

Abstract
Mammalian polo-like kinase 1 (Plk1) has been studied intensively as a key regulator of various cell cycle events that are critical for proper M-phase progression. The polo-box domain (PBD) present in Plk1’s C-terminal non-catalytic region has been shown to play a central role in targeting the N-terminal kinase domain of Plk1 to specific subcellular locations. Subsequent studies reveal that PBD binds to a phosphorylated motif generated by one of the two mechanisms - self-priming by Plk1 itself or non-self-priming by a Pro-directed kinase, such as Cdc2. Here, we comparatively review the differences in the biochemical steps of these mechanisms and discuss their physiological significance. Considering the diverse functions of Plk1 during the cell cycle, a better understanding of how the catalytic activity of Plk1 functions in concert with its cis-acting PBD and how this coordinated process is intricately regulated to promote Plk1 functions will be important for providing new insights into different mechanisms underlying various Plk1-mediated biological events that occur at the multiple stages of the cell cycle.
Keywords: non-self-priming, self-priming, Plk1, polo-box domain
INTRODUCTION
Reversible protein phosphorylation by protein kinases is one of the fundamental regulatory mechanisms that govern diverse cellular processes in eukaryotic organisms. The members of the polo subfamily of the Ser/Thr protein kinases [collectively, polo-like kinases (Plks)] have emerged as important regulators for cell cycle progression and cell proliferation [reviews, (Archambault and Glover, 2009; Barr et al., 2004; van de Weerdt and Medema, 2006)]. In humans, five Plk members (from Plk1 to Plk5; collectively, Plks) exhibiting differential functions and tissue distributions are known to exist today. These members are characterized by the presence of a highly homologous region, called polo-box domain (PBD) (Clay et al., 1993) that plays a critical role in targeting their kinase domains to specific subcellular structures (Lee et al., 1998). Except the most recently identified Plk5 (de Cárcer et al., 2011a), four of them (Plk1-4) also possess an N-terminal kinase domain with a high level of sequence homology (Fig. 1).
Fig. 1.
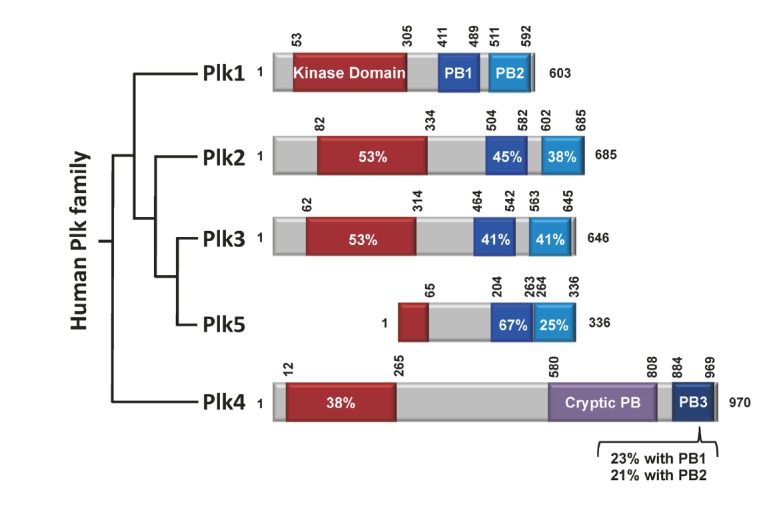
Schematic diagram of human Plk1 to Plk5. Plk1-4 contain the kinase domain (red) at their N-termini. The polo-box domains (PBDs) of Plk1-3 are composed of two structurally similar PB1 (blue) and PB2 (light blue) motifs that form a phospho-recognition module (Elia et al., 2003b). Whether the PBD of Plk5 also binds to a phospho-epitope is not known. In contrast to the PBDs of Plk1-3, the cryptic polo-box (CPB) of Plk4 homodimerizes to form a stable dimer (Leung et al., 2002; Slevin et al., 2012) and bind to a target in a manner that does not require a phosphorylated motif (Kim T.S. and K. S. Lee, unpublished data). In addition, PB3, which is thought to form a homodimer (Leung et al., 2002), exhibits a low level of homology with PB1 or PB2. Sequence identities with the corresponding domains in Plk1 are given in percentages. Numbers indicate the positions of amino acid residues in a given protein.
Plk1 is the most evolutionarily conserved member of the polo subfamily, whose orthologs have been isolated from budding yeast to mammals (de Cárcer et al., 2011b). In humans, Plk1 has been shown to regulate diverse biochemical and cellular processes at various stages of M phase. In line with the multiplicity of its functions, Plk1 has been shown to localize to important mitotic apparatuses, such as centrosomes, kinetochores, and midbodies (Arnaud et al., 1998; Golsteyn et al., 1995; Lee et al., 1995; Seong et al., 2002). Remarkably, the subcellular localization of Plk1 to all of these subcellular structures requires the function of PBD (Jang et al., 2002; Lee et al., 1998; Park et al., 2010; Seong et al., 2002), suggesting that PBD plays a central role in promoting Plk1-mediated events at these locations. Consistent with the importance of Plk1 in regulating the cell cycle and cell proliferation, Plk1 overexpression is tightly associated with oncogenesis of human cells (Strebhardt, 2010; Strebhardt and Ullrich, 2006; Takaki et al., 2008). Thus, understanding the mechanism by which Plk1 exerts its catalytic activity to its physiological substrates will be important for a better understanding of the biological process underlying cell division and proliferation.
Over the years, studies with various Plk1 PBD-binding proteins have yielded a plethora of information on how Plk1 regulates various biological processes. On the other hand, how other Plks operate remains largely unknown. Therefore, in this minireview, we will focus on providing a comprehensive view of how the N-terminal catalytic activity of Plk1 functions in concert with its cis-acting PBD to trigger Plk1-dependent biochemical steps, and how this event ultimately brings about the cellular processes required for normal mitotic progression. The mechanisms underlying Plk1-dependent events may serve as a template in understanding how other related Plks function during the cell cycle.
