

Abstract
Macrophages, found in circulating blood as well as integrated into several tissues and organs throughout the body, represent an important first line of defense against disease and a necessary component of healthy tissue homeostasis. Additionally, macrophages that arise from the differentiation of monocytes recruited from the blood to inflamed tissues play a central role in regulating local inflammation. Studies of macrophage activation in the last decade or so have revealed that these cells adopt a staggering range of phenotypes that are finely tuned responses to a variety of different stimuli, and that the resulting subsets of activated macrophages play critical roles in both progression and resolution of disease. This review summarizes the current understanding of the contributions of differentially polarized macrophages to various infectious and inflammatory diseases and the ongoing effort to develop novel therapies that target this key aspect of macrophage biology.
Keywords: cancer, infectious disease, M1 M2, macrophage polarization, NASH/Obesity
INTRODUCTION
Macrophages (Mϕ) represent a ubiquitous yet complex and nuanced population of immune cells that play essential roles in both disease and homeostasis throughout the body (Hume, 2008). In addition to monocytes and Mϕ circulating throughout the bloodstream, specialized tissue-resident Mϕ can be found in most major organs, including Kupffer cells in the liver, Langer-hans cells in the skin, microglia in the brain, splenic red pulp Mϕ, lung alveolar Mϕ, adipose tissue Mϕ, and bone osteoclasts, to name a few (Davies et al., 2013; Gautier et al., 2012; Ji et al., 2013; You et al., 2013). While some identify these populations as the endpoint of bone marrow monocyte maturation, several lines of evidence indicate that tissue resident Mϕ originate during embryogenesis in association with their specific tissue independently from blood monocytes and monocytes/Mϕ recruited to sites of inflammation (Davies et al., 2013; Gomez and Geissmann, 2013; Schulz et al., 2012). Regardless of their location, Mϕ are responsible for the maintenance of healthy tissues through phagocytic clearance of apoptotic cells and foreign materials and through tissue repair and remodeling during wound healing (Duffield, 2005; Ghavami et al., 2014; Majai et al., 2014; Mantovani et al., 2013). Mϕ are also major regulators of the inflammatory response to disease and infection, acting as a bridge between innate and adaptive immunity by monitoring the microenvironment through an array of surface receptors and secreting appropriate cytokines and chemokines (Heydtmann, 2009; Schwabe et al., 2006).
Depending on the stimuli they encounter, tissue resident and circulatory Mϕ populations can be directed to distinct phenotypic programs in a process known as Mϕ polarization (Fig. 1, Table 1). The diverse properties of different Mϕ subsets can have drastic effects on health and disease within the tissues where they reside; while the induction of a particular subset can be protective during homeostasis or disease, this process can be altered or subverted to enhance pathogenesis and disease progression (by, for example, inappropriately dampening the immune response or exacerbating harmful inflammation) (Murray and Wynn, 2011). Therefore, this review aims to summarize recent findings regarding the identity, properties, and roles of polarized Mϕ in various disease models and the development of therapeutic strategies that target both the process of Mϕ polarization and individual Mϕ subsets.
Fig. 1.
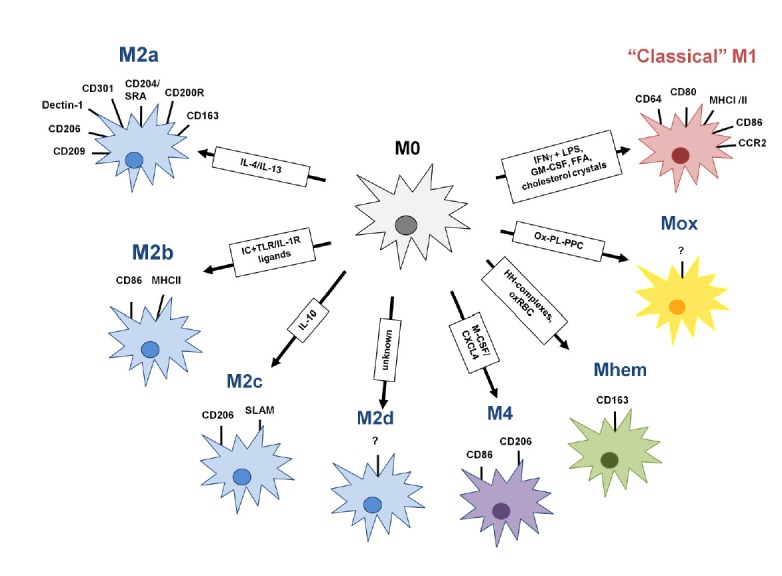
Schematic representation of Mϕ polarization. While M1/classically activated macrophages are typically induced by IFNγ and microbial products like LPS, different stimuli lead to the development of an array of finely tuned alternately activated states. IC, Immune complex; HH, Hapto-hemoglobin; OxRBC, oxidized red blood cells; Ox-PL-PPC, ox-PL 1-palmitoyl-2arachidonoyl-sn-glycero-3-phosphorycholine; FFA, free fatty acid.
Table 1.
SR, scavenger receptor; MR, mannose receptor; HO-1, heme oxygenase-1; VEGF, vascular endothelial growth factor; SD-1, sulfiredocin-1; TR-reductase, thioredoxin-reductase (Kadl et al., 2010; Leitinger and Schulman, 2013; Murray and Wynn, 2011)
| Polarization state | Gene expression | Cytokines | Chemokines |
|---|---|---|---|
| M1 | CD80, CD86, MHC I/II, IL-1R I, TLR2, TLR4, iNOS | TNFα, IL-1, IL-6, IL-12, IL-15, IL-23, ROS, iNOS, type I IFN | CXCL1-3, CXCL5, CXCL8-10, CXCL16, CCL2-5, CCL8, CCL11, CCL15, CCL20, CX3CL1 |
| M2a | CD163, MHC II, SR, CD206, MR, IL-1R II, YM-1, Fizz1, Arg-1 | IL-10, TGFβ, IL-12, IL-1Ra | CCL1, CCL2, CCL24, CCL22, CCL17, CCL18 |
| M2b | CD86, MHC II | IL-10, IL-1, TNFα, IL-6 | CCL1, CCL20, CXCL1, CXCL2, CXCL3 |
| M2c | CD163, TLR1, MR, Arg-1, YM-1, TLR8 | IL-10, TGFβ | CCL16, CCL18 |
| M2d | VEGF | IL-10, VEGF | CCL5, CXCL10, CXCL16 |
| M4 | ? | TNFα | CCL18, CCL20 |
| Mhem | HO-1 | IL-10 | ? |
| MOx | HO-1, SD-1, TR-reductase | ? | ? |
PHENOTYPIC POLARIZATION OF MΦ
The most well-described and commonly reported paradigm of Mϕ polarization is the M1/M2 polarization axis (Mantovani et al., 2004; Martinez et al., 2009; Sica et al., 2013). Originally named to reflect relationships to Th1/Th2 polarization of immune responses, M1 and M2 Mϕ are also referred to as classically or alternatively activated Mϕ, respectively (Gordon, 2003; Mills et al., 2000).
Classical activation is stimulated by microbial products and proinflammatory cytokines (IFNγ and/or LPS or TNF), and the resulting M1 Mϕ are characterized by high antigen presentation, high production of IL-12 and IL-23, and high production of nitric oxide (NO) and reactive oxygen intermediates (ROI) (Verreck et al., 2004). M1 Mϕ have been shown to produce several other inflammatory cytokines like TNFα; IL-1, -6, and -12; Type I IFN; CXCL1-3, 5 and 8–10; CCL2-5 and 11; CXCL16; and CX3CL1 (Mantovani et al., 2004; Sica and Mantovani, 2012).
By contrast, alternative/M2 activation is mediated by IL-4, IL-10, and IL-13, which were initially thought to produce “deactivated Mϕ” (Martinez et al., 2009). M2 Mϕ are marked by the upregulation of several surface molecules including Dectin-1, DC-SIGN, mannose receptor (MRC1/CD206), scavenger receptor A (CD204), scavenger receptor B-1, CD163, CCR2, CXCR1, and CXCR2 (Gordon, 2003; Mantovani et al., 2004; Martinez et al., 2009). M2 Mϕ exhibit altered cytokine and chemokine production, and typically produce high levels of IL-10 and low levels of IL-12 (Mosser, 2003). CCL1, CCL2, CCL17, CCL18, CCL22, CCL24, and IL-1Ra are also made by alternatively activated Mϕ (Mantovani et al., 2004). Genetic studies of M2 Mϕ in mouse models have identified additional signatures of alternative activation, including arginase 1 (Arg1), YM1 (a member of the chitinase family) and FIZZ1 (found in inflammatory zone 1, RETNLA) (Raes et al., 2002; 2005). Generally, the M2 polarization state is characterized by little to no secretion of proinflammatory cytokines, increased secretion of anti-inflammatory cytokines, enhanced scavenging of cellular debris, promotion of tissue remodeling and repair, and, in some cases, increased capacity to fight parasitic infections (Alfano et al., 2013). Additionally, the concept of resolution of inflammation has evolved and is no longer perceived as a passive process that simply occurs when the insult disappears, but rather as a highly orchestrated response coordinated by a complex regulatory network of cells and anti-M1 mediators called pro-resolving mediators (Rius et al., 2012).
