it? Int J Pharm. 2011;415(1–2):9–14. doi: 10.1016/j.ijpharm.2011.02.056. [DOI] [PubMed] [Google Scholar]
- 13.de Wildt SN. Profound changes in drug metabolism enzymes and possible effects on drug therapy in neonates and children. Expert Opin Drug Metab Toxicol. 2011;7(8):935–948. doi: 10.1517/17425255.2011.577739. [DOI] [PubMed] [Google Scholar]
- 14.Johnson TN, Rostami-Hodjegan A. Resurgence in the use of physiologically based pharmacokinetic models in paediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth. 2011;21(3):291–301. doi: 10.1111/j.1460-9592.2010.03323.x. [DOI] [PubMed] [Google Scholar]
- 15.Johnson TN, Thomson M. Intestinal metabolism and transport of drugs in children: the effects of age and disease. J Pediatr Gastroenterol Nutr. 2008;47(1):3–10. doi: 10.1097/MPG.0b013e31816a8cca. [DOI] [PubMed] [Google Scholar]
- 16.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–1167. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 17.Allegaert K, Rochette A, Veyckemans F. Developmental pharmacology of tramadol during infancy: ontogeny, pharmacogenetics and elimination clearance. Paediatr Anaesth. 2011;21(3):266–273. doi: 10.1111/j.1460-9592.2010.03389.x. [DOI] [PubMed] [Google Scholar]
- 18.Anderson BJ. Developmental pharmacology; filling one knowledge gap in paediatric anesthesiology. Paediatr Anaesth. 2011;21(3):179–182. doi: 10.1111/j.1460-9592.2011.03539.x. [DOI] [PubMed] [Google Scholar]
- 19.Allegaert K. Mechanism based medicine in infancy: complex interplay between developmental pharmacology and pharmacogenetics. Int J Clin Pharm. 2011;33(3):473–474. doi: 10.1007/s11096-011-9504-3. [DOI] [PubMed] [Google Scholar]
- 20.Reed MD, Besunder JB. Developmental pharmacology: ontogenic basis of drug disposition. Pediatr Clin N Am. 1989;36(5):1053–1074. doi: 10.1016/s0031-3955(16)36757-8. [DOI] [PubMed] [Google Scholar]
- 21.van den Anker JN. Developmental pharmacology. Dev Disabil Res Rev. 2010;16(3):233–238. doi: 10.1002/ddrr.122. [DOI] [PubMed] [Google Scholar]
- 22.Viergever RF, Rademaker CM, Ghersi D. Pharmacokinetic research in children: an analysis of registered records of clinical trials. BMJ Open. 2011;1(1):e000221. doi: 10.1136/bmjopen-2011-000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Jouan M, Jullien V, Tetanye E, Tran A, Rey E, Treluyer JM, et al. Quinine pharmacokinetics and pharmacodynamics in children with malaria caused by Plasmodium falciparum. Antimicrob Agents Chemother. 2005;49(9):3658–3662. doi: 10.1128/AAC.49.9.3658-3662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahlby U, Thomson AH, Milligan PA, Karlsson MO. Models for time-varying covariates in population pharmacokinetic-pharmacodynamic analysis. Br J Clin Pharmacol. 2004;58(4):367–377. doi: 10.1111/j.1365-2125.2004.02170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staatz CE, Byrne C, Thomson AH. Population pharmacokinetic modelling of gentamicin and vancomycin in patients with unstable renal function following cardiothoracic surgery. Br J Clin Pharmacol. 2006;61(2):164–176. doi: 10.1111/j.1365-2125.2005.02547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Hest RM, van Gelder T, Bouw R, Goggin T, Gordon R, Mamelok RD, et al. Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol. 2007;63(6):741–752. doi: 10.1111/j.1365-2125.2006.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wade KC, Wu D, Kaufman DA, Ward RM, Benjamin DK, Jr, Sullivan JE, et al. Population pharmacokinetics of fluconazole in young infants. Antimicrob Agents Chemother. 2008;52(11):4043–4049. doi: 10.1128/AAC.00569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Cock RF, Allegaert K, Sherwin CM, Nielsen EI, de Hoog M, van den Anker JN, et al. A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm Res. 2013;31:754–767. doi: 10.1007/s11095-013-1197-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cella M, Knibbe C, de Wildt SN, Van Gerven J, Danhof M, Della Pasqua O. Scaling of pharmacokinetics across paediatric populations: the lack of interpolative power of allometric models. Br J Clin Pharmacol. 2012;74(3):525–535. doi: 10.1111/j.1365-2125.2012.04206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cella M, Zhao W, Jacqz-Aigrain E, Burger D, Danhof M, Della Pasqua O. Paediatric drug development: are population models predictive of pharmacokinetics across paediatric populations? Br J Clin Pharmacol. 2011;72(3):454–464. doi: 10.1111/j.1365-2125.2011.03992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strougo A, Eissing T, Yassen A, Willmann S, Danhof M, Freijer J. First dose in children: physiological insights into pharmacokinetic scaling approaches and their implications in paediatric drug development. J Pharmacokinet Pharmacodyn. 2012;39(2):195–203. doi: 10.1007/s10928-012-9241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salem F, Johnson TN, Barter ZE, Leeder JS, Rostami-Hodjegan A. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol. 2013;53(8):857–865. doi: 10.1002/jcph.100. [DOI] [PubMed] [Google Scholar]
- 33.Edginton AN, Schmitt W, Willmann S. Development and evaluation of a generic physiologically based pharmacokinetic model for children. Clin Pharmacokinet. 2006;45(10):1013–1034. doi: 10.2165/00003088-200645100-00005. [DOI] [PubMed] [Google Scholar]
- 34.Anderson BJ, Woollard GA, Holford NH. A model for size and age changes in the pharmacokinetics of paracetamol in neonates, infants and children. Br J Clin Pharmacol. 2000;50(2):125–134. doi: 10.1046/j.1365-2125.2000.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharpe CM, Capparelli EV, Mower A, Farrell MJ, Soldin SJ, Haas RH. A seven-day study of the pharmacokinetics of intravenous levetiracetam in neonates: marked changes in pharmacokinetics occur during the first week of life. Pediatr Res. 2012;72(1):43–49. doi: 10.1038/pr.2012.51. [DOI] [PubMed] [Google Scholar]
- 36.Price PS, Conolly RB, Chaisson CF, Gross EA, Young JS, Mathis ET, et al. Modeling interindividual variation in physiological factors used in PBPK models of humans. Crit Rev Toxicol. 2003;33(5):469–503. doi: 10.1080/10408440390242324. [DOI] [PubMed] [Google Scholar]
- 37.British National Formulary for Children (BNFC) 2010-11 British National formulary publications L, 2010 (P. 182).
