

Abstract
The present study investigated the utility of in vitro lipolysis performance indicators drug solubilization and maximum supersaturation ratio (SRM) for their predictive use for the in vivo performance in a minipig model. The commercial Lipanthyl formulation and a series of LbDDS based on identical self-nanoemulsifying drug delivery systems (SNEDDS) containing 200 mg of fenofibrate, either dissolved or suspended, were subjected to combined gastric (pH 2) and intestinal (pH 6.5) in vitro lipolysis. Based on the solubilization profiles and SRM the rank-order SNEDDS (75% drug load) > super-SNEDDS (150% drug load, dissolved) = SNEDDS suspension (150% drug load, partially suspended) > Lipanthyl was established, with an increased likelihood of drug precipitation above SRM > 3. The in vitro performance, however, was not reproduced in vivo in a minipig model as the mean plasma concentration over time curves of all LbDDS were comparable, independent of the initial physical state of the drug. There was no correlation between the area under the solubilization-time curves (AUCin vitro) of the intestinal step and the AUCin vivo. The study suggests careful interpretation of in vitro performance criteria and revision of LbDDS optimization towards increased solubilization.
KEY WORDS: in vitro ipolysis, in vitro/in vivo correlation, lipids, SNEDDS suspensions, super-SNEDDS, supersaturation
INTRODUCTION
The oral route, as the preferred way of drug administration, has become increasingly challenging due to the vast number of drugs candidates with poor water solubility and/or slow dissolution rate (1). Drug solubilization is a prerequisite prior to drug absorption for an intended systemic effect of an orally administered drug (2). As the gastrointestinal permeability of many poorly water-soluble drugs is often sufficiently high to enable rapid absorption in the intestinal tract, formulation scientists have developed a number of drug delivery strategies that can facilitate problems associated with limited drug solubility and dissolution rate (3,4). Among these enabling formulation approaches lipid-based drug delivery systems (LbDDS), especially self-emulsifying drug delivery systems, have become a popular formulation option for poorly water-soluble, lipophilic drugs (5). Despite the success of some marketed LbDDS (e.g., Neoral®), the number of commercially available LbDDS is still limited. This reluctance might be attributed to the fact that despite intense research in the area, the factors governing the in vivo performance of LbDDS are still poorly understood. Moreover, the majority of in vivo studies have compared the performance of LbDDS with conventional solid dosage forms rather than evaluating differences between diverse LbDDS, which could help elucidate the critical factors governing the performance of LbDDS.
In LbDDS the drug is commonly dissolved in a single or, more frequently, a blend of excipients as in the case of self-nanoemulsifying drug delivery systems (SNEDDS) (6). SNEDDS are an isotropic mixture of lipids, surfactants, cosurfactants, and cosolvents that generate ultrafine, kinetically stable emulsions in aqueous media under conditions of gentle agitation (7). The delivery of the drug in the dissolved state is considered the major advantage of SNEDDS as the dissolution step of the crystalline material thereby is circumvented (8). However, there have been concerns on the potential negative impact on drug absorption due to drug precipitation in the small intestine resulting from the partitioning of hydrophilic formulation components (e.g., cosolvents and hydrophilic surfactants) during SNEDDS dispersion in the intestinal fluids and the subsequent enzymatic breakdown of digestible SNEDDS excipients (9,10). The development of SNEDDS has, therefore, been focused on the avoidance of drug precipitation during in vitro testing, e.g., by reducing drug loads in the formulations (11). Recent studies however, have shown that the precipitation of the poorly water-soluble drugs cinnarizine, simvastatin, and halofantrine observed during in vitro lipolysis was not reflected in a reduced area under the plasma concentration-time curve (AUC) in the corresponding in vivo studies carried out in a dog model (12–14). It is interesting to note that these drugs were also shown to precipitate in an amorphous form during in vitro lipolysis, with a faster dissolution rate compared with the crystalline form of the drugs (13–15). Although it is not known whether or not (and in which form) the drugs also precipitate in vivo, these studies point at the potential importance of the nature of the solid state of drug precipitates and could explain the poor predictive value of the in vitro model on the in vivo performance.
It has recently been suggested that the propensity of drug precipitation during in vitro dispersion and digestion of LbDDS can serve as a potential indicator for the in vivo performance (16). As a measure for the likelihood of drug precipitation, the maximum supersaturation ratio (SRM) has been suggested as a representation of the ratio of the theoretical drug concentration (in the absence of precipitation) and drug solubility in the aqueous phase (16,17). A value of SRM > 2.5 has been identified as the threshold above which drug precipitation is likely to occur. The majority of the studies evaluated the SRM precipitation parameter in vitro with only very limited in vivo data available (11). While the proposed precipitation parameter SRM reflected the in vivo performance of formulations containing moderate drug load (40% of the drug solubility in the formulation), the in vitro results failed to predict the in vivo performance at higher drug load (80% drug solubility) in a dog model.
From an industry perspective and due to patient compliance SNEDDS with drug loads high enough to reduce the pill burden are desirable. However, in conventional SNEDDS, the drug load is frequently limited by the drug solubility in the formulation. In an attempt to overcome this limitation, supersaturated SNEDDS (“super-SNEDDS”) containing drug loads in the formulation above equilibrium solubility have been described recently (13,14). Despite pronounced generation of (amorphous) drug precipitates during in vitro lipolysis, the super-SNEDDS were equivalent or superior to conventional SNEDDS in vivo (13,14).
So far, the number of studies evaluating the in vivo performance of SNEDDS based on precipitation parameters, such as the SRM, is very limited. The aims of the current study were, therefore, twofold: (1) to evaluate the in vitro and in vivo performance of commercial Lipanthyl and a series of SNEDDS, including super-SNEDDS, of the same relative composition containing a high drug load of fenofibrate, a compound previously shown to precipitate in a crystalline form during in vitro lipolysis (16) and (2) to evaluate the proposed in vitro precipitation parameter for its predictability on the in vivo performance. To facilitate the above study aims, an identical dose of 200 mg fenofibrate was used across all formulations by adjusting the overall mass of the respective formulation. A recently established in vitro lipolysis model was employed, combining consecutive gastric and intestinal in vitro lipolysis (18–20). Moreover, the in vitro lipolysis conditions (such as porcine bile salts and pancreatic lipase extract of porcine origin) were chosen to match the minipig model used for the subsequent in vivo study.
MATERIALS AND METHODS
Materials
Fenofibrate, fenofibric acid, clofibric acid, soybean oil (long-chain triglycerides), sodium hydroxide, porcine pancreatic lipase (≥3× USP activity), porcine bile extract (68% purity), sodium taurodeoxycholate, and calcium chloride were purchased from Sigma-Aldrich (St. Louis, MO, USA), and maleic acid and 4-bromobenzeneboronic acid (BBBA) from Fluka (Buchs, Switzerland). Kolliphor RH 40 (polyoxyl 40 hydrogenated castor oil) and Maisine 35-1 (long-chain mono-, di-, and triglycerides) were donated by BASF (Ludwigshafen, Germany) and Gattefossé (St. Priest, France), respectively. Soybean phospholipids (SPC; 99% purity) were supplied by Lipoid (Ludwigshafen, Germany). Candida antarctica lipase A (10 mg/mL) was a gift from Novozyme (Bagsvaerd, Denmark). HPLC-grade acetonitrile, methanol, and absolute ethanol were obtained from VWR (Herlev, Denmark). Purified water was obtained from a Siemens Ultra Clear water purification system (Guenzburg, Germany). All other chemicals were of analytical grade and were used as received unless specified otherwise.