M2 Mϕ can be further divided into subtypes according to their inductive stimuli and secreted chemokines (Martinez et al., 2008). M2a Mϕ are stimulated by IL-4 and IL-13 and produce CCL24, CCL22, CCL17, and CCL18, which are recognized by CCR3, CCR4, and CCR8 and promote recruitment of eosinophils, basophils, and Th2 cells. M2b Mϕ result from activation with immune complexes and TLR agonists (like LPS) and produce CCL1, which recruits Tregs. IL-10 drives Mϕ polarization to M2c cells, which produce CCL16 and 18, recruiting eosinophils and naïve T cells, respectively. M2d Mϕ accumulate in the tumor microenvironment and present an IL-10hiVEGFhi M2 profile, but also exhibit some M1 characteristics such as expression of INFγ-inducible chemokines CCL5, CXCL10, and CXCL16 (Duluc et al., 2009).
A full understanding of the M1/M2 paradigm of Mϕ polarization, however, contains some caveats. First, M1 and M2 Mϕ as defined in the foundational literature most likely do not exist as distinct categories, but rather should be considered as extremes at either end of a continuum of overlapping functional states (Mosser and Edwards, 2008). Indeed, Mϕ with combinations of M1 and M2 markers can be found in atherosclerotic plaques and some murine tumors (Kadl et al., 2010; Umemura et al., 2008). Second, unlike the irreversible phenotypic changes seen in lymphocytes after exposure to polarizing cytokines, Mϕ polarization is both transient and plastic (Biswas and Mantovani, 2010; Biswas et al., 2008; Sica et al., 2013; Stout and Suttles, 2004). For example, M2 Mϕ can be reprogrammed to express M1 genes following exposure to TLR ligands or IFNγ (Mylonas et al., 2009; Stout et al., 2005). Finally, while there is partial overlap of M1- and M2-identifying markers in murine and human studies, there are still markers in each system that fail to translate to the other. The chitinase-like proteins YM1 and YM2, along with FIZZ1, are markers of murine M2 polarization which lack human orthologs, while CCL14, CCL18, and CCL23 are human-restricted M2 markers with no murine orthologs (Chang et al., 2001; Martinez et al., 2009; Raes et al., 2002). Finally, there are other specially activated Mϕ (M4, Mhem, and Mox) that have been described in atherosclerosis and may lie on a separate activation axis from M1/M2 Mϕ (Fig. 1). These atherosclerotic Mϕ subsets have been discussed in recent reviews (Fenyo and Gafencu, 2013; Leitinger and Schulman, 2013), but will not be examined in detail here.
SIGNALING PATHWAYS INVOLVED IN MΦ POLARIZATION
The network of molecular mediators that regulate M1/M2 polarization in response to various stimuli is incompletely understood, but several signaling pathways have been implicated in this process (Fig. 2). One of the major pathways identified is the JAK/STAT pathway, which mediates responses to a collection of different cyotkines and growth factors and regulates processes from hematopoiesis and immune development to lactation and adipogenesis (Rawlings et al., 2004). Binding of IFNγ to its cell surface receptor leads to activation of receptor-associated JAKs, which in turn cause STAT1 to dimerize and translocate to the nucleus where it initiates transcription of genes that promote M1-associated functions like enhanced microbicidal activity and proinflammatory cytokine production (Hu et al., 2007; Rauch et al., 2013). Mϕ-specific deletion of SOCS3, an inhibitor of cytokine and JAK/STAT signaling, was found to increase levels of the M1 genes IL-1β, IL-6, IL-12, IL-23, and iNOS (Qin et al., 2012a) and increase phosphorylation of STAT1 and STAT3 (Qin et al., 2012b).
Fig. 2.
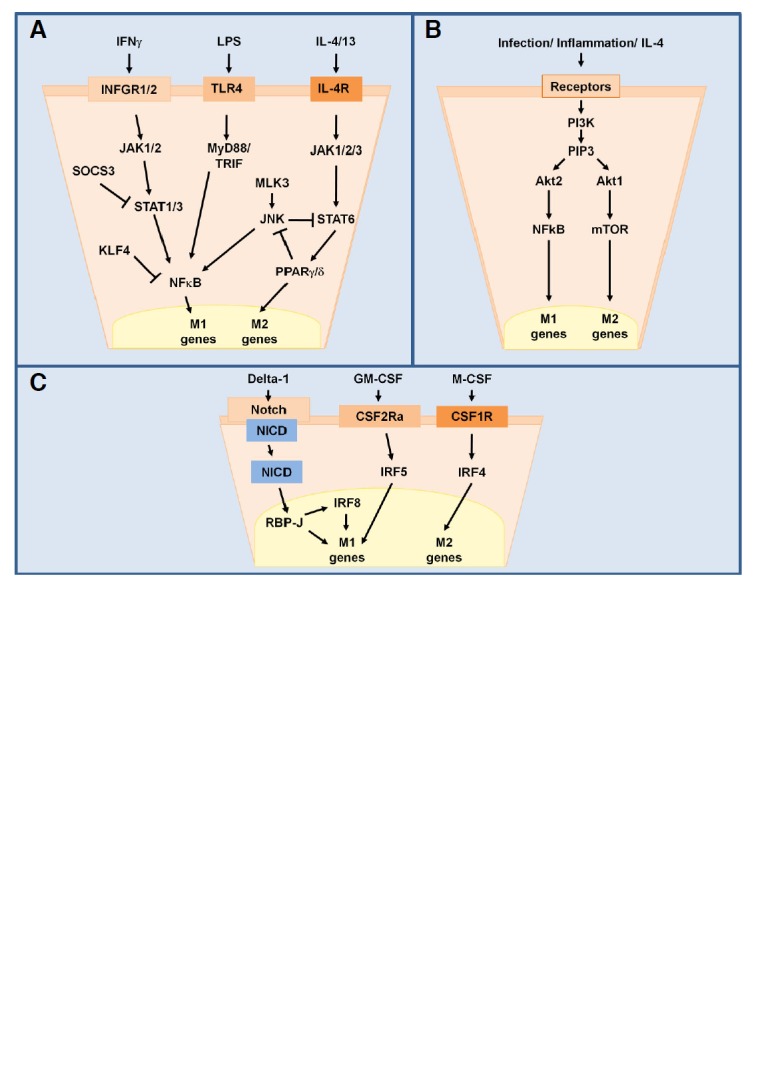
Major signaling pathways involved in M1/M2 Mϕ polarization. STAT1/3 and STAT6 regulate transcription of M1 and M2 genes following recognition of IFNγ or IL-4/13 by their surface receptors and activation of JAKs (A). Differential activation of Akt1 or Akt2 via PI3K and PIP3 leads to either M2 or M1 polarization, respecively (B). Upon activation, the intracellular domain of Notch (NICD) is cleaved and activates IRF8 to promote M1 polarization. GM-CSF triggers M1 polarization via IRF5 while M-CSF induces M2 polarization via IRF4 (C).
In contrast, STAT6 is activated by IL-4 and IL-13 to induce M2 polarization (Daley et al., 2009; Moreno et al., 2003; Stolfi et al., 2011). C-Jun N-terminal kinase (JNK), a mitogen-activated protein kinase (MAPK) involved in cell proliferation, transformation, differentiation, and apoptosis is likely involved in this pathway (Zhou et al., 2013). Upon activation, JNK can phosphorylate serine 707 on STAT6, thereby deactivating it (Shirakawa et al., 2011). A study of Mϕ polarization in obesity showed that mice lacking the JNK activator MLK3 were also deficient in M1 Mϕ polarization (Gadang et al., 2013). The transcription factors PPARγ and PPARδ are activated by STAT6 and necessary for M2 polarization, and PPARδ−/− Mϕ exhibit enhanced activation of JNK following treatment with adipocyte-conditioned medium, which contains the M2 cytokines IL-4 and IL-13 (Kang et al., 2008; Odegaard et al., 2007). The zinc-finger transcriptional regulator Krüppel-like factor 4 (KLF4) is involved in this pathway as well and cooperates with STAT6 to skew polarization towards M2 by sequestering coactivators of NF-κB (Liao et al., 2011).
Furthermore, the phosphoinositol-3-kinase (PI3K) signaling pathway, which activates multiple kinase cascades through the production of the second messenger PIP3, regulates Mϕ survival and gene expression via activation of the Akt family of serine/threonine protein kinases (Liu et al., 2001; Luyendyk et al., 2008). Knockout studies have demonstrated that M1 polarization depends on the activation of Akt2 while M2 polarization requires Akt1 (Arranz et al., 2012). In addition, the PI3K/Akt signaling pathway controls the activation of mTOR, which promotes M2 polarization (Byles et al., 2013; Mercalli et al., 2013; Weichhart and Säemann, 2008).