PBD: A PHOSPHO-RECOGNITION MODULE CRITICAL FOR PLK1 FUNCTION
A glimpse of the PBD function in targeting Plk1 to specific sub-cellular locations was first revealed by expressing various mammalian Plk1 PBD mutants in budding yeast bearing a polo mutation called cdc5-1 (Lee et al., 1998). Subsequent studies with a dominant-negative Plk1 PBD suggested that PBD is sufficient to localize to distinct subcellular structures, and that a PBD-dependent protein-protein interaction is critically required for proper M-phase progression (Hanisch et al., 2006; Seong et al., 2002).
A breakthrough in our understanding of the Plk1 PBD-dependent interaction came from the work of Michael Yaffe and his colleagues, which demonstrated that PBD recognizes a consensus phospho-Ser (p-Ser)/phospho-Thr (p-Thr) motif containing [Pro/Phe]-[Φ/Pro]-[Φ]-[Thr/Gln/His/Met]-Ser-[pThr/pSer]-[Pro/X] (Φ represents hydrophobic residues and X means any residues) (Elia et al., 2003a; 2003b). This seminal discovery, together with subsequent X-ray crystallographic analyses, provided the first insight into the molecular basis of how Plk1 PBD binds to its targets in a phospho-specific manner (Cheng et al., 2003; Elia et al., 2003a; García-Alvarez et al., 2007). It is now widely appreciated that the PBD functions as a molecular mediator that brings the kinase domain of Plk1 in proximity to its substrates by interacting with a phosphorylated motif on the substrate itself or its associated protein(s) (Lowery et al., 2004; Park et al., 2010). Thus, the PBD-dependent protein-protein interaction is likely central to various Plk1-mediated biological processes.
The PBD-dependent interaction with a phosphorylated target occurs at two distinct steps: 1) generation of a phosphorylated binding motif and 2) subsequent binding of Plk1 with the resulting phospho-target. In this sequence of events, it is critical to understand the mechanism by which priming phosphorylation occurs and which kinase is responsible for this event. Once a phospho-epitope is generated, Plk1 can bind to the phospho-motif in concentration- and affinity-dependent manners through a stochastic process. Therefore, the mechanism regulating the priming phosphorylation is discussed in detail below.
MECHANISMS OF GENERATING A PBD-BINDING MOTIF
Studies with various Plk1 PBD-binding proteins revealed that a priming phosphorylation site for PBD binding is generated by either Plk1 itself (i.e., self-priming) or a kinase other than Plk1 (e.g., non-self-priming) (Lee et al., 2008). Indeed, a large fraction of PBD-binding targets have been shown to employ either one of the two binding modes to interact with Plk1 (Park et al., 2010). The differences in these two binding mechanisms are explained below, using two well-characterized PBD-binding proteins, PBIP1 and hCenexin1, as examples for each mechanism (Fig. 2).
Fig. 2.
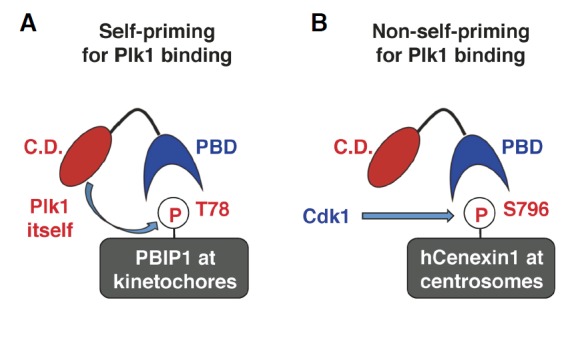
Schematic diagram illustrating the two distinct examples of PBD-dependent binding. (A) In this example, Plk1 phosphorylates and generates the p-T78 motif on a kinetochore protein, PBIP1, for its own binding. Since the Plk1 PBD-docking site is generated by Plk1 itself, this mechanism is called self-priming and binding. Although not depicted here, the Plk1 molecule binding to the p-T78 motif could be another Plk1 molecule present in close proximity to the newly generated p-T78 site. (B) In this example, Cdk1 phosphorylates a centrosomal protein, hCenexin1, and generates the p-S796 motif for Plk1 PBD binding. Since the Plk1 PBD-docking site is generated by a kinase other than Plk1, this mechanism is called non-self-priming and binding.
Self-priming model
It has been shown that inhibiting the catalytic activity of Plk1 induces delocalization of Plk1 from centrosomes, kinetochores, and midbodies (Lenart et al., 2007). This observation suggests that although other mechanisms may also exist, Plk1 promotes its own localization to these subcellular structures in a manner that requires its catalytic activity.
One of the examples elegantly demonstrating Plk1 activity-dependent subcellular localization came from the work that investigated the functional relationship between Plk1 and its kinetochore-binding protein PBIP1 (Kang et al., 2006), also called CENP-50/KLIP1/MLF1IP (Hanissian et al., 2004; Minoshima et al., 2005; Pan et al., 2003). Results showed that Plk1 efficiently phosphorylates the T78 residue of PBIP1 and binds to the resulting p-T78 motif to recruit itself to kinetochores (Kang et al., 2006) (Fig. 2A). Thus, the p-T78 motif serves as a self-generated tether for Plk1’s own localization. Intriguingly, this tethering step also enables Plk1 to further phosphorylate other PBIP1 molecules at kinetochores (discussed in detail below). The specificity of both Plk1-dependent PBIP1 T78 phosphorylation and the ensuing interaction between Plk1 PBD and PBIP1 p-T78 was verified by the observation that Plk2 and Plk3 failed to phosphorylate and bind to the T78 motif (Lee et al., 2008; Park et al., 2009). In addition to PBIP1, a self-priming and binding mechanism has been suggested for a midbody-associated protein (PRC1) (Neef et al., 2007) and mitotic kinesin-like protein 2 (MKLP-2) (Neef et al., 2003). Since the Plk1-PBIP1 interaction has been characterized extensively (Kang et al., 2006; Lee et al., 2008; Park et al., 2011), it was chosen for in-depth discussion of the self-priming model (see below).