Methods
Preparation of the Formulations
A previously characterized SNEDDS composed of 24% soybean oil, 32.2% Maisine 35-1, 30% Kolliphor RH 40, and 13.8% ethanol was used for the current study (21). The molten Maisine 35-1 and Kolliphor RH 40 were blended with soybean oil and, after cooling to room temperature, the required amount of ethanol was added before the mixture was stirred on a magnetic stirrer. Drug-loaded formulations were produced by weighing the required amount of fenofibrate directly into dust-free screw-top glass vials followed by the addition of appropriate amounts of drug-free SNEDDS. The following formulations were prepared using the same drug-free SNEDDS (Table I): (1) SNEDDS containing fenofibrate corresponding to 75% of the drug’s solubility in the SNEDDS; (2) supersaturated SNEDDS (super-SNEDDS) corresponding to 150% of the drug’s solubility in the SNEDDS. Supersaturation in the formulation was induced by consecutive heating and cooling cycles as described previously (13,14); (3) SNEDDS suspension containing a saturated solution of fenofibrate in SNEDDS (corresponding to 100% fenofibrate solubility in SNEDDS) plus suspended fenofibrate (corresponding to 50% of the drug’s solubility in the SNEDDS; “100 + 50%” drug load). The SNEDDS suspension was prepared analogous to the super-SNEDDS but without the heating/cooling cycle. Preliminary X-ray diffraction analyses had confirmed that fenofibrate precipitated in the same crystalline form as the starting material after inducing drug precipitation from a super-SNEDDS (data not shown). The SNEDDS suspension, therefore, resembled the likely composition of a super-SNEDDS after precipitation of excess fenofibrate within the formulation. The freshly prepared formulations were filled manually into hard gelatin capsules (AAEL DB, Capsugel, Strasbourg, France) in amounts corresponding to a dose of 200 mg fenofibrate before immediate use in the in vitro and in vivo studies. To allow reproducible filling of the hard gelatin capsules with SNEDDS suspension, the preconcentrate was continuously stirred to prevent sedimentation of undissolved drug. Commercially available Lipanthyl 200 M capsules containing micronized fenofibrate (Abbott AG, Baar, Switzerland), representing a conventional solid dosage form, were used as received.
Table I.
Drug Loads, Amounts of Formulation, and Number of Capsules used for In Vitro and In Vivo Studies
| Formulation (% drug load) | ||||
|---|---|---|---|---|
| SNEDDS (75%) | Super-SNEDDS (150%) | SNEDDS suspension (100 + 50%) | Lipanthyl | |
| Total dose (mg) | 200 | 200 | 200 | 200 |
| Total amount of lipid formulation (g) | 2.451 | 1.226 | 1.226 | n/a |
| Number of capsules | 3 | 2 | 2 | 1 |
SNEDDS self-nanoemulsifying drug delivery systems
Determination of Fenofibrate Saturation Solubility in Formulations and in Digestion Media
The equilibrium solubility of fenofibrate in anhydrous SNEDDS was determined after incubation of excess fenofibrate (0.2 g) in 1 g of drug-free SNEDDS at 37°C for up to 72 h using a tube shaker/rotator. The excess drug was removed in 24 h intervals by centrifugation at 14,000 rpm for 20 min at 37°C (Eppendorf 5804R, Eppendorf, Hamburg, Germany). Following appropriate dilution in methanol/chloroform aliquots of the clear supernatant were subsequently quantified for fenofibrate by HPLC as described below. Equilibrium solubility was assumed when values of two consecutive days varied less than 5%. The equilibrium solubilities of fenofibrate in 200 mL of lipase-free, gastric lipolysis medium, in 300 mL and in 350 mL of lipase-free, intestinal lipolysis media (both in the absence and presence of corresponding amounts of drug-free formulations) were determined analogous to the method described above after appropriate dilution in methanol. All the determinations were done in triplicates.
Combined Gastric and Intestinal In Vitro Lipolysis
For the current study a combined gastric/intestinal in vitro lipolysis protocol was employed with minor adaptations of the method recently described by Christophersen et al. (20). The freshly prepared capsules filled with formulations corresponding to a dose of 200 mg fenofibrate and commercial Lipanthyl capsules were immersed in a temperature-controlled reaction vessel containing 200 mL of gastric in vitro lipolysis medium (37°C, pH 2) using stainless steel sinkers to prevent floating of the capsules. For the preparation of gastric lipolysis medium sodium taurodeoxycholate was used due to the poor solubility of the porcine bile salts at pH 2. The pH was controlled by a Titrando 842 pH-stat and Tiamo software 1.3 (Metrohm, Zofingen, Switzerland). Gastric lipolysis was carried out for 30 min using the microbial lipase C. antarctica lipase A previously shown to have an activity-profile similar to human gastric lipase (Christophersen et al. (20,22)). Thereafter, 110.5 mL of intestinal lipolysis medium (pH 6.5) were added to the gastric medium and the pH was automatically adjusted to 6.5 by the pH-stat apparatus. Following a 3-min equilibration time, the intestinal in vitro lipolysis was initiated by the addition of 50 mL of freshly prepared pancreatic lipase solution resulting in a total volume of 350 mL at the beginning of intestinal lipolysis. Throughout the subsequent 60-min intestinal lipolysis, the pH was maintained at 6.5 by the titration of liberated fatty acids with 1 M NaOH, and the lipolysis rate was controlled by the constant addition of 0.5 M calcium chloride (0.09 mL/min) (23). Background titration experiments were carried out to determine the sodium hydroxide consumption during the unspecific digestion of phospholipids and impurities present in the formulation-free lipolysis media (24). The initial compositions of the gastric and intestinal lipolysis media are compiled in Table II.
Table II.
Composition of the Fasted State Lipolysis Media as Used for Gastric and Intestinal In Vitro Lipolysis
| pH | Pepsin (mg/mL) | Bile saltsa (mM) | SPC (mM) | Maleate (mM) | NaCl (mM) | Lipase activity (U/mL)b | |
|---|---|---|---|---|---|---|---|
| Gastric | 2 | 0.15 | 0.080 | 0.020 | 2 | 34.2 | 10 |
| Intestinal | 6.5 | – | 3 | 0.6 | 10 | 70 | 179 |
SPC soybean phospholipids
aSodium taurodeoxycholate was used for the preparation of gastric medium; porcine bile extract for intestinal medium
bLipase activity in the gastric step was determined as tributyrin units and the activity in the duodenal step according to USP method (Christophersen et al. (20))
Sample Preparation During In Vitro Lipolysis
Samples (3.5 mL) were withdrawn from the digestion media at predetermined time points (15 and 30 min after initiation of gastric lipolysis; 1 min after pH adjustment to 6.5 (pre-lipase addition); after 5, 15, 30, 45, and 60 min of intestinal lipolysis). Lipolysis in the samples was inhibited by immediate addition of 20 μL of the lipase inhibitor BBBA (1 M in methanol); 0.1 mL of the inhibited sample was diluted with 0.9 mL methanol and was quantified for the total fenofibrate content at the respective time points by HPLC using the method described below. Of the remaining sample, 3.0 mL was subjected to ultracentrifugation (30 min at 100,000 rpm, 37°C) in an Optima MAX-XP ultracentrifuge (Beckman Coulter, Brea, CA, USA) generating an aqueous phase and a pellet, which were both analyzed for fenofibrate by HPLC after appropriate dilution with methanol.
