

Abstract
Mitigating secondary delayed neuronal injury has been a therapeutic strategy for minimizing neurological symptoms after several types of brain injury. Interestingly, secondary neuronal loss appeared to be closely related to functional loss and/or death of astrocytes. In the brain damage induced by agonists of two glutamate receptors, N-ethyl-D-aspartic acid (NMDA) and kainic acid (KA), NMDA induced neuronal death within 3 h, but did not increase further thereafter. However, in the KA-injected brain, neuronal death was not obviously detectable even at injection sites at 3 h, but extensively increased to encompass the entire hemisphere at 7 days. Brain inflammation, a possible cause of secondary neuronal damage, showed little differences between the two models. Importantly, however, astrocyte behavior was completely different. In the NMDA-injected cortex, the loss of glial fibrillary acidic protein-expressing (GFAP+) astrocytes was confined to the injection site until 7 days after the injection, and astrocytes around the damage sites showed extensive gliosis and appeared to isolate the damage sites. In contrast, in the KA-injected brain, GFAP+ astrocytes, like neurons, slowly, but progressively, disappeared across the entire hemisphere. Other markers of astrocytes, including S100β, glutamate transporter EAAT2, the potassium channel Kir4.1 and glutamine synthase, showed patterns similar to that of GFAP in both NMDA- and KA-injected cortexes. More importantly, astrocyte disappearance and/or functional loss preceded neuronal death in the KA-injected brain. Taken together, these results suggest that loss of astrocyte support to neurons may be a critical cause of delayed neuronal death in the injured brain.
Keywords: astrogliosis, brain injury, delayed neuronal death
INTRODUCTION
In traumatic and ischemic injury, neurons die acutely in the injury core, and slowly in the penumbra. Thus, slowly occurring, delayed neuronal death has been a therapeutic target for reducing overall neuronal injury. Brain inflammation has long been considered to cause delayed neuronal death. In culture, neurons die in the presence of microglia (brain macrophages) challenged with inflammatory stimulators, such as lipopolysaccharide (LPS), interferon-γ and β-amyloid; similar results have been reported in the ischemic brain (Chao et al., 1992; Giulian et al., 1993; Kaul et al., 2004; Ryu et al., 2004; Scali et al., 2000). Among the potential toxic inflammatory mediators suggested to be derived from microglia are nitric oxide (NO), tumor necrosis factor (TNF)-α, and cyclooxygenase (COX) products (Chao et al., 1992; Giulian et al., 1993; Kaul et al., 2004; Ryu et al., 2004; Scali et al., 2000). However, recently, we and others have reported that inflammatory responses in the injured brain and spinal cord are not neurotoxic; instead, brain inflammation is neuroprotective and functions to repair the injured brain (Howe and Barres, 2012; Jeong et al., 2010; 2013a; 2013b; Min et al., 2012; Streit, 2005; Vinet et al., 2012). Therefore, there may be another mechanism responsible for causing secondary neuronal death.
Astrocytes, which constitute the majority of cells in the brain, support the well-being and function of neurons. Astrocytes regulate the microenvironment of the brain. For example, excitatory amino acid transporter (EAAT)-1/2 and the inward rectifying potassium channel Kir4.1 maintain extracellular homeostasis through uptake of glutamate and potassium ions, respectively (Olsen et al., 2006; Rothstein et al., 1996). Aquaporin-4 (AQP4) expressed in astrocytes regulates extracellular water content (Simard and Nedergaard, 2004). Astrocytes also provide neurons with glucose and neurotrophic factors, and protect neurons from oxidative stress (Badaut et al., 2002; Chih and Roberts Jr, 2003; Raps et al., 1989; Tsacopoulos and Magistretti, 1996). It has been reported that selective ablation of reactive astrocytes exacerbates traumatic neuronal damage, and that transplantation of astrocytes diminishes brain damage (Ermakova et al., 2005; Myer et al., 2006). Loss of EAAT2/GLT-1 has been found in human amyotrophic lateral sclerosis (ALS) (Rothstein, 1995; Rothstein et al., 1992), and brain damage due to ischemia is aggravated in AQP4-deficient mice (Haj-Yasein et al., 2012; Shi et al., 2012; Zeng et al., 2012). Recently, we reported that the delayed neuronal death in contusion-induced spinal cord injury is spatially and temporally correlated with astrocyte death, suggesting that loss of astrocyte function is a critical cause of neuronal death (Min et al., 2012).