Non-self-priming model
Distinct from the Plk1-PBIP1 interaction that requires Plk1-dependent generation of the p-T78 motif, Plk1 also binds to phosphorylated epitopes generated by kinases other than Plk1 (therefore, called non-self-priming as opposed to the self-priming described above). We have previously demonstrated that a cyclin-dependent kinase, Cdk1, phosphorylates the S796 residue of hCenexin1 and induces the interaction between the resulting p-S796 motif and Plk1 PBD during mitosis. This event was shown to be important to properly recruit Plk1 to hCenexin1-localized centrosomes and to promote normal M-phase progression (Soung et al., 2009) (Fig. 2B). Notably, using Cdk1 as a priming kinase appears to be necessary because the S796 residue is immediately followed by the Pro residue at the +1 position (i.e., Pro+1; numbered from the S796 residue), thus guiding a Pro-directed kinase, such as Cdk1, to specifically phosphorylate the S796 residue. In vitro studies confirmed that Cdk1, but not Plk1, phosphorylates the hCenexin1 S796 site (Soung et al., 2006) for Plk1 PBD binding.
A growing body of evidence suggests that a large number of Plk1 PBD-binding proteins follow the non-self-priming and binding mode. In addition to hCenexin1 (Soung et al., 2009), they include Cdc25C phosphatase (Elia et al., 2003a), Wee1A kinase (Watanabe et al., 2004), Bub1 (Qi et al., 2006) and BubR1 (Elowe et al., 2007) spindle checkpoint proteins, FoxM1 transcriptional factor (Fu et al., 2008), a microtubule-binding protein Nedd1 (Johmura et al., 2011; Zhang et al., 2009), a centromere-associated SNF2 family ATPase PICH (Baumann et al., 2007), and many other proteins involved in various cellular events (Park et al., 2010). This finding suggests that the non-self-priming and binding mechanism is likely more widespread than the self-priming and binding mechanism described above.
BIOCHEMICAL CHARACTERISTICS OF SELF- VS. NON-SELF-PRIMING AND BINDING
The finding that two distinct mechanisms operate to generate PBD-binding targets is intriguing. One of the significant questions is whether the mode of PBD binding has evolved into two distinct mechanisms to provide physiological advantages or it has randomly progressed in two directions to deal with diverse biochemical processes mediated by Plk1. As one of the approaches to investigating these possibilities, we converted the self-priming site of PBIP1 to a Cdk1-dependent, non-self-priming site by mutating the kinase-guiding residues immediately C-terminal to the T78 residue (Park et al., 2011). The resulting PBIP1-cdk mutant contains a Pro+1 (numbered from the T78 residue) and positively charged Arg+2, Lys+3, and Arg+4 residues to draw Cdk1-dependent phosphorylation to the T78 motif (Fig. 3). As a consequence, the T78 residue of the PBIP1-cdk mutant was specifically phosphorylated by Cdk1, but not by Plk1, thus strictly imposing Cdk1-dependent phosphorylation as a priming step for subsequent Plk1 binding (Park et al., 2011).
Fig. 3.
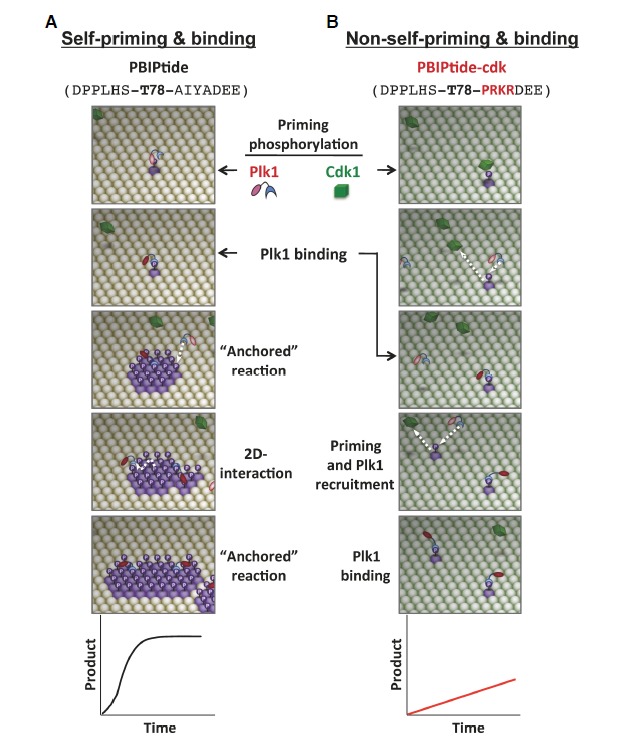
A comparative illustration of self-priming- versus non-self-priming-based biochemical processes. The schematic diagrams are based on the results obtained with a pair of ELISA-based kinase assays that utilize either Plk1-phosphorylatable PBIPtide (A) or Cdk1-phosphorylatable PBIPtide-cdk (B) as substrates (Park et al., 2011). The sequences for PBIPtide (A) and the PBIPtide-cdk mutant (B) are shown in parentheses with the mutations highlighted in red. The T78 residue is indicated in boldface type. Plk1 is initially at an inactive state (pink kinase domain), which becomes partially activated (red kinase domain) upon binding to a PBD-docking site. Phosphorylation of the T210 residue at the activation loop of Plk1 further activates the enzyme (Lee and Erikson, 1997; Xu et al., 2013). Straight arrows indicate the incoming or outgoing movement of a molecule, whereas the curved arrow in (A) indicates the lateral movement of a molecule on a two-dimensional surface. Graphs depict the reaction kinetics of each mechanism. See Supplementary Movie S1.