Interferon-regulatory factor (IRF) proteins are also regulators of Mϕ polarization. IRF5 is associated with M1 polarization and promotes the transcription of genes encoding IL-12 while repressing the gene that encodes IL-10 (Krausgruber et al., 2011). Notch signaling through the nuclear transducer RBP-J controls expression of IRF8, which induces M1 gene expression (Xu et al., 2012). IRF4 is highly expressed in adipose tissue Mϕ (ATM) and its deletion leads to increased production of IL-1β and TNFα and expression of M1 markers, indicating that IRF4 activation contributes to M2 polarization (Eguchi et al., 2013). The IRFs also underlie the ability of GM-CSF and M-CSF to induce polarization: GM-CSF leads to downstream activation of IRF5 (M1) while M-CSF leads to IRF4 (M2) activation (Lawrence and Natoli, 2011).
BACTERIAL INFECTIONS
Given the ability of Mϕ to acquire enhanced microbicidal abilities following stimulation with microbial products and the preeminent roles of Mϕ in both innate and adaptive immune responses, one might predict that pathogens would evolve strategies to redirect and alter Mϕ activation in their favor. Several transcriptome analysis studies have established that innate immune cells, particularly Mϕ, engage in a common response to pathogen challenge that involves a shared pattern of gene expression (Jenner and Young, 2005; Nau et al., 2002). A multi-study review of transcriptional responses of mononuclear phagocytes to bacteria and bacterial components focusing specifically on genes involved in Mϕ polarization identified a common response program that mainly involved the upregulation of M1-associated genes, including the cytokines TNF, IL-6, IL-12, IL-1β, the cytokine receptors IL-7R and IL-15RA, the chemokines CCL2, CCL5, and CXCL8, and the chemokine receptor CCR7 (Benoit et al., 2008). This M1 activation program is typically associated with protection against disease, and M1 polarization has been shown to aid host control of several bacteria, including Listeria monocytogenes, Salmonella typhimurium, Mycobacterium tuberculosis, Mycobacterium ulcerans, and Chlamydia infections (Benoit et al., 2008; Chacón-Salinas et al., 2005; Jouanguy et al., 1999; Kiszewski et al., 2006; Rottenberg et al., 2002; Shaughnessy and Swanson, 2007).
Consequently, several pathogenic bacteria, especially intracellular species, have developed mechanisms to interfere with Mϕ polarization in order to enhance their own survival. Some species accomplish this by blunting M1 polarization to reduce inflammation and microbicidal functions of Mϕ. The intracellular form of the enteropathogen Shigella flexneri produces an altered, hypoacetylated form of LPS that evades recognition by TLR4 and elicits decreased production of proinflammatory cytokines from murine bone marrow derived Mϕ (BMDM) (Paciello et al., 2013). During pulmonary infection in mice, Staphylococcus aureus induces Akt1 signaling to enhance SOCS1 activity and inhibit NF-κB activity, shifting Mϕ from an antimicrobial M1 phenotype to a functionally inert one (Xu et al., 2013). M. tuberculosis secretes the virulence factors lipoarabinomannan and early secretory antigenic target-6 (ESAT-6), which inhibit M1 activation by inhibiting phagosome maturation and NF-κB activation, respectively (Lugo-Villarino et al., 2011). M. tuberculosis also subverts the inflammatory response by stimulating Wnt6 signaling in infected Mϕ in granulomatous lesions in the lung, driving M2-like polarization (Schaale et al., 2013). S. aureus biofilms are resistant to Mϕ invasion, but those Mϕ that do successfully penetrate catheter-associated biofilms in vitro display decreased expression of IL-1β, TNFα, and iNOS expression but robust Arg1 expression, signifying an M2 profile (Thurlow et al., 2011). S. typhimurium has been shown to preferentially associate with M2 Mϕ, and PPARδ expression is upregulated in Salmonella-infected Mϕ while PPARδ deficiency severely inhibits bacterial replication and persistence (Eisele et al., 2013). Interestingly, the dependency of S. typhimurium on PPARδ expression was shown to be due to its metabolic effects rather than its ability to reduce production of antimicrobial mediators by promoting M2 polarization, and it remains to be determined whether S. typhimurium directly augments PPARδ activity to promote persistence.
VIRAL INFECTIONS
Similar to evasion strategies employed by bacterial pathogens, many viruses take advantage of the Mϕ polarization system to enhance their own growth and virulence. However, unlike bacterial pathogens, which generally tend to thrive within and encourage production of M2-polarized Mϕ, viral pathogens more commonly activate M1 polarization. This inflammatory phenotype is often correlated with disease severity. Hepatitis C virus preferentially infects hepatocytes and establishes a chronic inflammatory infection, often leading to fibrotic cirrhosis and hepatocellular carcinoma (HCC) (Lavanchy, 2011). It has been demonstrated that the viral protein NS3 enhances IL-12 and TNFα production by THP-1 Mϕ, implicating M1 polarization in sustaining inflammation (Hajizadeh et al., 2013). Furthermore, activation of Mϕ with TLR agonists triggers the secretion of TNFα, which promotes HCV entry into polarized hepatoma cells by relocalizing the tight junction protein and HCV entry factor occludin (Fletcher et al., 2013). Of the three common clades of avian H5N1 influenza virus circulating in poultry in China (2.3.2, 2.3.4, and 7), clade 2.3.4 is the most successful at infecting, replicating within, and inducing cytopathic effects in human monocyte-derived Mϕ (MDM) (Sun et al., 2013). H5N1 clade 2.3.4 also stimulated the highest expression of IL-1β, IL-6, IL-8, TNFα, IFNγ, and MCP-1 in MDMs (Sun et al., 2013). M2 Mϕ polarization by S. aureus, which is commonly present among the airway mucosal microbiota, inhibits influenza-mediated lung injury, implying that M1 Mϕ exacerbate flu infection (Wang et al., 2013).
Nonetheless, some viruses do benefit by skewing Mϕ polarization towards an M2 phenotype. During infection by severe acute respiratory syndrome coronavirus (SARS-CoV), lung damage resulting from both intrinsic viral infection and dysregulation of the host immune response rapidly progresses to diffuse alveolar damage, resulting in acute respiratory distress syndrome and pulmonary fibrosis (Franks et al., 2003; Peiris et al., 2003). A recent study revealed that SARS-CoV-infected mice lacking hematopoeitic STAT1 expression have greater weight loss and lung pathology associated with upregulation of the M2 indicators YM1, FIZZ1, IL-4, and IL-13 (Page et al., 2012). Absence of lung disease and prefibrotic lesions in infected STAT1/STAT6−/− double-knockout mice also supported the notion that M2 Mϕ contribute to SARS-CoV pathogenesis. Human cytomegalovirus (HCMV) has a more complex relationship with Mϕ polarization. The HCMV gene UL111A encodes a homolog of human IL-10 that is capable of polarizing monocytes towards an anti-inflammatory M2c phenotype including high expression of the scavenger receptor CD163, suppression of MHC expression, and exppression of heme oxygenase 1 (which suppresses TNFα and IL-1β) (Avdic et al., 2013). Additionally, HCMV optimally infects M2-but not M1-polarized Mϕ and late-phase HCMV infection is dependent on the M2-promoting activation of mTOR (Poglitsch et al., 2012). Despite this, HCMV-activated Mϕ have been shown to adopt an M1 transcriptome profile (Chan et al., 2008). HIV-1 similarly seems to benefit from M2 polarization: HIV-1 displays impaired viral DNA synthesis, delayed proviral integration, and reduced proviral transcription in M1 Mϕ, while the M2a surface receptor DC-SIGN facilitates HIV-1 entry, DNA synthesis, and transmission from infected Mϕ to CD4+ T cells (Cassetta et al., 2013; Cassol et al., 2010; 2013). Notably, clathrin-mediated endocytosis of HIV-1 is increased in M1 and decreased in M2 Mϕ, but this method of endocytosis leads to increased viral degradation and is unlikely to result in productive infection (Gobeil et al., 2012). Yet, like HCMV, HIV-1 infection of MDMs drives them toward M1 polarization, and the viral protein Nef is preferentially taken up by M2 Mϕ and stimulates an M2-to-M1 transition (Cassol et al., 2010; Chihara et al., 2012; Lugo-Villarino et al., 2011). These contradictions may be explained by a viral survival strategy that takes advantage of both M1 and M2 Mϕ as means to different ends: M2 Mϕ as a reservoir of replication and M1 Mϕ to recruit fresh immune cells to spread the infection. This can also be inferred from the ability of proinflammatory cytokines and chemokines from HCMV-infected Mϕ to enhance virus replication and dissemination (Alfano et al., 2013; Smith and Bentz, 2004a; 2004b).