In this study, we analyzed astrocyte behavior and neuronal death in kainic acid (KA)- and N-ethyl-D-aspartic acid (NMDA)-induced injury models. Interestingly, the former induced dramatic secondary neuronal death whereas the latter did not, despite a similar inflammatory response in the two models. These results suggest that there is little relationship between brain inflammation and secondary neuronal death. Importantly, loss of astrocyte support appeared to cause secondary neuronal death. These results suggest that protection of astrocytes and/or maintenance of astrocyte function could be new therapeutic strategies for protecting neurons from injury.
MATERIALS AND METHODS
Stereotaxic surgery and drug injection
Male Sprague-Dawley rats (230–250 g, 7 week old) were anesthetized by injection of chloral hydrate (360 mg/kg, i.p.) and positioned in a stereotaxic apparatus (David Kopf Instruments, USA). KA (20 nmole) and NMDA (100 nmole) in 2 μl sterile phosphate-buffered saline (PBS), were unilaterally administered into the cortex (AP, +0.7 mm; ML, −2.0 mm; DV, −2.0 mm from bregma), according to the atlas of Paxinos and Watson (Paxinos and Watson, 2005). All drugs were purchased from Sigma (USA) and were prepared in PBS, according to the manufacturers’ instructions. All animals were injected using a Hamilton syringe equipped with a 30-gauge blunt needle attached to a syringe pump (KD Scientific, USA). Drugs were infused at a rate of 0.4 μl/min. After injection, the needle was held in place for an additional 5 min before removal. Contralateral sides and PBS-injected cortexes were used as controls. We collected data from at least 5 animals for each insult (PBS, NMDA, and KA) and each time point (3 h–7 days) after the first author of this study had been trained to obtain consistent damage areas in consistent regions with each drug.
Tissue preparation
For immunohistochemistry, rats were anesthetized and transcardially perfused with saline solution containing 0.5% sodium nitrate and heparin (10 U/ml), followed by 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for tissue fixation. Brains were separated and post-fixed overnight at 4°C in 4% paraformaldehyde. Fixed brains were stored at 4°C in a 30% sucrose solution until they sank. Six separate series of 30-μm coronal brain sections were obtained using a sliding microtome (Microm, Germany) and stored in antifreeze stock solution (phosphate buffer pH 7.2 containing 30% glycerol, 30% ethylene glycol) at 4°C before use.
For RNA preparation, rats were anesthetized and transcardially perfused with saline solution only for 1 min. Brains were removed and sliced with a Rat Brain Slicer Matrix (1.0 mm slice intervals, RBM-4000C; ASI Instruments, USA) and a razor blade. A slice that included the needle injection spot was selected, and tissue blocks (2 × 2 × 2 mm) surrounding the needle track were collected and stored at −70°C until use.
Immunohistochemistry
For 3, 3′-diaminobenzidine (DAB) staining, every sixth serial section was selected, rinsed three times with PBS, treated with 3% H2O2 for 5 min, and rinsed with PBS containing 0.2% Triton X-100 (PBST). Non-specific binding was blocked with 1% bovine serum albumen (BSA) in PBST. Sections were incubated over night at room temperature with primary antibodies. Information on the primary antibodies used is shown in Table 1. After rinsing in PBST, sections were incubated with biotinylated secondary antibodies (Vector Laboratories, USA) for 1 h and the avidin/biotin system (Vector Laboratories) for 1 h, and then visualized using a DAB solution (0.05% DAB and 0.003% hydrogen peroxide in 0.1 M phosphate buffer). For double staining of the neuronal nuclear marker NeuN and glial fibrillary acidic protein (GFAP), sections were stained with anti-NeuN antibodies and visualized using DAB (brown products). The same sections were then washed with PBS, treated with 1% BSA, incubated with anti-GFAP antibodies, and visualized using DAB and nickel sulfate (dark purple products). After staining, sections were mounted on gelatin-coated slides and examined under a bright field microscope (Olympus Optical, BX51, Japan). Bright field images were obtained using PictureFrame Application 2.3 software.