Then, to comparatively investigate the biochemical characteristics of self-priming versus non-self-priming, a pair of enzyme-linked immunosorbent assay (ELISA)-based Plk1-binding as-says were generated with either a PBIP1 fragment (i.e., PBIP-tide containing the T78 motif) or the corresponding PBIP1-cdk mutant fragment (i.e., PBIPtide-cdk) immobilized on the ELISA plate, and the level of T78 phosphorylation on the PBIPtide or PBIPtide-cdk was quantified. Providing that PBIPtide or the PBIPtide-cdk mutant immobilized on the plate mimics PBIP1 or PBIP1-cdk proteins localized to a confined area of a mitotic kinetochore plate, the biochemical outcome of Plk1-dependent self-priming or Cdk1-dependent non-self-priming in the recruitment of Plk1 to PBIP1 or PBIP1-cdk-loaded kinetochores, respectively, could be indirectly assessed using these ELISA-based assays (Park et al., 2011). The differences in their reaction modes of self-priming and non-self-priming mechanisms are described in detail below.
Biochemical steps underlying self-priming and binding
In this mechanism, the catalytic activity of Plk1 first phosphorylates its binding target prior to the PBD-mediated interaction (Fig. 3A, first panel; See attached Supplementary Movie S1). However, in the absence of either an activational phosphorylation at the T210 of Plk1 or a phospho-peptide bound to its PBD, Plk1 is considered not activated (Elia et al., 2003b; Xu et al., 2013). Under these conditions, a basal level of Plk1 activity (pink-colored kinase domain in Fig. 3) must phosphorylate the T78 target through a stochastic process. Therefore, in the case of self-priming and binding, the most rate-limiting step is the generation of the initial p-T78 motif on the PBIPtide immobilized on the ELISA plate (which is analogous to PBIP1 localized on the kinetochore plate). Once a binding site is generated, either the same Plk1 molecule that phosphorylated this site or another Plk1 molecule near the newly generated p-T78 motif can bind to this site through its PBD (self-priming and binding) (Fig. 3A, first and second panels). Upon anchoring at the p-T78 motif of PBIPtide, Plk1 becomes partially activated (red-colored kinase domain in Fig. 3) through the relief of the autoinhibition imposed by the kinase domain-PBD interaction (Elia et al., 2003b). The full activation of Plk1 is thought to require both binding of PBD and phosphorylation of Plk1 at the T210 site (Xu et al., 2013). These anchored and partially activated Plk1 can efficiently phosphorylate neighboring PBIPtide molecules and generate p-T78 epitopes on them (PBD-dependent anchored reaction). Since this anchored state eliminates an inefficient stochastic process, which is obligatory in a three-dimensional solution environment, the PBD-mediated anchorage onto a phospho-T78 motif provides a great advantage for the catalytic activity of Plk1 to rapidly generate a patch of the p-T78 motifs around the initial PBD docking site.
The p-T78 patches present on the surface of the PBIPtide-coated plate (i.e., PBIP1-loaded kinetochores) generate a unique biochemical environment in which Plk1 can best utilize its intrinsic biochemical characteristics involving its cis-acting PBD. Since Plk1 PBD exhibits a high affinity (Kd of ∼10 nM) to the p-T78 motif (Liu et al., 2011), the p-T78 patch not only allows additional Plk1 molecules to be recruited to unoccupied p-T78 motifs (Fig. 3A, arrow in third panel), but also enables these molecules to maneuver around the surface of the p-T78 motif-loaded ELISA plate as they dissociate from the initial docking sites. The mode of the interaction between a Plk1 molecule and the surface of the p-T78 motif-loaded area is termed a “two-dimensional surface interaction” (Fig. 3A, a laterally moving dotted arrow in the fourth panel) to distinguish it from the stochastic biochemical process occurring between two freely moving molecules. The concept of a two-dimensional surface interaction was introduced by Dennis and his coworkers to explain the interaction mode between lipid-dependent enzymes, such as phospholipases, and their substrates on the surface of plasma membranes (Carman et al., 1995). As a result of both PBD-dependent anchored reactions on a confined surface and the ensuing two-dimensional surface interactions with the resulting p-T78 patches, Plk1 can cooperatively generate p-T78 motifs on the PBIPtide-immobilized ELISA plate (i.e., PBIP1-loaded kinetochore plate) (Fig. 3A, fifth panel). Through the repeated cycles of self-priming and binding, Plk1 can rapidly amass itself at kinetochore plates to promote M-phase progression (Park et al., 2011). Thus, as a result of self-priming and binding-induced cooperativity, the Plk1-mediated biochemical event exhibits sigmoidal kinetic properties (Fig. 3A, graph at the bottom). Notably, since the initial p-T78 product generated by unbound Plk1 serves as an anchoring site for subsequent Plk1-dependent phosphorylation onto neighboring T78 motifs, this is a “product-activated,” feed-forward mechanism designed to rapidly amplify ongoing biochemical and cellular processes.
Biochemical steps underlying non-self-priming and binding
The mechanism of non-self-priming has been the subject of previous reviews (Barr et al., 2004; Lowery et al., 2004; 2005). Unlike the self-priming and binding mechanism for wild-type PBIP1 (Fig. 3A), phosphorylation of the PBIP1-cdk mutant at the T78 residue requires another kinase, such as cyclin B-dependent Cdk1, that selectively phosphorylates a Pro-directed site (Fig. 3B). As Cdk1 dissociates from the PBIP1-cdk mutant after phosphorylating the T78 residue, a molecule of Plk1 can be recruited to the resulting p-T78 motif immobilized on the ELISA plate (Fig. 3B, white arrows in the second and fourth panels).