Solid State Characterization of Pellets
Additional samples were withdrawn after 60 min of intestinal in vitro lipolysis in order to characterize the solid state of the pellets generated after ultracentrifugation by X-ray powder diffraction (XRPD) using a X´Pert Pro X-ray diffractometer (MPD PW3040/60 XRD, PANalytical, Almelo, The Netherlands). The isolated pellets were placed on aluminum holders and were irradiated by a CuKα radiation source (λ = 1.542 Å) at 40 kV and 30 mA. The employed scan speed was 0.1285°(2Ө)/min with a step size of 0.0884° (2Ө) between 5° and 35° (2Ө) start and end angle, respectively.
Quantification of Fenofibrate from In Vitro Studies
The samples obtained from solubility and in vitro lipolysis studies were analyzed on a Summit HPLC system (Dionex, Germering, Germany) operated by Chromeleon Software 7 (Thermo Scientific, Germering, Germany). Injected samples (20 μL) were separated on a Waters X-Bridge C18 3.5 μm, 150 mm × 4.6 mm, column (Waters, Milford, MA, USA) using a previously reported method (25). In brief, a methanol: MilliQ–water mixture (85:15%, v/v) was isocratically pumped at a flow of 0.9 mL/min resulting in fenofibrate retention times of approximately 4.3 min detected at a wavelength of 288 nm. The peak areas of the samples were compared with a linear standard curve (R2 > 0.999) established between 0.5 and 100 μg/mL.
In Vitro Data Analysis
The obtained equilibrium solubilities of fenofibrate in the aqueous phase after 30 min gastric lipolysis and after 5 min of the intestinal lipolysis step were used to calculate the maximum SRM according to Eq. 1 (11,17):
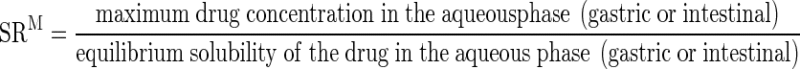 |
1 |
The SRM is directly related to the drug load since it reflects the maximum attainable degree of supersaturation in the absence of precipitation at a given time during digestion. The area under the fenofibrate solubilization-time curve during the course of intestinal lipolysis (AUCin vitro) was determined by the linear trapezoidal method for the correlation with the in vivo data (AUCin vivo).
In Vivo Study
The protocol for the in vivo study was approved by the Animal Welfare Committee, appointed by the Danish Ministry of Justice. All animal procedures were carried out in compliance with EC Directive 86/609/EEC and with the Danish law regulating experiments with animals and the NIH guidelines on animal welfare. Six male Göttingen minipigs (11.5–13.4 kg at study start) were obtained from Ellegaard Göttingen Minipigs A/S (Dalmose, Denmark) and acclimatized for 14 days before initiation of the study in an air-conditioned building with controlled environmental parameters (relative humidity, 50 ± 10%; temperature, 20 ± 1°C; light 06.00–18.00 h). Between the studies the animals had ad libitum access to standard pig food (Altromin 9023, Altromin Spezialfutter, Lage, Germany) and fresh tap water. The pigs were examined weekly by a veterinarian and observed closely after each experimental day. Before entering the experiment, the minipigs were fasted for 18–20 h with free access to water and had access to food 8 h post-dosing. In weekly intervals (wash out period seven days), each animal received 200 mg fenofibrate in freshly prepared capsules containing SNEDDS, super-SNEDDS, SNEDDS suspension, and Lipanthyl in a cross-over design. Before administration, the fenofibrate content in the formulations was confirmed by HPLC analyses. Administration of the capsules was facilitated by a biting block and subsequent administration of approximately 10 mL of tap water according to an internal standard protocol. Blood samples (2 mL) were collected at predetermined time points (predosing, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 24, 30, 48, and 72 h post-dosing) in heparinized tubes. Following centrifugation at 3,200 rpm (15 min, 4°C), the plasma was harvested and the samples were stored at −80°C until analyses.
Quantification of Plasma Samples
The plasma samples were analyzed for fenofibric acid, the main metabolite of fenofibrate, using the validated assay previously described by Hanafy et al. with minor modifications (26). For sample preparation, 50 μL of plasma was spiked with internal standard (40 μL, 200 μg/mL clofibric acid in methanol) before adding 410 μL of methanol. After brief whirl mixing the samples were sonicated for 10 min in a Corpax GS20 ultrasonic water bath (Corpax, Valby, Denmark) followed by storage for 10 min at −20°C. Thereafter, the samples were centrifuged for 14 min at 15,600 rpm at 4°C (Eppendorf 5804R, Eppendorf, Hamburg, Germany). Each sample was analyzed in duplicate by injection of the clear supernatant (50 μL) and separation on the identical chromatographic system as described above using methanol/MilliQ water (68%:32%, 0.1% formic acid, v/v) as mobile phase at a flow rate of 0.6 mL/min. Samples for the standard curve (0.05 to 3.0 μg/mL, R2 > 0.997) were prepared in a similar manner by spiking blank plasma with varying concentrations of fenofibric acid in methanol, 40 μL of internal standard, and 410 μL of methanol. The accuracy of the assay was between 94 and 103% and the precision between 4.2 and 1.6% at 0.1 and 1.0 μg/mL, respectively (n = 5).
Pharmacokinetics and Statistical Analyses
The pharmacokinetic parameters were determined by noncompartmental analyses using WinNonlin Professional 6.3 (Pharsight Corporation, Mountain View, USA). The AUC were calculated by the linear trapezoidal method from time zero to 8 h (AUC0–8 h), i.e., until the time of feeding post-dosing, and from zero to 72 h (AUC0–72 h). The peak plasma concentrations (Cmax) and their time of occurrence (tmax) were recorded directly from the individual plasma concentration-time curves. The oral bioavailability of fenofibrate after administration of SNEDDS, super-SNEDDS, and SNEDDS suspension relative to the commercial Lipanthyl capsules were calculated based on the AUC0–72 h obtained from individual animals. Analyses of variance (ANOVA) in combination with Tukey’s multiple comparisons test, and Pearson correlation coefficients were computed by GraphPad Prism Version 6.03 (La Jolla, CA, USA) to determine statistical significance between group means and to determine correlations between in vitro and in vivo data (p < 0.05).
RESULTS
Solubility Determination and Preparation of Formulations
The equilibrium solubility of fenofibrate in the SNEDDS preconcentrate at 37°C was attained within 48 h incubation time and totaled 108.8 ± 4.1 mg fenofibrate/g preconcentrate. At this value the equilibrium solubility represents 100% drug load. Based on the equilibrium solubility, drug loads of 75 and 150% were calculated and the formulations were prepared accordingly to achieve the target dose of 200 mg in each formulation. In the case of SNEDDS and super-SNEDDS, the entire drug load was dissolved in the formulation, whereas in the case of the SNEDDS suspension crystalline fenofibrate was partially suspended in the formulation as described above. As a consequence of the reduced drug load in the SNEDDS compared with the other formulations it was necessary to prepare a slightly greater number of capsules for the in vitro and in vivo studies (Table I).