Table 1.
Information for primary antibodies used in immunohistochemistry
| Protein | Dilution | Company |
|---|---|---|
| EAAT2 | 1:500 | Abcam |
| GFAP | 1:700 | Sigma |
| GS | 1:500 | BD Biosciences |
| Iba-1 | 1:1000 | Wako |
| iNOS | 1:200 | Upstate Biotechnology |
| Kir4.1 | 1:800 | Alomone |
| MPO | 1:1000 | Dako |
| NeuN | 1:300 | Millipore |
| S100β | 1:500 | Abcam |
| S100β | 1:800 | Swant |
For immunofluorescence staining, sections were washed twice in PBS, treated with 1% BSA, and incubated with combinations of primary antibodies. For visualization, Alexa Fluor 488- or Alexa Fluor 555-conjugated secondary antibodies (1:600 dilution; Invitrogen, USA) were used. Counterstaining with 4′,6-diamidino-2-phenylindol (DAPI; Vector Laboratories, USA) was used to detect nuclei. Sections were analyzed under a confocal microscope (Carl Zeiss, Germany) with 40× water-and 63× oil-immersion objectives at 20°C. Images were captured using Confocal software (LSM Image Browser).
Quantitative real-time polymerase chain reaction (Q-PCR)
Total RNA was isolated using an easy-BLUE RNA Extraction Kit (iNtRON, Korea), and cDNA was prepared using Super-Script III Reverse (Invitrogen, USA), according to the manufacturers’ instructions. For Q-PCR, approximately 50 ng cDNA was analyzed using a KAPA SYBR FAST qPCR Kit (KAPA Biosystems, USA) and a Corbett Rotor-Gene 6000 real-time rotary analyzer (Corbett Research, Australia). Specific primers for interleukin (IL)-1β, inducible nitric oxide synthase (iNOS), TNF-α, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) used in Q-PCR are shown in Table 2. Q-PCR conditions were as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 3 s (denaturation), 55°C for 20 s (annealing), and 72°C for 3 s (elongation). That a single product was amplified under the conditions used was confirmed by performing a melting curve analysis for each primer pair using a melt ramp of 72–95°C and raising the temperature by 1°C at each step (5 s/step). Amplified products were also verified by electrophoresis on 1.5% agarose gels with GelRed (Biotium, USA) staining. Results were normalized to GAPDH, used as a reference.
Table 2.
List of primer sequences for Q-PCR
| Gene | Sequence (5′-3′) |
|---|---|
| IL-1β | F: TGATGTTCCCATTAGACAGC R: GAGGTGCTGATGTACCAGTT |
| TNF-α | F: GTAGCCCACGTCGTAGCAAA R: CCCTTCTCCAGCTGGGAGAC |
| iNOS | F: GCAGAATGTGACCATCATGG R: ACAACCTTGGTGTTGAAGGC |
| GAPDH | F: TCCCTCAAGATTGTCAGCAA R: AGATCCACAACGGATACATT |
F, Forward primer; R, Reverse primer
Image analysis
Photographs of the most damaged sections were taken and analyzed using Image J. At least 3–5 different animals for each experimental conditions were quantified.
Statistical analysis
All values are expressed as means ± SEMs. The statistical significance of differences between mean values was assessed by Student’s t-test using the Statistical Package for Social Sciences 8.0 (SPSS Inc., USA). All data presented in this study are representative of at least three independent experiments.