Since Plk1 anchored on the Cdk1-generated p-T78 motif cannot phosphorylate the Pro-directed T78 residue of neighboring PBIP1-cdk mutant molecules (i.e., no PBD-dependent anchored reaction), Plk1 fails to generate additional p-T78 motifs for subsequent Plk1 binding. Rather, it may phosphorylate other residues on PBIPtide-cdk or other proteins associated with it. These two potential downstream events, which may occur in vivo, cannot be assessed by the assay shown in Fig. 3B. Under these circumstances, mutation of the self-priming site of PBIP1 to a Cdk1-dependent, non-self-priming site prevents Plk1 from performing a “product-activated” anchored reaction and also greatly limits the ability of Plk1 to carry out two-dimensional interactions with surface-restricted p-T78 motifs. Consequently, in sharp contrast to the cooperativity observed with the formation of the Plk1-PBIP1 complex in Fig. 3A, the level of Plk1 bound to PBIPtide-cdk molecules is only linearly proportional to the level of Cdk1 activity present in the lysates (Fig. 3B, graph at the bottom).
PBD-dependent anchorage and two-dimensional surface interaction-two key elements underpinning biochemical cooperativity
It is now clear that the self-priming and binding mechanism provides superior capacity over the non-self-priming and binding mechanism in the production of Plk1 PBD-binding motifs. If both the PBD-dependent anchored reaction and the two-dimensional surface interaction proposed in the self-priming and binding mechanism are indeed critically required for the observed cooperativity in Fig. 3, then preventing either one of these processes will lead to the loss of cooperativity in generating the p-T78 motif and subsequently recruiting Plk1 to this motif.
We demonstrated that interfering with the PBD function by providing a short, competitive p-T78 peptide in solution completely eliminated the cooperativity in the Plk1-dependent biochemical process (Park et al., 2011) (Fig. 4). Therefore, without PBD-dependent anchorage, the entire Plk1-dependent PBIP-tide phosphorylation becomes only linearly proportional to the degree of stochastic interactions between the kinase domain of Plk1 and PBIPtide. In addition, Plk1 efficiently phosphorylated the PBIPtide proteins immobilized on a two-dimensional surface rather than the same PBIPtide proteins provided in a three-dimensional solution (Park et al., 2011) (Fig. 4). This result was completely unexpected because conventional enzyme-substrate reactions occur best in freely moving, three-dimensional solutions rather than under immobilized conditions on a confined surface area. Collectively, since disrupting either the PBD-dependent anchorage or the two-dimensional surface interaction is sufficient for eliminating the sigmoidal reaction kinetics observed in Fig. 3A, both of these elements may orchestrate to induce the observed cooperativity in Plk1-dependent events. Indeed, PBD-dependent anchorage is an integral part of the two-dimensional surface interaction, which in turn promotes the generation of additional PBD-anchoring sites in a positive-feedback loop (see below). Thus, PBD-dependent anchorage and the two-dimensional surface interaction are mutually required to efficiently induce Plk1-mediated biochemical processes.
Fig. 4.
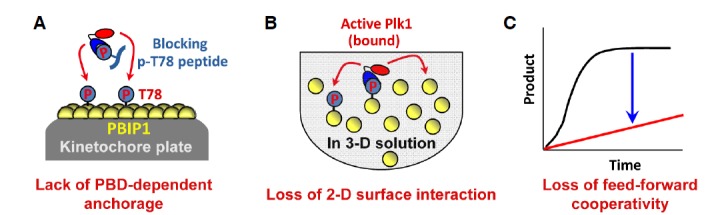
Loss of feed-forward cooperativity by either the interference of a PBD-dependent interaction or the abrogation of a two-dimentional surface interaction. (A) The provision of a PBD-binding p-T78 peptide prevents Plk1 from binding to the p-T78 motif of PBIP1 localized at the kinetochore plate. In the absence of PBD-dependent anchorage onto the p-T78 motif, Plk1 fails to efficiently phosphorylate PBIP1. (B) Under the conditions where two-dimensional surface interaction is abrogated, Plk1 is not able to take advantage of PBD-dependent anchored reactions onto neighboring substrates. Rather, it phosphorylates its substrates through a three-dimensional stochastic processes. (C) Loss of either one of the PBD-dependent anchorage and the two-dimensional surface interaction is sufficient to prevent Plk1 from phosphorylating its substrates in a cooperative manner.
PHYSIOLOGICAL SIGNIFICANCE AND EXTRA VIEWS ON SELF-PRIMING AND BINDING
Physiological significance
The self-priming and binding mechanism works in such a way that Plk1 creates a phospho-motif, which then serves as a PBD-docking site for itself or other Plk1 molecules present in proximity to the phosphomotif. The great advantage of this mechanism is that these initial events can be readily amplified through the repeated cycles of PBD-dependent anchored reactions and two-dimensional surface interactions, thus enabling Plk1 to rapidly accumulate its active forms at subcellular structures loaded with its self-primeable substrates (Fig. 3). Through the series of these events, the self-priming and binding-based biochemical cooperativity is efficiently converted into a PBD-dependent cellular process to promote Plk1-mediated mitotic events. Highlighting the importance of the self-priming and binding mechanism, the loss of the PBIP1 T78-dependent interaction with Plk1 resulted in impaired Plk1 recruitment to kinetochores, mitotic block, and chromosome missegregation, which ultimately leads to aneuploidy (Kang et al., 2006), a hallmark of cancer. Given that Plk1 is localized at distinct subcellular structures, such as centrosomes, kinetochores, and midbodies (Fig. 5), self-priming and binding could be a common mechanism that promotes Plk1 recruitment to these structures.
Fig. 5.
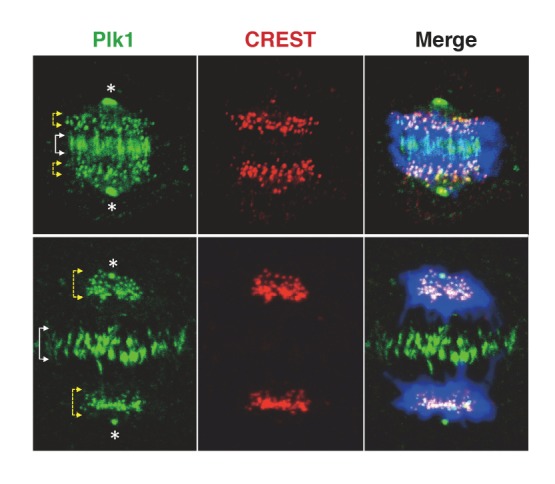
Subcellular localization of Plk1 at distinct subcellular structures. Immunostained HeLa cells show that Plk1 is abundantly localized at specific subcellular structures, such as centrosomes, kinetochores (marked with CREST), and midbodies. Like PBIP1-dependent Plk1 recruitment to kinetochores (Kang et al., 2006), whether the self-priming and binding mechanism operates to help promote Plk1 recruitment to centrosomes and midbodies remains to be further investigated. Merge, Plk1 (green) and CREST (red) signals overlaid with DAPI-stained chromosomes (blue); Asterisks, centrosomes; yellow-arrowed brac-ket, kinetochores; white-arrowed bracket, mizone.