- 38.Mukherjee A, Dombi T, Wittke B, Lalonde R. Population pharmacokinetics of sildenafil in term neonates: evidence of rapid maturation of metabolic clearance in the early postnatal period. Clin Pharmacol Ther. 2009;85(1):56–63. doi: 10.1038/clpt.2008.177. [DOI] [PubMed] [Google Scholar]
- 39.Loughnan PM, Greenwald A, Purton WW, Aranda JV, Watters G, Neims AH. Pharmacokinetic observations of phenytoin disposition in the newborn and young infant. Arch Dis Child. 1977;52(4):302–309. doi: 10.1136/adc.52.4.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jauregizar N, Quintana A, Suarez E, Raczka E, de la Fuente L, Calvo R. Age-related changes in pharmacokinetics and pharmacodynamics of lerisetron in the rat: a population pharmacokinetic model. Gerontology. 2003;49(4):205–214. doi: 10.1159/000070400. [DOI] [PubMed] [Google Scholar]
- 41.Mangoni AA, Jackson SH. Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 2004;57(1):6–14. doi: 10.1046/j.1365-2125.2003.02007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobias D. Age-related changes in pharmacokinetics and pharmacodynamics: a review. Consult Pharm. 2004;19(8):736–739. doi: 10.4140/TCP.n.2004.736. [DOI] [PubMed] [Google Scholar]
- 43.Shi S, Klotz U. Age-related changes in pharmacokinetics. Curr Drug Metab. 2011;12(7):601–610. doi: 10.2174/138920011796504527. [DOI] [PubMed] [Google Scholar]
- 44.Rogers AS. The role of cytochrome P450 in developmental pharmacology. J Adolesc Health. 1994;15(8):635–640. doi: 10.1016/S1054-139X(94)90630-0. [DOI] [PubMed] [Google Scholar]
- 45.Reed MD. Developmental pharmacology: relationship to drug use. DICP. 1989;23(7–8 Suppl):S21–S26. doi: 10.1177/106002808902300705. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez W, Selen A, Avant D, Chaurasia C, Crescenzi T, Gieser G, et al. Improving paediatric dosing through paediatric initiatives: what we have learned. Paediatrics. 2008;121(3):530–539. doi: 10.1542/peds.2007-1529. [DOI] [PubMed] [Google Scholar]
- 47.Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247(2):625–634. doi: 10.1111/j.1432-1033.1997.00625.x. [DOI] [PubMed] [Google Scholar]
- 48.Stevens JC, Hines RN, Gu C, Koukouritaki SB, Manro JR, Tandler PJ, et al. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307(2):573–582. doi: 10.1124/jpet.103.054841. [DOI] [PubMed] [Google Scholar]
- 49.Treluyer JM, Bowers G, Cazali N, Sonnier M, Rey E, Pons G, et al. Oxidative metabolism of amprenavir in the human liver. Effect of the CYP3A maturation. Drug Metab Dispos. 2003;31(3):275–281. doi: 10.1124/dmd.31.3.275. [DOI] [PubMed] [Google Scholar]
- 50.Johnson TN, Tucker GT, Rostami-Hodjegan A. Ontogeny of CYP2D6 and CYP3A4 in the first year of life. Clin Pharmacol Ther. 2008;83(5):670–671. doi: 10.1038/sj.clpt.6100327. [DOI] [PubMed] [Google Scholar]
- 51.Tateseishi T, Nakura H, Asoh M, et al. A comparison of hepatic cytochromes P450 protein expression between infancy and postinfancy. Life Sci. 1997;61(26):2567–2574. doi: 10.1016/S0024-3205(97)01011-4. [DOI] [PubMed] [Google Scholar]
- 52.Hines RN. Ontogeny of human hepatic cytochromes P450. Biochem Mol Toxicol. 2007;21(4):169–175. doi: 10.1002/jbt.20179. [DOI] [PubMed] [Google Scholar]
- 53.Anderson BJ, Larsson P. A maturation model for midazolam clearance. Pediatr Anesth. 2011;21(3):302–308. doi: 10.1111/j.1460-9592.2010.03364.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 35.3 kb)