RESULTS
Neurons underwent extensive delayed cell death in the KA-injected cortex, but not in the NMDA-injected cortex
Previously, we found that in contusion-induced spinal cord injury, astrocytes play a critical role in the secondary injury (Min et al., 2012). Here, we investigated neuronal death in two brain injury models-KA- and NMDA-induced injury-based on the idea that astrocytes are vulnerable to KA but not to NMDA (David et al., 1996).
Interestingly, in the NMDA-injected cortex, local NeuN+ neuronal loss was detected at 3 h in the injection site, and the damaged area did not increase further up to 7 days (Figs. 1B and 1D). We designated the area in which NeuN+ neurons disappeared at 3 h and 7 days as the ‘a’ region, and the area outside the ‘a’ region in the lateral direction as the ‘b’ region (Fig. 1A). We also examined neuronal behavior in the KA-injected cortex. NeuN+ neurons were still present, although not healthy, in the injection site at 3 h, but disappeared throughout the entire hemisphere at 7 days (Figs. 1C and 1D), indicating that KA injection induced extensive delayed neuronal death. In the PBS-injected brain, NeuN+ neurons were as healthy as those in the intact brain (Fig. 1A vs. Fig. 1C). These results demonstrate the presence and absence of delayed neuronal death in the KA- and NMDA-injected cortex, respectively.
Fig. 1.
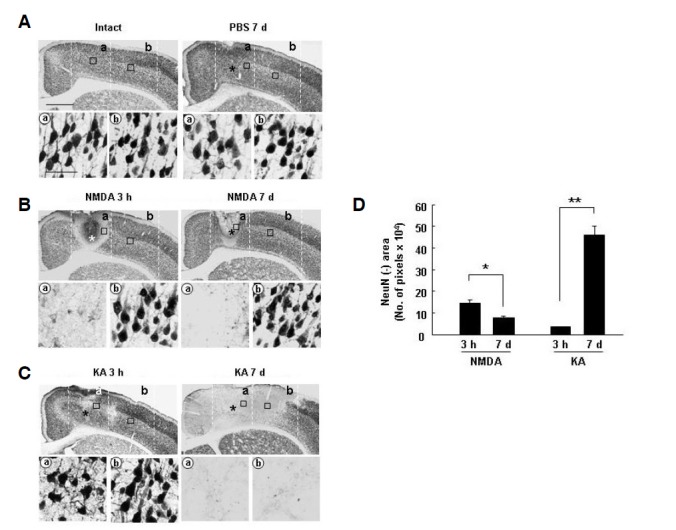
Neurons underwent extensive cell death in the KA-injected cortex, but not in the NMDA-injected cortex. (A, B, and C) Cortical brain sections (30 μm thickness) were obtained at the indicated times after injection of 100 nmole NMDA (in 2 μl PBS), 20 nmole KA, or PBS, and every sixth serial section was selected, labeled with NeuN antibodies, and visualized with peroxidase-conjugated secondary antibodies. Photographs of the most damaged sections were taken. The number of animals used in this study was at least 5 for each drug (PBS, NMDA, and KA) and each time point. (A, B, and C) Lower panels are higher magnification images of boxed areas in regions ‘a’ (core) and ‘b’ (penumbra) in upper panels. NeuN+ neuronal loss was detected in only the core region at 3 h to 7 days in the NMDA-injected cortex (B), but in both core and penumbra regions in the KA-injected cortex (C). Asterisks (*) indicate drug injection sites. Scale bars = 1 mm (upper panels) and 50 μm (lower panels). (D) Using Image J, NeuN-negative areas in the most damaged sections from at least 3 different animals were quantified at the indicated times. Values are means ± SEMs of more than three samples (*p < 0.01; **p < 0.001).