Potential regulatory mechanisms
If the self-priming and binding mechanism serves only to recruit Plk1 to specific subcellular locations, then how can the Plk1 recruitment process be reversed? Although it may not appear at first glance that this process can be regulated, there can be a number of different ways to modulate it. One possibility is that a gradual decrease in Plk1 activity by weakening its upstream activator(s) or strengthening its negative regulator(s) would proportionally decelerate the self-recruitment process, thus leading to the downregulation of the level of Plk1 bound to its self-primed targets. In this scenario, the self-priming and binding mechanism allows not only auto-amplification but also auto-downregulation, and makes the entire process self-regulatory.
An alternative possibility is the presence of a putative phosphatase that dephosphorylates the phosphotargets that Plk1 PBD binds to. Although a phosphatase responsible for this event has yet to be identified, an action of a phosphatase will directly antagonize that of a priming kinase, and the sum of these two counteracting activities will determine the outcome of PBD-dependent activities.
Aside from the two above-mentioned possibilities that regulate the level of available phospho-epitope for PBD binding, a third possibility is to modulate the amount of a PBD-binding target localized at a confined surface area (e.g., PBIP1 localized at kinetochores). This mechanism, which could be regulated at the level of either translation or subcellular localization of a target protein, may directly alter the efficiency of the PBD-dependent anchored reaction and two-dimensional surface interaction, which are critical for the observed cooperativity in the PBD-dependent biochemical reactions.
Unique aspects of the self-priming and binding mechanism
Positive feedback is a process that results in a gain in the output through an action around a feedback loop. This is somewhat analogous to a small panic situation among a herd of cattle, which can be amplified through the repeated cycles of a number of cattle running and the overall level of panic among the herd. In biological systems, mechanisms involving a positive-feedback loop have been well appreciated as a means to amplify biochemical signals through the action of a downstream component(s) that further activates an upstream component(s) in the same pathway. In some manner, the self-priming and binding mechanism is functionally analogous to the positive-amplification loop frequently observed in various signal transduction pathways. However, it is distinct from the positive-amplification loop in that Plk1 utilizes its own reaction product (i.e., self-primed motif) to promote its downstream phosphorylation events. Therefore, self-priming and binding is a “product-activated” mechanism that promotes downstream events in a feed-forward rather than feedback fashion. In addition, since the self-priming and binding-dependent Plk1 event is a self-propelled process that is autonomously regulated by the level of Plk1 activity itself, the self-priming and binding mechanism may help Plk1 maintain its intracellular equilibrium between the localized Plk1 population at subcellular structures and the free Plk1 population in its surrounding cytosol or nucleoplasm.
PHYSIOLOGICAL SIGNIFICANCE AND EXTRA VIEWS ON NON-SELF-PRIMING AND BINDING
As explained above, the non-self-priming and binding mechanism requires the action of a priming kinase other than Plk1 to phosphorylate and generate a phospho-motif prior to PBD binding. Under this scenario, the priming kinase must be activated early enough to generate a phosphorylated motif before Plk1 PBD is available for binding. As described in Fig. 2B, Plk1 binds to hCenexin1 only after a priming kinase, Cdk1, phosphorylates and generates the p-S796 motif for PBD binding. Consistent with the importance of this event, mutation of the S796 of hCenexin1 to Ala significantly delocalizes Plk1 from centrosomes, and, as a result, impairs the recruitment of γ-tubulin to centrosomes and the formation of bipolar spindles at these structures (Soung et al., 2009). Although a physiological substrate(s) phosphorylated by hCenexin1 p-S796-anchored Plk1 is yet to be identified, these observations suggest that the hCenexin1 function is intricately regulated by two sequentially acting kinases, Cdk1 and Plk1, and that this ordered event is important for properly promoting Plk1-dependent mitotic events at this location. Through inherent nature, this mechanism ensures that Plk1 PBD-dependent biochemical and cellular processes occur only after Cdk1 completes its actions in the early steps of these events, consequently imposing a layer of regulation on PBD-dependent Plk1 functions. Since the cell cycle pathway is a unidirectional process, sequential actions of both the priming kinase and Plk1 may help make cell cycle progression an irreversible process.
CONCLUSION
Plk1 dynamically localizes to multiple subcellular structures through the interactions with a large number of proteins in a temporally and spatially regulated manner. The C-terminal noncatalytic PBD has been well characterized as a protein-protein interaction domain that specifically recognizes the p-Ser-/p-Thr-containing motif. Now, it is clear that the kinase domain of Plk1 functions in concert with its cis-acting PBD to induce biochemical cooperativity in various Plk1-mediated biochemical and cellular processes, and that PBD lies at the center of these events.