Combined Gastric and Intestinal In Vitro Lipolysis
All capsules containing lipid formulations (SNEDDS, super-SNEDDS, and SNEDDS suspension) ruptured and fully dispersed their content within approximately 3 min in the low pH medium of the gastric lipolysis step. Similarly, Lipanthyl capsules raptured within the same time, but instead of forming a fine emulsion the powder content (mainly micronized fenofibrate, lactose, magnesium stearate, pregelatinized maize starch, sodium lauryl sulfate, and crospovidone, according to manufacturer’s information) was suspended in the medium. For all formulations, no consumption of sodium hydroxide was observed during the gastric step of in vitro lipolysis (data not shown). The addition of pancreatic lipase solution for the intestinal lipolysis step at pH 6.5 triggered the hydrolysis of digestible excipients and resulted in the continuous consumption of sodium hydroxide over 60 min of intestinal in vitro lipolysis and correlated inversely with the drug load of the formulations (R2 = 0.96, p = 0.02). Consistent with this observation the sodium hydroxide consumption during the digestion of super-SNEDDS (150% drug load) and SNEDDS suspension (“100 + 50%” drug load) were comparable. As expected for the Lipanthyl capsules, devoid of SNEDDS excipients, only small amounts of sodium hydroxide were released (<0.2 mL), comparable to those observed during the digestion of lipolysis medium in the absence of formulations.
The solubilization of fenofibrate in the aqueous phase during consecutive gastric and intestinal in vitro lipolysis showed pronounced differences across the different formulations (Fig. 1). For the SNEDDS (75% drug load) almost the entire dose of fenofibrate was maintained solubilized in the aqueous phase, throughout 30 min of the gastric step. However, after initiation of the intestinal step the amount of fenofibrate solubilized in the aqueous phase continuously decreased to approximately 60% at 60 min.
Fig. 1.
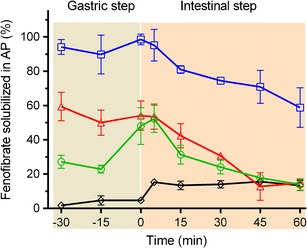
Relative distribution of fenofibrate in the aqueous phase during 30 min of gastric (pH 2, gray shaded) and 60 min intestinal (pH 6.5, orange shaded) in vitro lipolysis of SNEDDS (75% drug load, squares), 150% drug-loaded super-SNEDDS (triangles), “100 + 50%” drug-loaded SNEDDS suspension (circles), and Lipanthyl (diamonds), mean ± SD, n = 3. The drug load in each formulation was 200 mg of fenofibrate
For the super-SNEDDS (150% drug load), considerably less fenofibrate was recovered from the aqueous phase (approximately 50–60%) reflecting the greater degree of drug precipitation prior to the first sampling time point during the gastric lipolysis step. In line with the SNEDDS data, the intestinal digestion step resulted in further reduction of fenofibrate in the aqueous phase to approximately 15% of the fenofibrate dose after 45 min of intestinal in vitro lipolysis. Fenofibrate solubilization was reduced during gastric lipolysis for the SNEDDS suspension (“100 + 50%” drug load) in which approximately one third of the entire fenofibrate dose was present in the formulation as suspended, crystalline drug. During the initial gastric step, approximately 25% of the fenofibrate dose was solubilized in the aqueous phase. The addition of intestinal medium and adjustment of the digestion medium to pH 6.5 (pre-lipase) facilitated an increased fenofibrate solubilization to levels comparable with the super-SNEDDS. The subsequent digestion of the SNEDDS suspension triggered a decline in the fenofibrate solubilization analogous to the super-SNEDDS. By contrast, during the gastric in vitro lipolysis step of the conventional Lipanthyl formulation comparably small amounts of the drug (5%) were solubilized in the aqueous phase, indicating that the presence of sodium lauryl sulfate in the commercial product contributes little to drug solubilization. The addition of the intestinal medium caused an increase of solubilized fenofibrate to approximately 15%. As expected, and in contrast to the lipid formulations, the initiation of intestinal lipolysis had no negative impact on the solubilization of fenofibrate throughout the remaining intestinal digestion step.
Solubilization of Fenofibrate in the Digestion Media
In the presence of blank SNEDDS 646.9 ± 6.7 μg/mL of fenofibrate was solubilized in the gastric medium whereas in the intestinal medium (adjusted to pH 6.5, pre-lipase) the fenofibrate solubilization was reduced to 430.0 ± 6.8 μg/mL and decreased only slightly to 389.1 ± 5.7 μg/mL in the total volume (350 mL) of intestinal medium at the beginning of intestinal lipolysis. The absolute amounts of lipid formulation excipients present in the media decreased in the case of super-SNEDDS and SNEDDS suspension due to the higher drug loading, hence the solubilization of fenofibrate was determined in the presence of the corresponding, reduced amount of excipients for these formulations. As expected, the solubilization capacity for fenofibrate was lower than for the SNEDDS (291.6 ± 7.0 μg/mL in the gastric medium, 219.7 ± 6.3 μg/mL and 205.2 ± 8.6 μg/mL in 300 and 350 mL of intestinal medium, respectively). By contrast, the solubilization capacities of fenofibrate in the lipid-free media (representing the powder formulation present in Lipanthyl) were considerably lower at 4.9 ± 0.5 and 19.2 ± 1.6 μg/mL for the gastric and intestinal media, respectively.
Maximum Supersaturation Ratio
Based on the equilibrium solubilities and the measured concentrations in the aqueous phase, the maximum SRM across the different formulations during the gastric and intestinal in vitro lipolysis steps were calculated and are compiled in Table III.
Table III.
Maximum Supersaturation Ratios (SRM)a During the Gastric and the Intestinal Lipolysis Step Across the Different Formulations
| Condition | Time (min) | SNEDDS | super-SNEDDS | SNEDDS suspension | Lipanthyl |
|---|---|---|---|---|---|
| Gastric (pH 2) | 30 | 1.5 | 3.4 | 3.4 | 203.4 |
| Intestinal (pH 6.5, pre-lipase 300 mL) | 0 | 1.6 | 3.0 | 3.0 | 34.8 |
| Intestinal (pH 6.5, post-lipase 350 mL) | 5 | 1.5 | 2.8 | 2.8 | 29.8 |
The SRM was calculated as the maximum drug concentration in the aqueous phase (gastric or intestinal) divided by the corresponding equilibrium solubility of the drug in the aqueous phase (gastric or intestinal)
SNEDDS self-nanoemulsifying drug delivery systems
The attainable maximum SRM was moderate for the 75% drug-loaded SNEDDS (approximate SRM = 1.5) but was increased for the super-SNEDDS (approximate SRM = 2.8) and SNEDDS suspension (approximate SRM = 3.4). The shift from gastric to intestinal lipolysis medium was not reflected in substantially different SRM, independent from the formulation and the relative drug load. The very high SRM calculated for Lipanthyl reflected the fact that the same amount of drug was present in this formulation as in the lipid formulations. However, as Lipanthyl is devoid of the excipients that were present in SNEDDS (e.g., Kolliphor RH40) the commercial product could not enhance the low solubilization capacity of the blank digestion medium.
Solid State Characterization of the Pellets
The solid state properties of the precipitated fenofibrate were investigated by XRPD. The diffractograms of the isolated pellets obtained after 60 min of intestinal in vitro lipolysis and ultracentrifugation are depicted in Fig. 2. Consistent with previous results the characteristic diffraction patterns of crystalline fenofibrate were visible in all pellets except the negative control, in which drug-free SNEDDS were subjected to in vitro lipolysis (27).
Fig. 2.