Inflammatory responses occurred similarly in both NMDA-and KA-injected cortexes
It has been suggested that brain inflammation that accompanies brain injury is harmful to neurons (Chao et al., 1992; Giulian et al., 1993; Kaul et al., 2004; Ryu et al., 2004; Scali et al., 2000). Thus, we analyzed where inflammatory responses in NMDA- and KA- injected cortex occurred in relation to the presence and absence of progressive delayed neuronal death using the marker protein Iba-1 to identify resident microglia and blood-derived macrophages (Jeong et al., 2010; 2013b; Min et al., 2012). Iba-1+ ramified resident microglia appeared to be dead or undergoing the process of dying at 3 h in both NMDA- and KA-injection cores (Fig. 2A, 3 h, region ‘a’). However, at 7 days, NMDA and KA injection produced dramatically different results. In the NMDA model, Iba-1+ round cells appeared in the injection core (Fig. 2A, NMDA 7 days, region ‘a’), but Iba-1+ ramified cells were in the penumbra region (Fig. 2A, NMDA 7 days, region ‘b’). However, in the KA model, Iba-1+ round cells filled the entire hemisphere (Fig. 2A, KA 7 days, regions ‘a, b’). Previously, we reported that Iba-1+ round cells are monocytes infiltrated from blood to repair damaged brain (Jeong et al., 2010; 2013b; Ji et al., 2007; Min et al., 2012). In the PBS-injected brain, Iba-1+ ramified resident microglia showed a morphology similar morphology to those in the intact brain (Fig. 2A). Behavior of Iba-1+ cells in intact, NMDA-, KA, and PBS-injected brain at indicated times were quantified by measuring intensities of Iba-1 using Image J (Fig. 2B). We measured mRNA expression of TNF-α and IL-1β increased in NMDA- and KA-models. Quantitative PCR analyses showed little difference in the inflammatory responses in the injury core (Fig. 2C). mRNA expression of TNF-α and IL-1β increased at 3–12 h, and decreased at 1 day in both NMDA- and KA-injected cortexes compared to the PBS-injected cortex (Fig. 2C). The mRNA levels of TNF-α and IL-1β were little different in the NMDA-, and KA-injected brain (Fig. 2C), although these differences did not reach statistical significance. mRNA expression of iNOS increased to similar levels at 12 h to 1 day in both NMDA- and KA-injected cortexes (Fig. 2C), suggesting that iNOS does not contribute to delayed neuronal death in the KA model. We also examined infiltration of myeloperoxidase-positive (MPO+) neutrophils, which could produce cytotoxic inflammatory mediators (Ji et al., 2007; 2008). At 1 day, MPO+ neutrophils were readily detectable in both core and penumbra regions of the KA-injected cortex, whereas much fewer neutrophils were detectable in only core regions in the NMDA-injected cortex (arrows in Fig. 2D). Some neutrophils also expressed iNOS in both NMDA and KA models (arrows in Fig. 2D). Relative levels of MPO and iNOS in intact, NMDA-, KA, and PBS-injected brain at 1 day after injection were quantified using Image J (Fig. 2D). Although the number of MPO+ neutrophils in area ‘b’ slightly higher in KA-injected brain than in PBS-, and NMDA-injected brain (Fig. 2D), neutrophils may not kill neurons in the KA model since LPS recruited many more neutrophils, but barely damaged neurons (Supplementary Fig. 1), as we previously reported (Ji et al., 2007; 2008). Furthermore, iNOS levels were not different in PBS-, NMDA-, and KA-injected brain (Fig. 2D). Therefore, neutrophils are not likely the cause of delayed neuronal death in the KA model. Taken together, these results suggest that there was little difference in inflammatory responses between KA- and NMDA-injected cortexes although the territories of microglial cell death and monocytes infiltration were different.
Fig. 2.