DIABETES, OBESITY, AND NON-ALCOHOLIC STEATOHEPATITIS
Type 1 diabetes is an autoimmune disease that results in high blood sugar following the destruction of insulin-producing pancreatic beta cells via activation of innate immunity and expansion of auto-reactive T cells and autoantibody-producing B cells. Monocytes/Mϕ from patients with Type 1 diabetes present a proinflammatory profile (high levels of TNFα, IL-6 and IL-1β) when compared to normal subjects (Bradshaw et al., 2009; Devaraj et al., 2006; Shanmugam et al., 2004). Moreover, elevated levels of glucose and islet amyloid polypeptide (IAPP) deposition lead to the activation of TLRs and inflammasomes, resulting in beta cell death and decreased insulin secretion (Henao-Mejia et al., 2013). Recently, it has been suggested that M1 Mϕ may contribute to diabetes-related complications such as cardiovascular diseases by altering the immune system of type 1 diabetics (Burke and Kolodgie, 2004). Furthermore, the sustained increase of growth hormone in murine models of type 1 diabetes leads to a reduction of diabetes symptoms by attenuating the apoptosis and increasing the expansion of beta cells (Villares et al., 2013). Growth hormone also triggers M2 polarization of Mϕ via modulation of the cytokine milieu, stimulating the activity of suppressor T cells and limiting Th17 cell activation (Villares et al., 2013).
Obesity is a major health problem in western countries and a risk factor for insulin resistance, type 2 diabetes, hepatic steatosis, and artherosclerosis. Obesity is closely associated with chronic inflammation in adipose tissues, suggesting that the chronic excess of nutrients triggers an immune response in adipose tissues (Goh et al., 2011; Hotamisligil, 2006; Kanneganti and Dixit, 2012). White adipose tissues store energy in the form of fat and regulate systemic metabolism through the release of adipokines by adipocytes that control insulin sensitivity in the liver and skeletal muscle (Sun et al., 2011; Tateya et al., 2013). In lean subjects and mice, adipose tissue Mϕ (ATM) present an M2 phenotype and are critical to maintaining insulin sensitivity in adipocytes through IL-10 production (Liao et al., 2011; Lumeng et al., 2007a; 2007b). In metabolic homeostasis, M2 ATMs are maintained by IL-4 and IL-13 secreted by adipocytes in a PPARγ/δ/β-and KLF4-dependent manner (Wynn et al., 2013; Zhou et al., 2013). In obese subjects and mice, adipocytes release proinflammatory mediators (i.e. CCL2/MCP-1, TNFα, CCL5, CCL8 and free fatty acid), promoting the infiltration of Ly6Chi inflammatory monocytes which differentiate into M1-polarized ATMs that express high levels of TNFα, iNOS, IL-6 and IL-1β (Cinti et al., 2005; Lumeng et al., 2007a; Olefsky and Glass, 2010; Tateya et al., 2013; Weisberg et al., 2003, 2006). Therefore, the severity of obesity-related metabolic dysfunctions correlates with M1 ATM infiltration whereas chronic inflammation in adipose tissue inhibits the production of adiponectin, thus contributing to the development of insulin resistance in surrounding adipocytes (Lumeng et al., 2007a; Weisberg et al., 2003; 2006).
Recently NAFLD (Non-alcoholic fatty liver disease) has emerged as an obesity-related health problem characterized by steatosis (accumulation of lipids in hepatocytes). Hepatic steatosis can evolve to non-alcoholic steatohepatitis (NASH) when accompanied by liver injury (ballooning hepatocytes) and hepatic inflammation, which may be associated with fibrosis and eventually culminates in cirrhosis and HCC. The development of the complex pathology of NASH involves a variety of liver cells including hepatocytes, hepatic Mϕ, and stellate cells. Inflammatory mediators, especially those derived from adipose tissues, the gut, and the liver, have recently been reported to play a major role in initiating and controlling the progression of NASH by regulating lipid metabolism (Day and James, 1998; Racanelli and Rehermann, 2006; Tilg and Moschen, 2010). In particular, the activation of innate immune cells such as Kupffer cells and infiltrating blood-derived monocytes is a major event of NASH development. In homeostatic conditions, Kupffer cells perform immune surveillance by removing pathogens and toxins from the circulation and maintain liver tolerance through IL-10 secretion (Thomson and Knolle, 2010). Kupffer cells communicate with a variety of hepatic immune cells and interact directly with hepatocytes passing through the space of Disse (Racanelli and Rehermann, 2006). In early mouse models of diet-induced steatohepatitis, Kupffer cells are the first innate cells responding to injured hepatocytes and differentiate toward M1 Mϕ, promoting the recruitment of blood-derived CD11bint Ly6Chi monocytes through secretion of TNFα and chemokines (MCP-1 and IP-10) (Tosello-Trampont et al., 2012). The recruitment of these inflammatory M1-polarized Ly6C+ blood-derived monocytes is dependent of CCR2 and MCP1 (Karlmark et al., 2009; Klein et al., 2007; Miura et al., 2012; Obstfeld et al., 2010). The hallmarks of NASH (i.e. steatosis, low-grade inflammation, and hepatic recruitment of M1-polarized Mϕ) are reduced/delayed following specific depletion of Kupffer cells or by silencing of TNFα in myeloid cells (Tosello-Trampont et al., 2012). Moreover, M1-polarized Kupffer cells also produce inflammatory mediators such as IL-1β and ROS, which induce hepatic steatosis and fibrosis (Miura et al., 2012; Schwabe and Brenner, 2006). NF-κB and JNK activation in Kupffer cells may contribute to the development of hepatic inflammation by promoting M1-like Mϕ polarization (Zhou et al., 2013).
Liver Mϕ are also implicated in the severity of NASH via the expression of Toll-like receptors (TLR2, TLR4, TLR9, MyD88) and scavenger receptors (scavenger receptor A and CD36) (Bieghs et al., 2010; Miura et al., 2010; 2012; 2013). TLRs and scavenger receptors trigger proinflammatory responses following recognition of hepatic free fatty acids, damage-associated molecular pattern (DAMPs) expressed by steatotic hepatocytes, and/or bacterial products derived from the gut (Farrell et al., 2012; Roh and Seki, 2013). NASH patients show increased intestinal permeability, resulting in greater hepatic abundance of bacterial products and other TLR ligands derived from the gut via portal vein circulation (Zhu et al., 2013). The imbalance of gut flora may influence liver disease by activating TLRs expressed on liver cells and leading to the activation of NLPR3 (Csak et al., 2011; Farrell et al., 2012; Henao-Mejia et al., 2013; Roh and Seki, 2013). In models of diet-induced NASH and obesity, inflammasome-deficient mice develop more severe NASH which is fully transferable to WT mice upon prolonged cohousing, suggesting that commensal bacteria in the GI tract play an important role in NASH disease progression (Csak et al., 2011; Henao-Mejia et al., 2012; 2013; Weisberg et al., 2003).
CANCER
Mϕ are a highly influential cell type in most varieties of cancer and are recruited to all solid tumors (Cassetta et al., 2011). The contributions of different subsets of polarized Mϕ to the tumor microenvironment and cancer progression are therefore a subject of great interest. M1 Mϕ are generally considered to be beneficial to the host, and peritumoral Mϕ that express M1 cytokines like IFNγ, IL-1β, and IL-6 have been shown to have antitumoral effects and are associated with improved prognoses (Dumont et al., 2008; Klimp et al., 2002; Niino et al., 2010; Öberg et al., 2002; Zhou et al., 2010). M1 Mϕ may have the opposite effect in virally induced cancers, however: administration of IFNγ or TNFα to patients infected with Kaposi sarcoma virus enhances disease progression (Monini et al., 1999). Proinflammatory Mϕ are also harmful in intraocular tumors, where TNFα- and iNOS-dependent antitumor responses lead to necrosis of bystander cells and destruction of the eye (Coursey et al., 2012).
M2-polarized tumor-associated Mϕ (TAM), on the other hand, have been repeatedly and consistently associated with unfavorable effects like tumor growth, angiogenesis, and metastasis in malignant cancers (Alfano et al., 2013). The M2 cytokines IL-4, IL-13, and IL-10 are present within the tumor microenvironment and TAMs from various cancer models have been shown to express an M2 activation profile that includes enhanced expression of CD163, MRC-1, c-type lectins, IL-10, and Arg-1 and decreased production of IL-12 (Beck et al., 2009; Biswas et al., 2006; Schmieder et al., 2012; Sica et al., 2002). The small secretory lectin Reg3β is an important inhibitor of inflammation in pancreatic and intestinal tissues, and deficiency of Reg3β (an activator of the STAT3 pathway) drastically impairs pancreatic tumor growth by skewing Mϕ polarization away from M2 and towards M1 (Gironella et al., 2013). M2 TAMs have also been shown to increase fibroblastic morphology, vimentin and snail expression, metalloproteinase activity, and proliferation and migration of pancreatic cancer cells, implicating them in the development of eptihelial-mesenchymal transition and metastasis (Liu et al., 2013). In HCC, high expression of the heparinsulfate proteoglycan glypican-3 (GPC3) on the surface of cancer cells is associated with increased Mϕ infiltration in human patients, and human/mouse xenograft transplantation with a GPC3-overexpressing cell line leads to infiltration by Mϕ expressing M2-specific markers (Takai et al., 2009a; 2009b). M2 TAMs worsen HCC both by promoting tumor growth and angiogenesis and by encouraging liver fibrosis through IL-13 and TGFβ secretion (Sica et al., 2013).