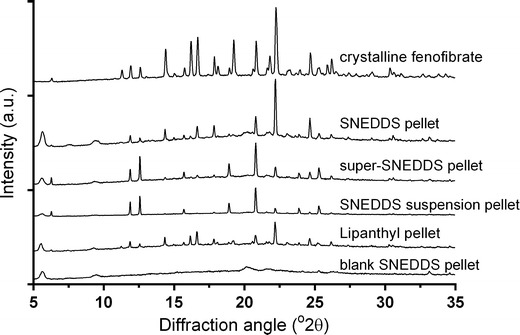
Diffractograms of the isolated pellets obtained after 60 min of intestinal in vitro lipolysis
In Vivo Study
Following oral administration fenofibrate is extensively metabolized in the enterocytes to fenofibric acid, hence the plasma concentration vs time curves of fenofibric acid are shown in Fig. 3, and the corresponding pharmacokinetic parameters are tabulated in Table IV.
Fig. 3.
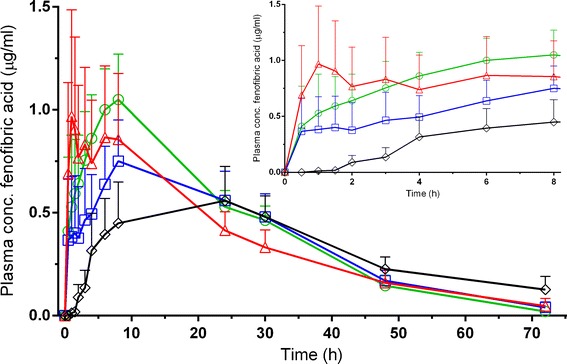
Plasma concentration of fenofibric acid over 72 h and the initial 8 h (insert) after oral administration of SNEDDS (squares), super-SNEDDS (triangles), SNEDDS suspension (circles), and Lipanthyl (diamonds) to six minipigs (mean ± SEM)
Table IV.
Pharmacokinetic Parameters for Fenofibric Acid After Administration of 200 mg Fenofibrate to Six Minipigs Are Reported as Mean ± SD Unless Indicated Differently
| C max (μg/mL)a | t max (h)a | AUC0–8 h (μg h/mL) | AUC0-72h (μg h/mL) | Rel. BA (%) | |
|---|---|---|---|---|---|
| SNEDDS | 0.9 (0.2–1.8) | 8.0 (0.5–30) | 4.1 ± 3.7 | 25.7 ± 15.5 | 116.7 ± 51.2 |
| Super-SNEDDS | 1.0 (0.1–2.7) | 8.0 (1–30) | 6.4 ± 6.6 | 26.1 ± 12.3 | 114.9 ± 63.6 |
| SNEDDS suspension | 1.3 (0.4–2.2) | 8.0 (1–30) | 6.3 ± 4.0 | 26.4 ± 9.0 | 158.2 ± 139.5 |
| Lipanthyl | 0.9 (0.4–1.2) | 24 (8–72) | 1.9 ± 2.3 | 23.7 ± 7.5 | 100 |
SNEDDS self-nanoemulsifying drug delivery systems, C max peak plasma concentrations, t max time of occurrence, AUC areas under the plasma concentration-time curves
aMedian and range in brackets
In general, there was considerable inter- and intra-individual variability in the plasma concentrations, complicating interpretation of the data. Where considered appropriate the data in Table IV is presented either as the mean ± SD or the median and the range (in addition the mean values are reported in this section).
After administration of 200 mg fenofibrate as lipid-based formulation the course of the plasma concentration profiles was comparable, regardless of the drug being initially completely dissolved (75% drug load SNEDDS and super-SNEDDS) or (partly) suspended (SNEDDS suspension). The mean maximum plasma concentrations (Cmax) were 1.0 μg/mL (SNEDDS) and 1.3 μg/mL both for super-SNEDDS and SNEDDS suspension. It is noteworthy that the upper range of the maximum plasma concentration for the SNEDDS formulation was 1.8 μg/mL, whereas 2.7 and 2.2 μg/mL were observed for super-SNEDDS and SNEDDS suspension, respectively (Table IV). By contrast, the maximum plasma concentration did not exceed 1.2 μg/mL after the administration of Lipanthyl. The area under the plasma concentration-time curve was calculated both for the time until the minipigs had access to food post-dosing (AUC0–8 h) and the last sampling time point (AUC0-72h). The AUC0-72h was comparable across the tested formulations which also resulted in no significant difference in the relative bioavailability between the formulations. With regard to the initial AUC0–8 h (Fig. 3, insert; Table IV), all SNEDDS showed a trend towards increased AUC0–8 h, which was more pronounced for the super-SNEDDS and SNEDDS suspension compared with SNEDDS, whereas the treatment with Lipanthyl resulted in a comparably low initial AUC0–8 h.
Correlation of In Vitro and In Vivo Data
The areas under plasma concentration-time curves (AUCin vivo), both for the initial 8 h (AUC0–8 h) and the last sampling time point (AUC0–72 h) post-dosing, were plotted against the area under the solubilization-time curves (AUCin vitro) of the intestinal step (Fig. 4). As illustrated and confirmed by analysis of correlation (Pearson correlation coefficient, r = 0.15 and r = 0.18, for AUC0–8 h, and AUC0–72 h, respectively, p > 0.8), no correlation was found between the AUCin vitro and the AUCin vivo data.
Fig. 4.
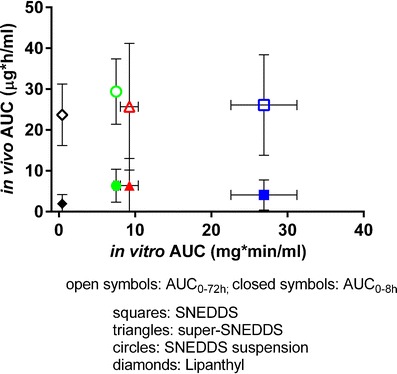
Relationship between the areas under the plasma concentration-time curves (AUCin vivo) and the area under the solubilization-time curves (AUCin vitro) during intestinal lipolysis. Open symbols represent the AUC0–8 h, closed symbols AUC0–72 h. Formulations include SNEDDS (squares), super-SNEDDS (triangles), SNEDDS suspension (circles), and Lipanthy (diamonds)
DISCUSSION
Lipid-based drug delivery systems, such as SNEDDS, have been investigated for the effects of droplet size, lipid composition, the degree of solubilization/precipitation, and the ability to maintain supersaturation during in vitro dispersion and in vitro digestion on the in vivo performance (9,11,12). Moreover, recent advances in the field have highlighted some critical factors during in vitro dispersion and in vitro digestion (16,17,28). It should be noted that in these previous studies the investigated formulations were rather diverse with respect to the nature of the lipid, and the relative lipid, surfactant, and cosolvent composition. By contrast, less literature is available regarding the evaluation of in vitro performance criteria and their ability to predict in vivo results for formulations of the same relative composition, which might aid in the identification of critical in vitro parameters (29,30). Therefore, the current study set out to compare the in vitro and in vivo performance of a series of SNEDDS of the same relative composition containing high drug loads. Fenofibrate was used as a model compound as the drug precipitates in crystalline rather than in an amorphous form facilitating the evaluation of recently proposed in vitro performance parameters, such as solubilization and SRM for their in vivo relevance.