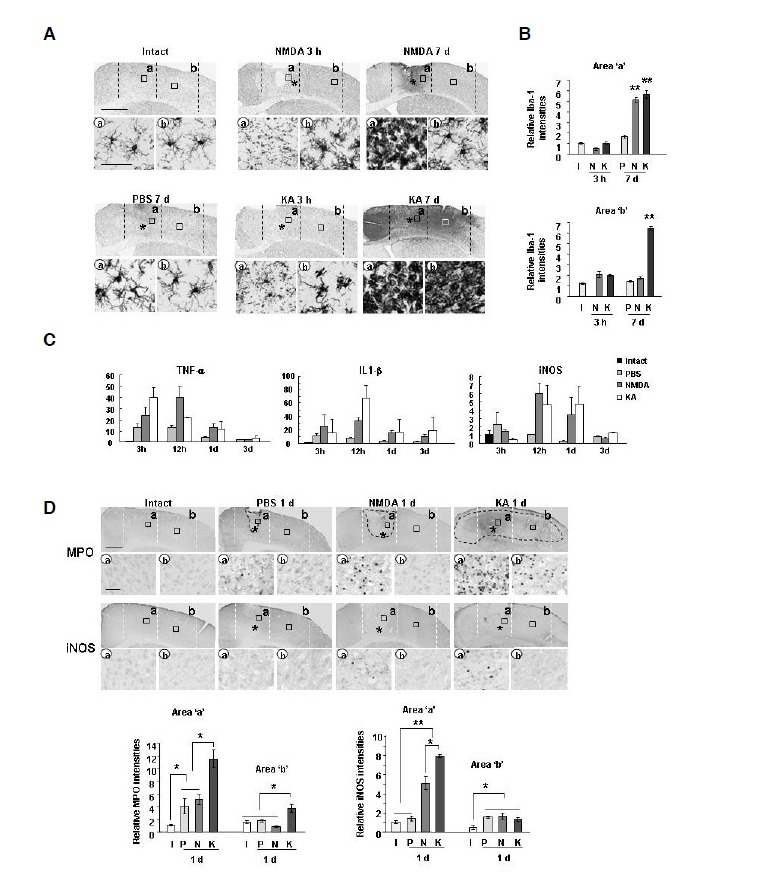
Similar inflammatory responses occurred in NMDA- and KA-injected brains. (A, D) Sections were obtained from intact brain (I) or at the indicated times after NMDA (N), KA (K), or PBS (P) injection were labeled with primary antibodies against Iba-1 (A), MPO (D), or iNOS (D) and visualized using peroxidase-conjugated secondary antibodies. Photographs of the most damaged sections were taken. Lower panels are higher magnification images of boxed areas in upper panels. Dotted lines indicate neuron damage areas. *, drug injection sites. (B) Relative Iba-1 intensities were measured using Image J as described in Fig. 1. (C) Quantitative analysis of mRNA expression of inflammatory mediators. At the indicated times after NMDA, KA, or PBS injection, tissue blocks were obtained and TNF-α, IL-1β, and iNOS mRNA levels were analyzed using Q-PCR. Values (B, C, and D) are means ± SEMs of more than three samples (*p < 0.01; **p < 0.001). Scale bars = 1 mm [upper panels in (A, D)] and 50 μm [lower panels in (A, D)].
Fig. 3.