THERAPIES TARGETING MΦ POLARIZATION
Given that Mϕ play important roles in maintaining tissue homeostasis and fighting disease, polarized Mϕ subsets that specifically contribute to the pathogenesis or amelioration of various diseases present themselves as attractive targets for therapeutic intervention. Different therapeutic strategies include either targeting the polarized Mϕ themselves or manipulating the signaling pathways involved in the process of Mϕ polarization to a desirable outcome.
Bacterial biofilms that form on body surfaces or on surgical implants lead to chronic and recurrent infections, and are difficult to treat with antibiotics (Donlan and Costerton, 2002; Otto, 2008). Early, local administration of M1 Mϕ or the C5a receptor agonist EP67, which stimulates M1 polarization, significantly attenuated biofilm formation in a mouse model (Hanke et al., 2013). Furthermore, treatment of established biofilms significantly reduced bacterial burden compared to antibiotic treatment, suggesting the potential of a therapeutic alternative (Hanke et al., 2013). Microbes themselves may also prove to be useful sources of therapeutics that modulate Mϕ polarization. In vitro application of extracellular polysaccharide secreted by an oligotrophic bacteria found in Lop Nur Desert, Bacillus sp. LBP32, was found to limit LPS-induced inflammation in the Mϕ cell line RAW 264.7 by inhibiting NF-κB and JNK activation, and may prove useful in diseases characterized by excessive M1 polarization (Diao et al., 2013). Similarly, the small-molecule compound bis-N-norgliovictin isolated from the marine-derived endophytic fungus S3-1-c inhibits LPS-induced M1 polarization of RAW 264.7 cells and murine peritoneal Mϕ, and improves survival in mouse models of sepsis (Song et al., 2013). As a proof of concept for treating inflammatory gastrointestinal diseases, a lab strain of E. coli was created that secretes a Herpes virus homolog of IL-10 via a Sec-dependent transporter construct. Viral IL-10 delivered in this manner was shown to activate STAT3 and suppress TNFα production in the J774.1 murine Mϕ cell line (Förster et al., 2013).
IKKβ, a downstream mediator of insulin resistance and activator of the NF-κB pathway (and therefore of M1 polarization), is inhibited by anti-inflammatory salicylates like aspirin, which attenuates hyperglycemia and hyperinsulinemia in obese rodents (Yin et al., 1998; Yuan et al., 2001). Several small trials in patients with type 2 diabetes have demonstrated that treatment with salicylates results in a marked reduction of diabetic metabolic parameters and improved glycemic control (Tateya et al., 2013).
Apolipoprotein A-I mimetics are a class of therapeutic molecules that attempt to modulate HDL to treat atherosclerosis and are the subject of extensive clinical and mechanistic study, as reviewed in Leman et al. (2013). Interestingly, the mimetic D4F also has potential for cancer therapy: D4F inhibits the M2-associated scavenger receptor CD204/SRA on TAMs, preventing metastatic spread (Neyen et al., 2013).
Anticancer therapies also seek to convert protumoral M2 Mϕ into M1 Mϕ. M2 Mϕ generated by IL-6 and prostaglandin E2 secreted by cervical cancer cells can be repolarized to M1 by coculture with Th1 cells, and this interaction could possibly be reproduced by activation with CD40L and IFNγ (Heusinkveld et al., 2011). Moreover, IFNγ was shown to successfully switch M2 TAMs purified from human ovarian tumors to an M1 phenotype, and the addition of IFNγ skewed de novo tumor-induced M2 differentiation of monocytes to favor M1 polarization (Duluc et al., 2009). Other potentially therapeutic molecules found to repolarize TAMs from an M2 to an M1 phenotype include zoledronic acid, CpG oligonucleotide, and histidine-rich glycoprotein (Coscia et al., 2010; Huang et al., 2012; Rolny et al., 2011).
CONCLUSION
The integral importance of Mϕ in the maintenance of nearly every tissue throughout the body and their position as the first line of defense against many diseases guarantees that they play critical roles in both disease progression and in resolution, and that altering the behavior of these cells can mean the difference between healthy recovery and severe illness. Mϕ polarization itself is an extremely nuanced and fine-tuned process and can produce nearly infinite variations of endpoint phenotypes, each of which has the potential to affect various diseases in different ways. While polarized Mϕ subsets and the polarization process itself are attractive and novel therapeutic targets in both infectious and inflammatory disease, better understanding of how polarization is controlled and how polarized Mϕ modulate specific diseases is necessary to fully harness the potential of these strategies.
Acknowledgments
This work was supported by NIH grants R01AI098126 and U19AI066328.
REFERENCES
- Alfano M., Graziano F., Genovese L., Poli G. Macrophage polarization at the crossroad between HIV-1 infection and cancer development. Arterioscler. Throm. Vas. Biol. 2013;33:1145–1152. doi: 10.1161/ATVBAHA.112.300171. [DOI] [PubMed] [Google Scholar]
- Arranz A., Doxaki C., Vergadi E., de la Torre Y.M., Vaporidi K., Lagoudaki E.D., Ieronymaki E., Androulidaki A., Venihaki M., Margioris A.N., et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA. 2012;109:9517–9522. doi: 10.1073/pnas.1119038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdic S., Cao J.Z., McSharry B.P., Clancy L.E., Brown R., Steain M., Gottlieb D., Abendroth A., Slobedman B. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J. Virol. 2013;87:10273–1082. doi: 10.1128/JVI.00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A.H., Espinosa I., Edris B., Li R., Montgomery K., Zhu S., Varma S., Marinelli R.J., van de Rijn M., West R.B. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin. Cancer Res. 2009;15:778–787. doi: 10.1158/1078-0432.CCR-08-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit M., Desnues B., Mege J.-L. Macrophage polarization in bacterial infections. J. Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- Bieghs V., Wouters K., van Gorp P.J., Gijbels M.J.J., de Winther M.P.J., Binder C.J., Lütjohann D., Febbraio M., Moore K.J., van Bilsen M., et al. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. 2010;138:2477–2486. doi: 10.1053/j.gastro.2010.02.051. 2486.e1-e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.K., Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat. Immunol. 2010;11:889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- Biswas S.K., Gangi L., Paul S., Schioppa T., Saccani A., Sironi M., Bottazzi B., Doni A., Vincenzo B., Pasqualini F., et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation) Blood. 2006;107:2112–22. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- Biswas S.K., Sica A., Lewis C.E. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J. Immunol. 2008;180:2011–2017. doi: 10.4049/jimmunol.180.4.2011. [DOI] [PubMed] [Google Scholar]
- Bradshaw E.M., Raddassi K., Elyaman W., Orban T., Gottlieb P. A., Kent S.C., Hafler D.A. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J. Immunol. 2009;183:4432–4439. doi: 10.4049/jimmunol.0900576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke A.P., Kolodgie F.D., Zieske A., Fowler D.R., Weber D.K., Varghese P.J., Farb A., Virmani R. Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler. Throm. Vas. Biol. 2004;24:1266–1271. doi: 10.1161/01.ATV.0000131783.74034.97. [DOI] [PubMed] [Google Scholar]
- Byles V., Covarrubias A.J., Ben-Sahra I., Lamming D.W., Sabatini D.M., Manning B.D., Horng T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013;4:2834. doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L., Cassol E., Poli G. Macrophage polarization in health and disease. ScientificWorldJournal. 2011;11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L., Kajaste-Rudnitski A., Coradin T., Saba E., Della Chiara G., Barbagallo M., Graziano F., Alfano M., Cassol E., Vicenzi E., et al. M1 polarization of human monocyte-derived macrophages restricts pre and postintegration steps of HIV-1 replication. AIDS. 2013;27:1847–1856. doi: 10.1097/QAD.0b013e328361d059. [DOI] [PubMed] [Google Scholar]
- Cassol E., Cassetta L., Alfano M., Poli G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010;87:599–608. doi: 10.1189/jlb.1009673. [DOI] [PubMed] [Google Scholar]
- Cassol E., Cassetta L., Rizzi C., Gabuzda D., Alfano M., Poli G. Dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin mediates HIV-1 infection of and transmission by M2a-polarized macrophages in vitro. AIDS. 2013;27:707–716. doi: 10.1097/QAD.0b013e32835cfc82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacón-Salinas R., Serafín-López J., Ramos-Payán R., Méndez-Aragón P., Hernández-Pando R., Van Soolingen D., Flores-Romo L., Estrada-Parra S., Estrada-García I. Differential pattern of cytokine expression by macrophages infected in vitro with different Mycobacterium tuberculosis genotypes. Clin. Exp. Immunol. 2005;140:443–449. doi: 10.1111/j.1365-2249.2005.02797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G., Bivins-Smith E.R., Smith M.S., Smith P.M., Yurochko A.D. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J. Immunol. 2008;181:698–711. doi: 10.4049/jimmunol.181.1.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang N.C., Hung S.I., Hwa K.Y., Kato I., Chen J.E., Liu C.H., Chang A.C. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. J. Biol. Chem. 2001;276:17497–17506. doi: 10.1074/jbc.M010417200. [DOI] [PubMed] [Google Scholar]