The incorporation of a gastric step in current lipolysis models, which are commonly limited to an intestinal step, is desirable as gastric digestion can contribute up to approximately 20% of the total lipid digestion (31). However, availability and economic considerations have hampered the use of, e.g., recombinant human gastric lipase for in vitro lipolysis. In lieu of recombinant human gastric lipase, C. antarctica lipase A has recently been used successfully in combination with intestinal in vitro lipolysis to predict the in vivo performance of SNEDDS in beagle dogs (20). In agreement with the previous study under fasted conditions, no consumption of sodium hydroxide was observed in the current study during 30 min of gastric in vitro lipolysis. It should be noted that the titration method is an indirect way to monitor hydrolysis of triglycerides that depends on the ionization of fatty acids. At pH 2, the fatty acids are protonated (pKa approximately 4–5), thus they are not available for titration. The lack of sodium hydroxide consumption during the gastric step, therefore, does not imply the absence of hydrolysis but rather demonstrates that the degree of gastric lipolysis is not directly accessible using the currently established titration method.
The solubilization of fenofibrate remained largely unchanged during the gastric step for each formulation suggesting that any occurring lipolysis did not significantly affect the solubilization of the drug. Fenofibrate is a neutral drug and its solubility in biorelevant media has been shown to depend on the amount and the type of surfactants used (25). Since the concentration of bile salts in the gastric medium was low, the high solubilization of fenofibrate in the dispersed lipid formulations can mainly be attributed to the excipients present in the formulations. Consistent with this the solubilization of powdered fenofibrate increased fivefold in the intestinal medium compared to the low levels in the gastric medium in the absence of formulation- derived excipients, but increased dramatically in the presence of SNEDDS (60- to 90-fold). Starting from an initially high solubilization capacity in the gastric medium the solubilization capacity of fenofibrate decreased in the intestinal medium in spite of the increased bile salt levels present in the intestinal medium. This can be attributed to the larger media volumes in the intestinal step and the concomitant dilution of the SNEDDS excipients as the solubilization of fenofibrate is primarily based on formulation excipients and not on bile salts. As an example, the 1.5-fold dilution of the digestion media following the switch from the gastric (200 mL) to the intestinal step (300 mL) corresponded well with the reduction in fenofibrate solubility by the same factor.
During intestinal in vitro lipolysis at pH 6.5 the continuous digestion of the three lipid formulations could be monitored by the consumption of sodium hydroxide (data not shown). In line with an earlier study, the drug load inversely correlated with the sodium hydroxide consumption, reflecting the displacement of digestible lipid components by the incorporated drug (32). For each lipid formulation, the proceeding digestion was reflected in a continuously decreasing fenofibrate solubilization. At the same time the SRM changed only moderately, again due to the dilution of the medium. In contrast, for Lipanthyl the change from gastric to intestinal medium, in the absence of SNEDDS excipients, gave rise to improved fenofibrate solubilization mediated by the elevated bile salt concentration which significantly reduced the SRM.
Based on the assumption that only drug in solution is available for absorption, the considerably enhanced fenofibrate solubilization generated by 75% drug load SNEDDS (Fig. 1) compared with the other formulations during the intestinal in vitro lipolysis step suggested that SNEDDS would also be superior in the in vivo performance, whereas the super-SNEDDS and SNEDDS suspension could be expected to yield similar plasma concentrations, and Lipanthyl would show the poorest in vivo performance. In agreement with previous studies drug precipitation was evident in cases where the SRM exceeded values of around 3 (super-SNEDDS and SNEDDS suspension), indicating a high “precipitation pressure” of the systems (16,17). Conversely, the low SRM of approximately 1.5 generated by 75% drug load SNEDDS was reflected in larger amounts of solubilized fenofibrate. Thus, based on the SRM values the same in vitro rank-order of the formulations (SNEDDS > super-SNEDDS/SNEDDS suspension > Lipanthyl) could be established as obtained from the drug solubilization curves. The evaluation of the formulations in the minipig model, however, poorly reflected the in vitro solubilization data. The administration of the various lipid formulations resulted in very similar in vivo performance (Cmax, tmax, AUC0–72 h, and relative bioavailability), whether the drug was initially present in solution (either below or above saturation solubility), suspended in the formulation, or whether the drug was maintained in solution or precipitated during in vitro lipolysis. Importantly, the enhanced solubilization of fenofibrate and the lower SRM calculated for SNEDDS was not reflected in enhanced in vivo performance, contrasting the in vitro results. Compared with 75% drug load SNEDDS there was a trend to faster absorption and increased plasma concentrations during the initial 8 h following administration of super-SNEDDS and SNEDDS suspension which was not expected according to the in vitro solubilization data of these formulations. Some agreement between the in vitro and in vivo performance was observed for Lipanthyl, at least when comparing the initial AUC0-8h and the slow and incomplete solubilization during in vitro lipolysis.
Compared with Lipanthyl, all SNEDDS showed an increased rate of drug absorption, as indicated by the higher Cmax, and partial AUC (AUC0–8 h). However, the initial benefits of faster onset and higher AUC0-8h were not sufficient to result in an improved relative bioavailability of fenofibrate after 72 h from SNEDDS compared to Lipanthyl. The faster initial drug absorption in the present investigation is in agreement with an earlier study by Hanafy et al. where a nanosuspension and solid lipid nanoparticle formulation of fenofibrate showed enhanced AUC0–8 h and Cmax in a rat model compared to micronized drug while the performance of the two colloidal formulations was comparable (26). The authors concluded that particle size reduction from the micro- to the nanometer range were likely to explain the observed differences between the formulations. In the current study, fenofibrate was, at least initially, molecularly dissolved in the 75% drug load SNEDDS and super-SNEDDS formulation. The particle size of drug precipitating during in vitro digestion is not known, neither is it accessible to which extend the drug precipitates in vivo. It might be possible that in vivo fenofibrate precipitates in particles of similar size from all the investigated SNEDDS leading to similar drug absorption during the transit in the large gastrointestinal tract of the pig (33). The anatomic peculiarities of the pig, e.g., the reported long gastric emptying time (34), might also facilitate complete absorption of fenofibrate from the slowly dissolving Lipanthyl formulation resulting in no apparent differences in the overall extent of bioavailability between all investigated formulations. Similar to the current study a discrepancy between in vitro and in vivo performance has recently also been reported by Thi et al. (35). The authors found marked differences during in vitro dissolution studies of fenofibrate-loaded SNEDDS of various lipid and surfactant composition in biorelevant media, whereas identical in vivo performances across the investigated SNEDDS were observed in a rat model. In contrast to the current findings, however, the in vivo performance of the SNEDDS was significantly enhanced compared to commercial Lipanthyl (i.e., the same micronized drug as used in the current study). The large variations between individual animals observed in the present in vivo study precluded the detection of significant differences between the treatments and might reflect pharmaceutical (e.g., the degree and extent of precipitation in the intestine) and physiological differences (e.g., gastric emptying, metabolism, and enterohepatic circulation) between animals (33,34,36,37). The gastric emptying might explain, e.g., the variation in tmax within each treatment (Table IV) but requires further investigation with regard to the comparably late occurrence of tmax in the minipig model, which to the authors best knowledge, has not been reported for other species (35,38–40). It might be possible that the evaluation of the in vivo performance by the quantification of the fenofibrate metabolite fenofibric acid is affected by the metabolic conversion of the parent compound to fenofibric acid and the subsequent enterohepatic circulation of the metabolite, which has been shown to be species dependent (36). Nevertheless, the discrepancies between the in vitro and in vivo results in current and aforementioned studies suggest that the administration of fenofibrate as SNEDDS is more relevant for the in vivo performance than the initial physical state of the drug (dissolved or suspended) and the solubilization profile obtained from in vitro lipolysis. This contrasts previous studies correlating the amount of solubilized drug in the aqueous phase after in vitro lipolysis with the in vivo performance (11,41,42). These studies used danazol as a model drug, and further differed from the current study in terms of the employed formulations, digestion protocols, and animal models. With regard to the digestion protocols, currently two methods are being followed: either the continuous addition of calcium ions over the course of in vitro lipolysis to precipitate liberated fatty acids as calcium soaps; or the bolus addition of a relatively large amount of calcium ions (typically, 5 mM) at the beginning of the digestion experiment (24,43). In the past, both approaches were not able to consistently correlate in vitro solubilisation data with in vivo performance. It is, therefore unlikely that the calcium addition in the digestion protocols could account for the limited correlation. In fact, a very recent study by Griffin et al. in which fenofibrate-containing LbDDS of various complexity were investigated the bolus addition of calcium was employed for the in vitro lipolysis protocol (44). Similar to the findings in the current study the group was not able to correlate solubilization data from in vitro lipolysis with in vivo data obtained in Landrace pigs. Given the pronounced differences in the physicochemical and biopharmaceutical properties of currently available drugs caution should be exercised when extrapolating the findings of the current study using fenofibrate A growing number of evidence, however, suggests that formulation optimization towards increased drug solubilization during in vitro lipolysis might be misleading for the successful development of SNEDDS (12–14,29,30). It would be desirable to extend the currently employed in vitro digestion models by an absorption step which could provide valuable information beyond drug solubilization. One possible way might be the utilization of recently described cell co-cultures capable to mimic the absorption barrier for drugs while being potentially more tolerant towards the presence of formulation-derived components and simulated intestinal media (45,46). Physicochemical alternatives to cell culture models include phospholipid vesicle-based barriers (47) and ultrafiltration (48), which could also help to improve the prediction of the in vivo performance from in vitro digestion data and, ultimately, lead to a more rational development of SNEDDS.