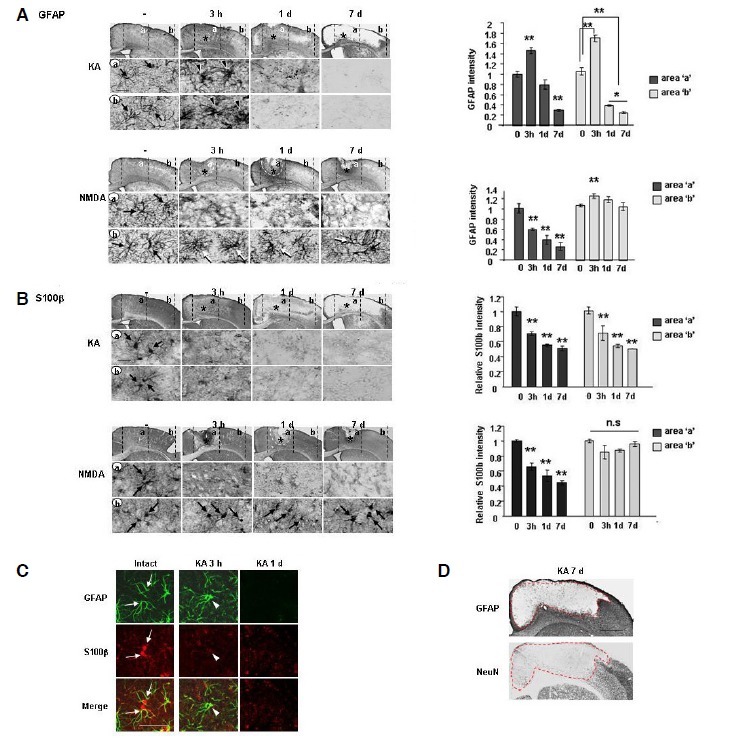
Extensive loss of GFAP+- and S100β+-astrocyte occurred in the KA-injected brain but not in the NMDA-injected brain. (A, B) Sections were obtained at the indicated times after NMDA or KA injection, labeled with primary antibodies against GFAP (A) or S100β (B), and visualized using peroxidase-conjugated secondary antibodies. *, drug injection sites. Photographs of the most damaged sections were taken. Relative intensities of GFAP (A) and S100β (B) were measured using Image J and plotted. (A) In the KA-injected brain, GFAP+ astrocytes became swollen at 3 h (arrowheads) compared to intact GFAP+ astrocytes (arrows), and disappeared at 1 day. In the NMDA-injected brain, GFAP+ astrocytes disappeared in the core region at 3 h and 1 day, but were healthy in the penumbra region for up to 7 days (arrows). Dotted lines in the NMDA model at 3 h and 1 day indicate neuron damage areas. (B) S100β+ cells rapidly disappeared (3 h) and did not reappear in both KA-and NMDA-injection cores. However, in the penumbra region, S100β+ cells were not detected in the KA model, but in the NMDA model, they were as healthy as they were in the intact brain (black arrows). (C) Brain sections obtained at 3 h and 1 day were double labeled with GFAP and S100β antibodies, and visualized using Alexa Fluor 488- and 555-conjugated antibodies, respectively. In the intact cortex, GFAP+ cells were co-labeled with S100β (white arrows). S100β immunoreactivity disappeared at 3 h, which was earlier than the disappearance of GFAP+ immunoreactivity (white arrowheads). (D) Astrocyte- and neuron-negative areas were compared by staining immediately adjacent serial sections with GFAP and NeuN antibodies. Dotted lines indicate the borders where GFAP+ and NeuN+ cells were absent. Scale bars = 1 mm [upper panels in (A, D)], 50 μm (C), and 20 μm [lower panels in (A)]. Values are means ± SEMs of more than three samples (*p < 0.01; **p < 0.001). n.s, not significant.
Severe astrocyte loss occurred in the KA-injected cortex
Next, we examined whether astrocyte behavior was related to delayed neuronal death based on the hypothesis that neurons may not survive without the support of astrocytes, which maintain homeostasis of the brain microenvironment and supply nutrients and growth factors to neurons (Badaut et al., 2002; Chih and Roberts Jr, 2003; Müller and Seifert, 1982; Olsen et al., 2006; Raps et al., 1989; Rothstein et al., 1996; Simard and Nedergaard, 2004; Tsacopoulos and Magistretti, 1996). Interestingly, the behavior of astrocytes demonstrated by several markers of astrocytes including GFAP, S100β, glutamate transporters (EAAT2; GLT-1), potassium channels (Kir4.1), and glutamate synthetase (GS) was different in NMDA and KA models (Figs. 3 and 4). GFAP+ astrocytes became swollen in the core and penumbra in the KA-injected brain at 3 h (black arrowheads in Fig. 3A, regions ‘a, b’) compared with astrocytes in intact (black arrows in Fig. 3A, regions ‘a, b’) and PBS-injected (Supplementary Fig. 2) brains. In the KA-injected brain, GFAP+ astrocytes eventually disappeared starting at 1 day, and the GFAP+ astrocyte-absent area increased thereafter up to 7 days (Fig. 3A). However, in the NMDA-injected cortex, GFAP+ astrocytes were disappeared in the injection site at 3 h (Fig. 3A, region ‘a’), but did not further disappeared between 1 and 7 days (Fig. 3A, white dotted lines in region ‘a’ and white arrows in region ‘b’). GFAP intensities in regions ‘a’ and ‘b’ in KA- and NMDA-injected brain at the indicated times were quantified (Fig. 3A). It is noticeable that KA significantly decreased GFAP intensities in region ‘b’ at 1 and 7 days after the injection but NMDA did not (Fig. 3A).