Over the years, various Plk1 PBD-binding proteins have been identified, and the physiological significance of these interactions has been characterized. Here, we discussed the two distinct modes of PBD-dependent mechanisms - self-priming and non-self-priming - that operate to promote Plk1-mediated events. Given that Plk1 mediates diverse events at the multiple stages of the cell cycle, the different modes of PBD-dependent interactions undoubtedly provide additional means for Plk1 to regulate each distinct process with finesse. Moreover, since Plk1 deregulation is thought to promote tumorigenesis (Strebhardt, 2010; Strebhardt and Ullrich, 2006; Takai et al., 2005), tight regulation of Plk1-dependent processes could be important for preventing genomic instability and cancer formation. Intriguingly, blocking PBD-dependent protein-protein interactions is sufficient to induce mitotic arrest and apoptotic cell death in cultured cancer cells (Liu et al., 2011; Yun et al., 2009), and the PBD of Plk1 is thought to be an attractive target for anti-Plk1 cancer therapy (Park et al., 2010). Therefore, future investigations of how PBD differentially mediates Plk1-dependent processes in normal and cancer cells will likely be critical to a better understanding of the effect of Plk1 deregulation in the promotion of human cancers.
Supplementary Data
Acknowledgments
We are grateful to all the past and present members of the Lee laboratory for stimulating discussions about self-priming and non-self-priming mechanisms for PBD binding. This work was supported in part by a National Cancer Institute intramural grant (K.S.L.) and Korea Basic Science Institute’s research grant T33418 (J.K.B).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Archambault V., Glover D.M. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- Arnaud L., Pines J., Nigg E.A. GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma. 1998;107:424–429. doi: 10.1007/s004120050326. [DOI] [PubMed] [Google Scholar]
- Barr F.A., Sillje H.H., Nigg E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- Baumann C., Körner R., Hofmann K., Nigg E.A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. doi: 10.1016/j.cell.2006.11.041. [DOI] [PubMed] [Google Scholar]
- Carman G.M., Deems R.A., Dennis E.A. Lipid signaling enzymes and surface dilution kinetics. J. Biol. Chem. 1995;270:18711–18714. doi: 10.1074/jbc.270.32.18711. [DOI] [PubMed] [Google Scholar]
- Cheng K.Y., Lowe E.D., Sinclair J., Nigg E.A., Johnson L.N. The crystal structure of the human polo-like kinase-1 polo box domain and its phospho-peptide complex. EMBO J. 2003;22:5757–5768. doi: 10.1093/emboj/cdg558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay F.J., McEwen S.J., Bertoncello I., Wilks A.F., Dunn A.R. Identification and cloning of a protein kinase-encoding mouse gene, Plk, related to the polo gene of Drosophila. Proc. Natl. Acad. Sci. USA. 1993;90:4882–4886. doi: 10.1073/pnas.90.11.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cárcer G., Escobar B., Higuero A.M., García L., Ansón A., Pérez G., Mollejo M., Manning G., Meléndez B., Abad-Rodríguez J., et al. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol. Cell. Biol. 2011a;31:1225–1239. doi: 10.1128/MCB.00607-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cárcer G., Manning G., Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle. 2011b;10:2255–2262. doi: 10.4161/cc.10.14.16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A.E., Cantley L.C., Yaffe M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003a;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- Elia A.E., Rellos P., Haire L.F., Chao J.W., Ivins F.J., Hoepker K., Mohammad D., Cantley L.C., Smerdon S.J., Yaffe M.B. The molecular basis for phospho-dependent substrate targeting and regulation of Plks by the polo-box domain. Cell. 2003b;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- Elowe S., Hümmer S., Uldschmid A., Li X., Nigg E.A. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007;21:2205–2219. doi: 10.1101/gad.436007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Malureanu L., Huang J., Wang W., Li H., van Deursen J.M., Tindal D.J., Chen J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008;10:1076–1082. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alvarez B., de Cárcer G., Ibañez S., Bragado-Nilsson E., Montoya G. Molecular and structural basis of polo-like kinase 1 substrate recognition: implications in centrosomal localization. Proc. Natl. Acad. Sci. USA. 2007;104:3107–3112. doi: 10.1073/pnas.0609131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golsteyn R.M., Mundt K.E., Fry A.M., Nigg E.A. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J. Cell Biol. 1995;129:1617–1628. doi: 10.1083/jcb.129.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch A., Wehner A., Nigg E.A., Sillje H.H. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol. Biol. Cell. 2006;17:448–459. doi: 10.1091/mbc.E05-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanissian S.H., Akbar U., Teng B., Janjetovic Z., Hoffmann A., Hitzler J.K., Iscove N., Hamre K., Du X., Tong Y., et al. cDNA cloning and characterization of a novel gene encoding the MLF1-interacting protein MLF1IP. Oncogene. 2004;23:3700–3707. doi: 10.1038/sj.onc.1207448. [DOI] [PubMed] [Google Scholar]
- Jang Y.J., Lin C.Y., Ma S., Erikson R.L. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl. Acad. Sci. USA. 2002;99:1984–1989. doi: 10.1073/pnas.042689299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johmura Y., Soung N.K., Park J.E., Yu L.R., Zhou M., Bang J.K., Kim B.Y., Veenstra T.D., Erikson R.L., Lee K.S. Regulation of microtubule-based microtubule nucleation by mammalian polo-like kinase 1. Proc. Natl. Acad. Sci. USA. 2011;108:11446–11451. doi: 10.1073/pnas.1106223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.H., Park J.-E., Yu L.-R., Soung N.-K., Yun S.-M., Bang J.K., Seong Y.S., Yu H., Veenstra T.D., Lee K.S. Self-regulation of Plk1 recruitment to the kinetochores is critical for chromosome congression and spindle checkpoint signaling. Mol. Cell. 2006;24:409–422. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Lee K.S., Erikson R.L. Plk is a functional homolog of Saccharomyces cerevisiae Cdc5, and elevated Plk activity induces multiple septation structures. Mol. Cell. Biol. 1997;17:3408–3417. doi: 10.1128/mcb.17.6.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S., Yuan Y.-L., Kuriyama R., Erikson R.L. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol. Cell. Biol. 1995;15:7143–7151. doi: 10.1128/mcb.15.12.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S., Grenfell T.Z., Yarm F.R., Erikson R.L. Mutation of the polo-box disrupts localization and mitotic functions of the mammalian polo kinase Plk. Proc. Natl. Acad. Sci. USA. 1998;95:9301–9306. doi: 10.1073/pnas.95.16.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S., Park J.E., Kang Y.H., Zimmerman W., Soung N.K., Seong Y.S., Kwak S.J., Erikson R.L. Mechanisms of mammalian polo-like kinase 1 (Plk1) localization: Self- versus non-self-priming. Cell Cycle. 2008;7:141–145. doi: 10.4161/cc.7.2.5272. [DOI] [PubMed] [Google Scholar]