CONCLUSIONS
The evaluation of three SNEDDS with identical lipid composition and a solid dosage form during combined gastric and intestinal in vitro lipolysis revealed differences with regard to the amount of solubilized drug in the aqueous phase. The likelihood of drug precipitation was correctly predicted by the maximum SRM. However, based on drug solubilization profiles and SRM as in vitro performance criteria it was not possible to predict formulation performance in a minipig model suggesting to exercise caution when interpreting in vitro data obtained from in vitro lipolysis.
Acknowledgment
The authors would like to thank the personnel at the Lundbeck animal facilities for their skillful handling of the animals and support for this study.
References
- 1.Williams RO III, Watts AB, Miller DA, editors. Formulating poorly water soluble drugs. New York: Springer; 2012. [Google Scholar]
- 2.Stegemann S, Leveiller F, Franchi D, de Jong H, Linden H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31(5):249–261. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 3.Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65(1):315–499. doi: 10.1124/pr.112.005660. [DOI] [PubMed] [Google Scholar]
- 4.Li P, Zhao L. Developing early formulations: practice and perspective. Int J Pharm. 2007;341(1–2):1–19. doi: 10.1016/j.ijpharm.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 5.Hauss DH, editor. Oral lipid-based formulations: enhancing the bioavailability of poorly water-soluble drugs. New York: Informa Healthcare; 2007. [Google Scholar]
- 6.Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and 'self-microemulsifying' drug delivery systems. Eur J Pharm Sci. 2000;11(Supplement 2):S93–S98. doi: 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 7.Anton N, Vandamme T. Nano-emulsions and micro-emulsions: clarifications of the critical differences. Pharm Res. 2011;28(5):978–985. doi: 10.1007/s11095-010-0309-1. [DOI] [PubMed] [Google Scholar]
- 8.Porter CJH, Pouton CW, Cuine JF, Charman WN. Enhancing intenstinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi: 10.1016/j.addr.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Cuiné JF, Charman WN, Pouton CW, Edwards GA, Porter CJH. Increasing the proportional content of surfactant (Cremophor® EL) relative to lipid in self-emulsifying lipid-based formulations of danazol reduces oral bioavailability in beagle dogs. Pharm Res. 2004;24(4):748–757. doi: 10.1007/s11095-006-9194-z. [DOI] [PubMed] [Google Scholar]
- 10.Mohsin K, Long MA, Pouton CW. Design of lipid-based formulations for oral administration of poorly water-soluble drugs: Precipitation of drug after dispersion of formulations in aqueous solution. J Pharm Sci. 2009;98(10):1–14. doi: 10.1002/jps.21659. [DOI] [PubMed] [Google Scholar]
- 11.Anby MU, Williams HD, McIntosh M, Benameur H, Edwards GA, Pouton CW, et al. Lipid digestion as a trigger for supersaturation: evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol Pharm. 2012;9(7):2063–2079. doi: 10.1021/mp300164u. [DOI] [PubMed] [Google Scholar]
- 12.Larsen AT, Ohlsson AG, Polentarutti B, Barker RA, Phillips AR, Abu-Rmaileh R, et al. Oral bioavailability of cinnarizine in dogs: relation to SNEDDS droplet size, drug solubility and in vitro precipitation. Eur J Pharm Sci. 2013;48(1–2):339–350. doi: 10.1016/j.ejps.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Thomas N, Holm R, Garmer M, Karlsson JJ, Müllertz A, Rades T. Supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2013;15(1):219–227. doi: 10.1208/s12248-012-9433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) J Control Release. 2012;160(1):25–32. doi: 10.1016/j.jconrel.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 15.Sassene PJ, Knopp MM, Hesselkilde JZ, Koradia V, Larsen A, Rades T, et al. Precipitation of a poorly soluble model drug during in vitro lipolysis: characterization and dissolution of the precipitate. J Pharm Sci. 2010;99(12):4982–4991. doi: 10.1002/jps.22226. [DOI] [PubMed] [Google Scholar]
- 16.Williams H, Sassene P, Kleberg K, Calderone M, Igonin A, Jule E, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 3: understanding supersaturation versus precipitation potential during the in vitro digestion of Type I, II, IIIA, IIIB and IV lipid-based formulations. Pharm Res. 2013 2013/05/10:1–18. [DOI] [PubMed]
- 17.Williams HD, Anby MU, Sassene P, Kleberg K, Bakala-N’Goma J-C, Calderone M, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations. 2. The effect of bile salt concentration and drug loading on the performance of Type I, II, IIIA, IIIB, and IV formulations during in vitro digestion. Mol Pharm. 2012;9(11):3286–3300. doi: 10.1021/mp300331z. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez S, Chevrier S, Ritter N, Mahler B, Demarne F, Carrière F, et al. In vitro gastrointestinal lipolysis of four formulations of piroxicam and cinnarizine with the self emulsifying excipients Labrasol® and Gelucire® 44/14. Pharm Res. 2009;26(8):1901–1910. doi: 10.1007/s11095-009-9906-2. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez S, Jannin V, Chevrier S, Chavant Y, Demarne F, Carrière F. In vitro digestion of the self-emulsifying lipid excipient Labrasol® by gastrointestinal lipases and influence of its colloidal structure on lipolysis rate. Pharm Res. 2013;30(12):3077–3087. doi: 10.1007/s11095-013-1053-0. [DOI] [PubMed] [Google Scholar]
- 20.Christophersen PC, Christiansen ML, Holm R, Kristensen J, Jacobsen J, Abrahamsson B, et al. Fed and fasted state gastro-intestinal in vitro lipolysis: in vitro in vivo relations of a conventional tablet, a SNEDDS and a solidified SNEDDS. Eur J Pharm Sci. 2013;in press. [DOI] [PubMed]
- 21.Ren S, Ogbonna A, Koradia V, Sassene P, Rades T, Müllertz A. Redissolution and polymorph characterization of a poorly water-soluble drug precipitated during in vitro lipolysis from a lipid-based formulation. New Orleans, LA, USA: American Association of Pharmaceutical Sciencits (AAPS) Annual Meeting; 2010. [Google Scholar]
- 22.Mercuri A, Passalacqua A, Wickham MJ, Faulks R, Craig DM, Barker S. The effect of composition and gastric conditions on the self-emulsification process of ibuprofen-loaded self-emulsifying drug delivery systems: a microscopic and dynamic gastric model study. Pharm Res. 2011;28(7):1540–1551. doi: 10.1007/s11095-011-0387-8. [DOI] [PubMed] [Google Scholar]
- 23.Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–122. doi: 10.1016/S0928-0987(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 24.Thomas N, Holm R, Rades T, Müllertz A. Characterising lipid lipolysis and its implication in lipid-based formulation development. AAPS J. 2012;14(4):860–871. doi: 10.1208/s12248-012-9398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleberg K, Jacobsen F, Fatouros DG, Müllertz A. Biorelevant media simulating fed state intestinal fluids: colloid phase characterization and impact on solubilization capacity. J Pharm Sci. 2010;99(8):3522–3532. doi: 10.1002/jps.22122. [DOI] [PubMed] [Google Scholar]
- 26.Hanafy A, Spahn-Langguth H, Vergnault G, Grenier P, Tubic Grozdanis M, Lenhardt T, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–426. doi: 10.1016/j.addr.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Sassene P, Michelsen J, Ren S, Koradia V, Larsen A, Rades T, et al. Characterization of precipitates formed during in vitro lipolysis of SMEDDS containing 3 poorly soluble model drugs. Innovation in Drug Delivery: From Preformulation to Development through Innovative Evaluation Process; 3.-6. October, Aix-en Provence, France2010.