- Chihara T., Hashimoto M., Osman A., Hiyoshi-Yoshidomi Y., Suzu I., Chutiwitoonchai N., Hiyoshi M., Okada S., Suzu S. HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J. Immunol. 2012;188:3620–3627. doi: 10.4049/jimmunol.1101593. [DOI] [PubMed] [Google Scholar]
- Cinti S., Mitchell G., Barbatelli G., Murano I., Ceresi E., Faloia E., Wang S., Fortier M., Greenberg A.S., Obin M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- Coscia M., Quaglino E., Iezzi M., Curcio C., Pantaleoni F., Riganti C., Holen I., Mönkkönen H., Boccadoro M., Forni G., et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell. Mol. Med. 2010;14:2803–2815. doi: 10.1111/j.1582-4934.2009.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coursey T.G., Chen P.W., Niederkorn J.Y. Abrogating TNF-α expression prevents bystander destruction of normal itssues diruing iNOS-mediated elimination of intraocular tumors. Cancer Res. 2012;71:2445–2454. doi: 10.1158/0008-5472.CAN-10-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csak T., Ganz M., Pespisa J., Kodys K., Dolganiuc A., Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–144. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley J.M., Brancato S.K., Thomay A.A., Reichner J.S., Albina J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2009;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L.C., Jenkins S.J., Allen J.E., Taylor P.R. Tissue-resident macrophages. Nat. Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day C.P., James O.F. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Devaraj S., Glaser N., Griffen S., Wang-Polagruto J., Miguelino E., Jialal I. Increased monocytic activity and bio-markers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55:774–779. doi: 10.2337/diabetes.55.03.06.db05-1417. [DOI] [PubMed] [Google Scholar]
- Diao Y., Xin Y., Zhou Y., Li N., Pan X., Qi S., Qi Z., Xu Y., Luo L., Wan H., et al. Extracellular polysaccharide from Bacillus sp. strain LBP32 prevents LPS-induced inflammation in RAW 264.7 macrophages by inhibiting NF-κB and MAPKs activation and ROS production. Int. Immunopharmacol. 2013;18:12–19. doi: 10.1016/j.intimp.2013.10.021. [DOI] [PubMed] [Google Scholar]
- Donlan R., Costerton J. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield J. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duluc D., Corvaisier M., Blanchard S., Catala L., Descamps P., Gamelin E., Ponsoda S., Delneste Y, Hebbar M., Jeannin P. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int. J. Cancer. 2009;125:367–373. doi: 10.1002/ijc.24401. [DOI] [PubMed] [Google Scholar]
- Dumont P., Berton A., Nagy N., Sandras F., Tinton S., Demetter P., Mascart F., Allaoui A., Decaestecker C., Salmon I. Expression of galectin-3 in the tumor immune response in colon cancer. Lab. Invest. 2008;88:896–906. doi: 10.1038/labinvest.2008.54. [DOI] [PubMed] [Google Scholar]
- Eguchi J., Kong X., Tenta M., Wang X., Kang S., Rosen E. D. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. 2013;62:3394–3403. doi: 10.2337/db12-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele N.A, Ruby T., Jacobson A., Manzanillo P.S., Cox J.S., Lam L., Mukundan L., Chawla A., Monack D.M. Salmonella require the fatty acid regulator PPARδ for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe. 2013;14:171–182. doi: 10.1016/j.chom.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell G.C., Chitturi S., Gan L., van Rooyen D. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo I.M., Gafencu A.V. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology. 2013;218:1376–1384. doi: 10.1016/j.imbio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Fletcher N., Sutaria R., Jo J., Barnes A. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology. 2013. (in press). [DOI] [PMC free article] [PubMed]
- Förster S., Brandt M., Mottok D.S., Zschüttig A., Zimmermann K., Blattner F.R., Gunzer F., Pöhlmann C. Secretory expression of biologically active human Herpes virus interleukin-10 analogues in Escherichia coli via a modified Sec-dependent transporter construct. BMC Biotechnol. 2013;13:82. doi: 10.1186/1472-6750-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks T.J., Chong P.Y., Chui P., Galvin J.R., Lourens R.M., Reid A.H., Selbs E., Mcevoy P.L., Hayden D.L., Fukuoka J., et al. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum. Pathol. 2003;34:743–748. doi: 10.1016/S0046-8177(03)00367-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadang V., Kohli R., Myronovych A., Hui D.Y., Perez-Tilve D., Jaeschke A. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am. J. Physiol. Endoc. Metab. 2013;305:E549–556. doi: 10.1152/ajpendo.00197.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier E. L., Shay T., Miller J., Greter M., Jakubzick C., Ivanov S., Helft J., Chow A., Elpek K. G., Gordonov S., et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami S., Shojaei S., Yeganeh B., Ande S.R., Jangamreddy J.R., Mehrpour M., Christoffersson J., Chaabane W., Moghadam A.R., Kashani H.H., et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Gironella M., Calvo C., Fernández A., Closa D., Iovanna J.L., Rosello-Catafau J., Folch-Puy E. Reg3β deficiency impairs pancreatic tumor growth by skewing macrophage polarization. Cancer Res. 2013;73:5682–5694. doi: 10.1158/0008-5472.CAN-12-3057. [DOI] [PubMed] [Google Scholar]
- Gobeil L.A., Lodge R., Tremblay M.J. Differential HIV-1 endocytosis and susceptibility to virus infection in human macrophages correlate with cell activation status. J. Virol. 2012;86:10399–10407. doi: 10.1128/JVI.01051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh Y.P.S., Chawla A., Nguyen K.D. Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E., Geissmann F. Myb-independent macrophages: a family of cells that develops with their tissue of residence and is involved in its homeostasis. Cold Spring Harb. Symp. Quant. Biol. 2013 doi: 10.1101/sqb.2013.78.020032. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hajizadeh M.R., Mokarram P., Kamali-Sarvestani E., Bolhassani A., Mostafavi-Pour Z. Recombinant nonstructural 3 protein, rNS3, of hepatitis C virus along with recombinant GP96 induce IL-12, TNFα and α5 integrin expression in antigen presenting cells. Hepat. Mon. 2013;13:e8104. doi: 10.5812/hepatmon.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke M.L., Heim C.E., Angle A., Sanderson S.D., Kielian T. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J. Immunol. 2013;190:2159–2168. doi: 10.4049/jimmunol.1202348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J., Elinav E., Jin C., Hao L., Mehal W.Z., Strowig T., Thaiss C.A., Kau A.L., Eisenbarth S.C., Jurczak M.J., et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J., Elinav E., Thaiss C.A., Flavell R.A. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2013 doi: 10.1146/annurev-physiol-021113-170324. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Heusinkveld M., de Vos van Steenwijk P.J., Goedemans R., Ramwadhdoebe T.H., Gorter A., Welters M.J.P., van Hall T., van der Burg S.H. M2 macrophages induced by prosta-glandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+Th1 cells. J. Immunol. 2011;187:1157–1165. doi: 10.4049/jimmunol.1100889. [DOI] [PubMed] [Google Scholar]
- Heydtmann M. Macrophages in hepatitis B and hepatitis C virus infections. J. Virol. 2009;83:2796–2802. doi: 10.1128/JVI.00996-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hu X., Chen J., Wang L., Ivashkiv L.B. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J. Leukoc. Biol. 2007;82:237–243. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- Huang Z., Zhang Z., Jiang Y., Zhang D., Chen J., Dong L., Zhang J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J. Control Release. 2012;158:286–292. doi: 10.1016/j.jconrel.2011.11.013. [DOI] [PubMed] [Google Scholar]
- Hume D.A. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal Immunol. 2008;1:432–441. doi: 10.1038/mi.2008.36. [DOI] [PubMed] [Google Scholar]
- Jenner R.G., Young R.A. Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 2005;3:281–294. doi: 10.1038/nrmicro1126. [DOI] [PubMed] [Google Scholar]
- Ji W.J., Ma Y.Q., Zhou X., Zhang Y.D., Lu R.Y., Sun H.Y., Guo Z.Z., Zhang Z., Li Y.M., Wei L.Q. Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J. Immunol. Methods. 2013. pii: S0022-1759(13)00328-1. [DOI] [PubMed]
- Jouanguy E., Döffinger R., Dupuis S., Pallier A., Altare F., Casanova J.L. IL-12 and IFN-γ in host defense against mycobacteria and salmonella in mice and men. Curr. Opin. Immunol. 1999;11:346–351. doi: 10.1016/s0952-7915(99)80055-7. [DOI] [PubMed] [Google Scholar]