- Lenart P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J.J., Hoffmann M., Rettig W.J., Kraut N., Peters J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Leung G.C., Hudson J.W., Kozarova A., Davidson A., Dennis J.W., Sicheri F. The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat. Struct. Biol. 2002;9:719–724. doi: 10.1038/nsb848. [DOI] [PubMed] [Google Scholar]
- Liu F., Park J.E., Qian W.J., Lim D., Gräber M., Berg T., Yaffe M.B., Lee K.S., Burke T.R.J. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery D.M., Mohammad D.H., Elia A.E., Yaffe M.B. The Polo-box domain: a molecular integrator of mitotic kinase cascades and Polo-like kinase function. Cell Cycle. 2004;3:128–131. [PubMed] [Google Scholar]
- Lowery D.M., Lim D., Yaffe M.B. Structure and function of polo-like kinases. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- Minoshima Y., Hori T., Okada M., Kimura H., Haraguchi T., Hiraoka Y., Bao Y.C., Kawashima T., Kitamura T., Fukagawa T. The constitutive centromere component CENP-50 is required for recovery from spindle damage. Mol. Cell. Biol. 2005;25:10315–10328. doi: 10.1128/MCB.25.23.10315-10328.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef R., Preisinger C., Sutcliffe J., Kopajtich R., Nigg E.A., Mayer T.U., Barr F.A. Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J. Cell Biol. 2003;162:863–875. doi: 10.1083/jcb.200306009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef R., Gruneberg U., Kopajtich R., Li X., Nigg E.A., Sillje H., Barr F.A. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat. Cell. Biol. 2007;9:436–444. doi: 10.1038/ncb1557. [DOI] [PubMed] [Google Scholar]
- Pan H.Y., Zhang Y.J., Wang X.P., Deng J.H., Zhou F.C., Gao S.J. Identification of a novel cellular transcriptional repressor interacting with the latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 2003;77:9758–9768. doi: 10.1128/JVI.77.18.9758-9768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-E., Li L., Park J., Knecht R., Strebhardt K., Yuspa S.H., Lee K.S. Direct quantification of polo-like kinase 1 activity in cells and tissues using a highly sensitive and specific ELISA assay. Proc. Natl. Acad. Sci. USA. 2009;106:1725–1730. doi: 10.1073/pnas.0812135106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.E., Soung N.K., Johmura Y., Kang Y.H., Liao C., Lee K.H., Park C.H., Nicklaus M.C., Lee K.S. Polo-box domain: a versatile mediator of polo-like kinase function. Cell. Mol. Life Sci. 2010;67:1957–1970. doi: 10.1007/s00018-010-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.E., Erikson R.L., Lee K.S. Feed-forward mechanism of converting biochemical cooperativity to mitotic processes at the kinetochore plate. Proc. Natl. Acad. Sci. USA. 2011;108:8200–8205. doi: 10.1073/pnas.1102020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W., Tang Z., Yu H. Phosphorylation- and Polo-Box-dependent Binding of Plk1 to Bub1 Is Required for the Kinetochore Localization of Plk1. Mol. Biol. Cell. 2006;17:3705–3716. doi: 10.1091/mbc.E06-03-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong Y.S., Kamijo K., Lee J.S., Fernandez E., Kuriyama R., Miki T., Lee K.S. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem. 2002;277:32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- Slevin L.K., Nye J., Pinkerton D.C., Buster D.W., Rogers G.C., Slep K.C. The structure of the plk4 cryptic polo box reveals two tandem polo boxes required for centriole duplication. Structure. 2012;20:1905–1917. doi: 10.1016/j.str.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung N.K., Kang Y.H., Kim K., Kamijo K., Yoon H., Seong Y.S., Kuo Y.L., Miki T., Kim S.R., Kuriyama R., et al. Requirement of hCenexin for proper mitotic functions of polo-like kinase 1 at the centrosomes. Mol. Cell. Biol. 2006;26:8316–8335. doi: 10.1128/MCB.00671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soung N.K., Park J.E., Yu L.R., Lee K.H., Lee J.M., Bang J.K., Veenstra T.D., Rhee K., Lee K.S. Plk1-dependent and -independent roles of an ODF2 splice variant, hCenexin1, at the centrosome of somatic cells. Dev. Cell. 2009;16:539–550. doi: 10.1016/j.devcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- Strebhardt K., Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- Takai N., Hamanaka R., Yoshimatsu J., Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- Takaki T., Trenz K., Costanzo V., Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr. Opin. Cell Biol. 2008;20:650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- van de Weerdt B.C., Medema R.H. Polo-like kinases: a team in control of the division. Cell Cycle. 2006;5:853–864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- Watanabe N., Arai H., Nishihara Y., Taniguchi M., Watanabe N., Hunter T., Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Shen C., Wang T., Quan J. Structural basis for the inhibition of Polo-like kinase 1. Nat. Struct. Mol. Biol. 2013;20:1047–1053. doi: 10.1038/nsmb.2623. [DOI] [PubMed] [Google Scholar]
- Yun S.M., Moulaei T., Lim D., Bang J.K., Park J.E., Shenoy S.R., Liu F., Kang Y.H., Liao C., Soung N.K., et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Chen Q., Feng J., Hou J., Yang F., Liu J., Jiang Q., Zhang C. Sequential phosphorylation of Nedd1 by Cdk1 and Plk1 is required for targeting of the gammaTuRC to the centrosome. J. Cell Sci. 2009;122:2240–2251. doi: 10.1242/jcs.042747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.