- 28.Williams HD, Sassene P, Kleberg K, Bakala-N'Goma J-C, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012;101(9):3360–3380. doi: 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- 29.Larsen A, Holm R, Pedersen M, Müllertz A. Lipid-based formulations for danazol containing a digestible surfactant, Labrafil® M2125CS: in vivo bioavailability and dynamic in vitro lipolysis. Pharm Res. 2008;25(12):2769–2777. doi: 10.1007/s11095-008-9641-0. [DOI] [PubMed] [Google Scholar]
- 30.Larsen A, Åkesson P, Juréus A, Saaby L, Abu-Rmaileh R, Abrahamsson B, et al. Bioavailability of cinnarizine in dogs: Effect of SNEDDS loading level and correlation with cinnarizine solubilization during in vitro lipolysis. Pharm Res. 2013 2013/08/15;in press:1–13. [DOI] [PubMed]
- 31.Carriere F, Barrowman JA, Verger R, Laugier R. Secretion and contribution to lipolysis of gastric and pancreatic lipases during a test meal in humans. Gastroenterology. 1993;105(3):876–888. doi: 10.1016/0016-5085(93)90908-u. [DOI] [PubMed] [Google Scholar]
- 32.Thomas N, Müllertz A, Graf A, Rades T. Influence of lipid composition and drug load on the in vitro performance of self-nanoemulsifying drug delivery systems. J Pharm Sci. 2012;101(5):1721–1731. doi: 10.1002/jps.23054. [DOI] [PubMed] [Google Scholar]
- 33.Bode G, Clausing P, Gervais F, Loegsted J, Luft J, Nogues V, et al. The utility of the minipig as an animal model in regulatory toxicology. J Pharmacol Toxicol Methods. 2010;62(3):196–220. doi: 10.1016/j.vascn.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Hossain M, Abramowitz W, Watrous BJ, Szpunar GJ, Ayres JW. Gastrointestinal transit of nondisintegrating, nonerodible oral dosage forms in pigs. Pharm Res. 1990;7(11):1163–1166. doi: 10.1023/A:1015936426906. [DOI] [PubMed] [Google Scholar]
- 35.Thi TD, Van Speybroeck M, Mols R, Annaert P, Martens J, Van Humbeeck J, et al. The conflict between in vitro release studies in human biorelevant media and the in vivo exposure in rats of the lipophilic compound fenofibrate. Int J Pharm. 2011;414(1–2):118–124. doi: 10.1016/j.ijpharm.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Weil A, Caldwell J, Strolin-Benedetti M. The metabolism and disposition of fenofibrate in rat, guinea pig, and dog. Drug Metab Dispos. 1988;16(2):302–309. [PubMed] [Google Scholar]
- 37.Davis SS, Illum L, Hinchcliffe M. Gastrointestinal transit of dosage forms in the pig. J Pharm Pharmacol. 2001;53(1):33–39. doi: 10.1211/0022357011775163. [DOI] [PubMed] [Google Scholar]
- 38.Fei Y, Kostewicz ES, Sheu M-T, Dressman JB. Analysis of the enhanced oral bioavailability of fenofibrate lipid formulations in fasted humans using an in vitro–in silico–in vivo approach. Eur J Pharm Biopharm. 2013;85(3):1274–1284. doi: 10.1016/j.ejpb.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 39.He H, Yang R, Tang X. In vitro and in vivo evaluation of fenofibrate solid dispersion prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2010;36(6):681–687. doi: 10.3109/03639040903449720. [DOI] [PubMed] [Google Scholar]
- 40.Linn M, Collnot E-M, Djuric D, Hempel K, Fabian E, Kolter K, et al. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur J Pharm Sci. 2012;45(3):336–343. doi: 10.1016/j.ejps.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Porter CJH, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. doi: 10.1023/B:PHAM.0000036914.22132.cc. [DOI] [PubMed] [Google Scholar]
- 42.Cuiné JF, Claire L, McEvoy CL, Charman WN, Pouton CW, Edwards GA, et al. Evaluation of the impact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs. J Pharm Sci. 2008;97(2):995–1012. doi: 10.1002/jps.21246. [DOI] [PubMed] [Google Scholar]
- 43.Larsen AT, Sassene P, Müllertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–255. doi: 10.1016/j.ijpharm.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Griffin B, Kuentz M, Vertzoni M, Kostewicz E, Fei Y, Faisal W, et al. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: case studies with fenofibrate. Eur J Pharm Biopharm. 2013 doi: 10.1016/j.ejpb.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 45.Araújo F, Sarmento B. Towards the characterization of an in vitro triple co-culture intestine cell model for permeability studies. Int J Pharm. 2013;458(1):128–134. doi: 10.1016/j.ijpharm.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Antunes F, Andrade F, Ferreira D, Nielsen HM, Sarmento B. Models to predict intestinal absorption of therapeutic peptides and proteins. Curr Drug Metab. 2013;14(1):4–20. doi: 10.2174/138920013804545160. [DOI] [PubMed] [Google Scholar]
- 47.Fischer SM, Flaten GE, Hagesæther E, Fricker G, Brandl M. In-vitro permeability of poorly water soluble drugs in the phospholipid vesicle-based permeation assay: the influence of nonionic surfactants. J Pharm Pharmacol. 2011;63(8):1022–1030. doi: 10.1111/j.2042-7158.2011.01301.x. [DOI] [PubMed] [Google Scholar]
- 48.Boyd BJ. Characterisation of drug release from cubosomes using the pressure ultrafiltration method. Int J Pharm. 2003;260(2):239–247. doi: 10.1016/S0378-5173(03)00262-X. [DOI] [PubMed] [Google Scholar]