- Kadl A., Meher A.K., Sharma P.R., Lee M.Y., Doran A.C., Johnstone S.R., Elliot M.R., Gruber F., Han J., Chen W., et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Cir. Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang K., Reilly S.M., Karabacak V., Gangl M.R., Fitzgerald K., Hatano B., Lee C.-H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008;7:485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti T.D., Dixit V.D. Immunological complications of obesity. Nat. Immunol. 2012;13:707–712. doi: 10.1038/ni.2343. [DOI] [PubMed] [Google Scholar]
- Karlmark K.R., Weiskirchen R., Zimmermann H.W., Gassler N., Ginhoux F., Weber C., Merad M., Luedde T., Trautwein C., Tacke F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- Kiszewski A.E., Becerril E., Aguilar L.D., Kader I.T.A., Myers W., Portaels F., Hernàndez Pando R. The local immune response in ulcerative lesions of Buruli disease. Clin. Exp. Immunol. 2006;143:445–451. doi: 10.1111/j.1365-2249.2006.03020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein I., Cornejo J.C., Polakos N.K., John B., Wuensch S.A., Topham D.J., Pierce R.H., Crispe I.N. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimp A.H., de Vries E.G.E., Scherphof G.L., Daemen T. A potential role of macrophage activation in the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002;44:143–161. doi: 10.1016/s1040-8428(01)00203-7. [DOI] [PubMed] [Google Scholar]
- Krausgruber T., Blazek K., Smallie T., Alzabin S., Lockstone H., Sahgal N., Hussell T., Feldmann M., Udalova I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Leitinger N., Schulman I.G. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler. Throm. Vas. Biol. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman L.J., Maryanoff B.E., Ghadiri M.R. Molecules that mimic apolipoprotein A-I: potential agents for treating Atherosclerosis. J. Med. Chem. 2013. (in press). [DOI] [PMC free article] [PubMed]
- Liao X., Sharma N., Kapadia F. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 2011;121:2736–2749. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Perlman H., Pagliari L.J., Pope R.M. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages. Role of Mcl-1, independent of nuclear factor (NF)-kappaB, Bad, or caspase activation. J. Exp. Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.Y., Xu J.Y., Shi X.Y., Huang W., Ruan T.Y., Xie P., Ding J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 2013;93:844–854. doi: 10.1038/labinvest.2013.69. [DOI] [PubMed] [Google Scholar]
- Lugo-Villarino G., Vérollet C., Maridonneau-Parini I., Neyrolles O. Macrophage polarization: convergence point targeted by Mycobacterium tuberculosis and HIV. Front. Immunol. 2011;2:43. doi: 10.3389/fimmu.2011.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng C.N., Bodzin J.L., Saltiel A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007a;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng C.N., Deyoung S.M., Bodzin J.L., Saltiel A.R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007b;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- Luyendyk J.P., Schabbauer G.A., Tencati M., Holscher T., Pawlinski R., Mackman N. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 2008;180:4218–4226. doi: 10.4049/jimmunol.180.6.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majai G., Kiss E., Tarr T., Zahuczky G., Hartman Z., Szegedi G., Fésüs L. Decreased apopto-phagocytic gene expression in the macrophages of systemic lupus erythematosus patients. Lupus. 2014;23:133–145. doi: 10.1177/0961203313511557. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Biswas S.K., Galdiero M.R., Sica A., Locati M. Macrophage plasticity and polarization in tissue repair and remodelling\ J. Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- Martinez F.O., Helming L., Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- Mercalli A., Calavita I., Dugnani E., Citro A., Cantarelli E., Nano R., Melzi R., Maffi P., Secchi A., Sordi V., et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology. 2013;140:179–190. doi: 10.1111/imm.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills C.D., Kincaid K., Alt J.M., Heilman M.J., Hill A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000;164:6166–6173. [Google Scholar]
- Miura K, Kodama Y., Inokuchi S., Schnabl B., Aoyama T., Ohnishi H., Olefsky J.M., Brenner D.A., Seki E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1 beta in mice. Gastroenterology. 2010;139:323–U453. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K., Yang L., van Rooijen N., Ohnishi H., Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;302:G1310–G1321. doi: 10.1152/ajpgi.00365.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K., Yang L., van Rooijen N., Brenner D.A., Ohnishi H., Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–589. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monini P., Colombini S., Stürzl M., Goletti D., Cafaro A., Sgadari C., Buttò S., Franco M., Leone P., Fais S., et al. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi’s sarcoma. Blood. 1999;93:4044–4058. [PubMed] [Google Scholar]
- Moreno J.L., Kaczmarek M., Keegan A.D., Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102:1078–1086. doi: 10.1182/blood-2002-11-3437. [DOI] [PubMed] [Google Scholar]
- Mosser D. The many faces of macrophage activation. J. Leukoc. Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Wynn T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonas K.J., Nair M.G., Prieto-Lafuente L., Paape D., Allen J.E. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J. Immunol. 2009;182:3084–3094. doi: 10.4049/jimmunol.0803463. [DOI] [PubMed] [Google Scholar]
- Nau G.J., Richmond J.F.L., Schlesinger A., Jennings E.G., Lander E.S., Young R.A. Human macrophage activation programs induced by bacterial pathogens. Proc. Natl. Acad. Sci. USA. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyen C., Mukhopadhyay S., Gordon S., Hagemann T. An apolipoprotein A-I mimetic targets scavenger receptor A on tumor-associated macrophages: a prospective anticancer treatment? Oncoimmunology. 2013;2:e24461. doi: 10.4161/onci.24461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niino D., Komohara Y., Murayama T., Aoki R., Kimura Y., Hashikawa K., Kiyasu J., Takeuchi M., Suefuji N., Sugita Y., et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL) Pathol. Int. 2010;60:278–283. doi: 10.1111/j.1440-1827.2010.02514.x. [DOI] [PubMed] [Google Scholar]
- Öberg Å., Samii S., Stenling R., Lindmark G. Different occurrence of CD8+, CD45R0+, and CD68+ immune cells in regional lymph node metastases from colorectal cancer as potential prognostic predictors. Int. J. Colorectal Dis. 2002;17:25–29. doi: 10.1007/s003840100337. [DOI] [PubMed] [Google Scholar]
- Obstfeld A.E., Sugaru E., Thearle M., Francisco A.M., Gayet C., Ginsberg H.N., Ables E.V., Ferrante A.W. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes. 2010;59:916–925. doi: 10.2337/db09-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard J.I., Ricardo-Gonzalez R.R., Goforth M.H., Morel C.R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A.W., et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky J.M., Glass C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Otto M. Staphylococcal biofilms. Curr. Top. Microbiol. Immunol. 2008;322:207–228. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciello I., Silipo A., Lembo-fazio L., Curcurù L., Zumsteg A., Noël G., Ciancarella V., Sturiale L., Molinaro A., Bernardini M.L. Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc. Natl. Acad. Sci. USA. 2013;110:E4345–4354. doi: 10.1073/pnas.1303641110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page C., Goicochea L., Matthews K., Zhang Y., Klover P., Holtzman M.J., Hennighausen L., Frieman M. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J. Virol. 2012;86:13334–13349. doi: 10.1128/JVI.01689-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S.M., Chu C.M., Cheng V.C.C., Chan K.S., Hung I.F.N., Poon L.L.M., Law K.I., Tang B.S., Hon T.Y., Chan C.S., et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poglitsch M., Weichhart T., Hecking M., Werzowa J., Katholnig K., Antlanger M., Krmpotic A, Jonjic S., Hörl W.H., Zlabinger G.J., et al. CMV late phase-induced mTOR activation is essential for efficient virus replication in polarized human macrophages. Am. J. Transplant. 2012;12:1458–1468. doi: 10.1111/j.1600-6143.2012.04002.x. [DOI] [PubMed] [Google Scholar]
- Qin H., Holdbrooks A.T., Liu Y., Reynolds S.L., Yanagisawa L.L., Benveniste E.N. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J. Immunol. 2012a;189:3439–3448. doi: 10.4049/jimmunol.1201168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Yeh W.I., De Sarno P., Holdbrooks A.T., Liu Y., Muldowney M.T., Reynolds S.L., Yanagisawa L.L., Fox T.H., Park K., et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc. Natl. Acad. Sci. USA. 2012b;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racanelli V., Rehermann B. The liver as an immunological organ. Hepatology. 2006;43:S54–S62. doi: 10.1002/hep.21060. [DOI] [PubMed] [Google Scholar]
- Raes G., De Baetselier P., Noël W., Beschin A., Brombacher F., Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- Raes G., Van den Bergh R., De Baetselier P., Ghassabeh G.H., Scotton C., Locati M., Mantovani A., Sozzani S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol. 2005;174:6561–6562. doi: 10.4049/jimmunol.174.11.6561. [DOI] [PubMed] [Google Scholar]
- Rauch I., Müller M., Decker T. The regulation of inflammation by interferons and their STATs. JAKSTAT. 2013;2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings J.S., Rosler K.M., Harrison D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- Rius B., López-Vicario C., González-Périz A., Morán-Salvador E., García-Alonso V., Clária J., Titos E. Resolution of inflammation in obesity-induced liver disease. Front. Immunol. 2012;3:257. doi: 10.3389/fimmu.2012.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh Y.S., Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013;28:38–42. doi: 10.1111/jgh.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolny C., Mazzone M., Tugues S., Laoui D., Johansson I., Coulon C., Sguadrito M.L., Segura I., Li X., Knevels E., et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Rottenberg M.E., Gigliotti-Rothfuchs A., Wigzell H. The role of IFN-gamma in the outcome of chlamydial infection. Curr. Opin. Immunol. 2002;14:444–451. doi: 10.1016/s0952-7915(02)00361-8. [DOI] [PubMed] [Google Scholar]
- Schaale K., Brandenburg J., Kispert A., Leitges M., Ehlers S., Reiling N. Wnt6 is expressed in granulomatous lesions of mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J. Immunol. 2013;191:5182–5195. doi: 10.4049/jimmunol.1201819. [DOI] [PubMed] [Google Scholar]
- Schmieder A., Michel J., Schönhaar K., Goerdt S., Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer. Biol. 2012;22:289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S.E., Pollard J.W., et al. A lineage of myeloid cells independent of Myb and Hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- Schwabe R.F., Brenner D.A. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- Schwabe R.F., Seki E., Brenner D.A. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–900. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Shanmugam N., Gaw Gonzalo I.T., Natarajan R. Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes. 2004;53:795–802. doi: 10.2337/diabetes.53.3.795. [DOI] [PubMed] [Google Scholar]
- Shaughnessy L.M., Swanson J.A. The role of the activated macrophage in clearing Listeria monocytogenes infection. Front. Biosci. 2007;12:2683–2692. doi: 10.2741/2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa T., Kawazoe Y., Tsujikawa T., Jung D., Sato S., Uesugi M. Deactivation of STAT6 through serine 707 phosphorylation by JNK. J. Biol. Chem. 2011;286:4003–4010. doi: 10.1074/jbc.M110.168435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;22:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A., Saccani A., Mantovani A. Tumor-associated macrophages: a molecular perspective. Int. Immunopharmacol. 2002;2:1045–1054. doi: 10.1016/s1567-5769(02)00064-4. [DOI] [PubMed] [Google Scholar]
- Sica A, Invernizzi P., Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2013. [In Press]. [DOI] [PubMed]
- Smith M., Bentz G. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 2004a;78:4444–4453. doi: 10.1128/JVI.78.9.4444-4453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M., Bentz G. HCMV activates PI (3) K in monocytes and promotes monocyte motility and transendothelial migration in a PI (3) K-dependent manner. J. Leukoc. Biol. 2004b;76:65–76. doi: 10.1189/jlb.1203621. [DOI] [PubMed] [Google Scholar]
- Song Y., Dou H., Gong W., Liu X., Yu Z., Li E., Tan R., Hou Y. Bis-N-norgliovictin, a small-molecule compound from marine fungus, inhibits LPS-induced inflammation in macrophages and improves survival in sepsis. Eur. J. Pharmacol. 2013;705:49–60. doi: 10.1016/j.ejphar.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Stolfi C., Caruso R., Franzè E., Sarra M., De Nitto D., Rizzo A., Pallone F., Monteleone G. Interleukin-25 fails to activate STAT6 and induce alternatively activated macrophages. Immunology. 2011;132:66–77. doi: 10.1111/j.1365-2567.2010.03340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout R.D., Jiang C., Matta B., Tietzel I., Watkins S.K., Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- Stout R.D., Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J. Leukoc. Biol. 2004;76:509–513. doi: 10.1189/jlb.0504272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. J. Clin. Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Sun Y., Pu J., Zhang Y., Zhu Q., Li J., Gu J., Chang K. C., Liu J. Comparative virus replication and host innate response in human cells infected with 3 prevalent clades (2.3.4, 2.3.2 and 7) of highly pathogenic avian influenza H5N1 viruses. J. Virol. 2013;88:725–729. doi: 10.1128/JVI.02510-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H., Ashihara M., Ishiguro T., Terashima H., Watanabe T., Kato A., Suzuki M. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol. Ther. 2009a;8:2329–2338. doi: 10.4161/cbt.8.24.9985. [DOI] [PubMed] [Google Scholar]
- Takai H., Kato A., Kato C., Watanabe T., Matsubara K., Suzuki M., Kataoka H. The expression profile of glypican-3 and its relation to macrophage population in human hepatocellular carcinoma. Liver Int. 2009b;29:1056–1064. doi: 10.1111/j.1478-3231.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- Tateya S., Kim F., Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front. Endocrinol. 2013;4:93. doi: 10.3389/fendo.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson A.W., Knolle P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010;10:753–766. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- Thurlow L.R., Hanke M.L., Fritz T., Angle A., Aldrich A., Williams S.H., Engebretsen I.L., Bayles K.W., Horswill A.R., Kielian T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011;186:6585–6596. doi: 10.4049/jimmunol.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H., Moschen A.R. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- Tosello-Trampont A.C., Landes S.G., Nguyen V., Novobrantseva T.I., Hahn Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012;287:40161–40172. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura N., Saio M., Suwa T., Kitoh Y., Bai J., Nonaka K., Ouyang G.F., Okada M., Balazs M., Adany R., et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008;83:1136–1144. doi: 10.1189/jlb.0907611. [DOI] [PubMed] [Google Scholar]
- Verreck F.A., de Boer T., Langenberg D.M., Hoeve M.A., Kramer M., Vaisberg E., Kastelein R., Kolk A., de Wall-Malefyt R., Ottenhoff T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA. 2004;101:4560–4565. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villares R., Kakabadse D., Juarranz Y., Gomariz R.P., Martínez A.C., Mellado M. Growth hormone prevents the development of autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 2013;101:4619–4627. doi: 10.1073/pnas.1314985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Li F., Sun R., Gao X., Wei H., Li L.-J., Tian Z. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat. Commun. 2013;4:2106. doi: 10.1038/ncomms3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T., Säemann M.D. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann. Rheum. Dis. 2008;67(Suppl 3):iii70–4. doi: 10.1136/ard.2008.098459. [DOI] [PubMed] [Google Scholar]
- Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg S.P., Hunter D., Huber R., Lemieux J., Slaymaker S., Vaddi K., Charo I., Leibel R.L., Ferrante A.W. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn T.A., Chawla A., Pollard J.W. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Zhu J., Smith S., Foldi J., Zhao B., Chung A.Y., Oultz H., Kitajewski J., Shi C., Weber S., et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 2012;13:642–650. doi: 10.1038/ni.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F., Kang Y., Zhang H., Piao Z., Yin H., Diao R., Xia J., Shi L. Akt1-mediated regulation of macrophage polarization in a murine model of Staphylococcus aureus pulmonary infection. J. Infect. Dis. 2013;208:528–538. doi: 10.1093/infdis/jit177. [DOI] [PubMed] [Google Scholar]
- Yin M.J., Yamamoto Y., Gaynor R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- You Q., Holt M., Yin H., Li G., Hu C.-J., Ju C. Role of hepatic resident and infiltrating macrophages in liver repair after acute injury. Biochem. Pharmacol. 2013;86:836–843. doi: 10.1016/j.bcp.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Konstantopoulos N., Lee J., Hansen L., Li Z.W., Karin M., Shoelson S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- Zhou Q., Peng R.-Q., Wu X.-J., Xia Q., Hou J.-H., Ding Y., Zhou Q.M., Zhang X., Pang Z.Z., Wan D.S., et al. The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J. Transl. Med. 2010;8:13. doi: 10.1186/1479-5876-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Huang C., Lin Z., Zhan S., Kong L., Fang C., Li J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 2013;26:192–197. doi: 10.1016/j.cellsig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Zhu L., Baker S.S., Gill C., Liu W., Alkhouri R., Baker R.D., Gill S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]