Fig. 4.
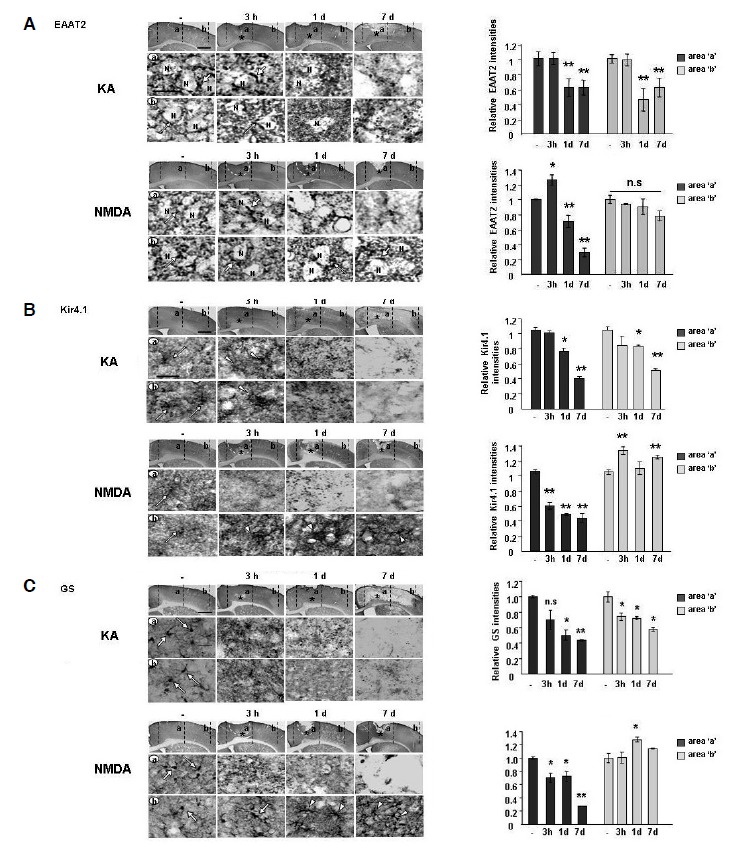
Astrocyte functional loss was severe in the KA-injected brain but not in the NMDA-injected brain. Sections obtained at the indicated times after NMDA or KA injection were labeled with primary antibodies against EAAT2 (A), Kir4.1 (B) or GS (C), and visualized using peroxidase-conjugated secondary antibodies. Relative intensities of EAAT2 (A), Kir4.1 (B) or GS (C) were measured using Image J and plotted. Lower panels are higher magnification images of upper panels. EAAT2 (A), Kir4.1 (B) and GS (C) all showed similar patterns, although EAAT2 was detected in the astrocyte processes (white arrows in A) that surround neuronal cell bodies (N), and Kir4.1 and GS were detected in the astrocyte cell bodies [white arrows and (B, C), respectively]. In the core region, EAAT2 (A), Kir4.1 (B) and GS (C) entirely disappeared from 3 h and/or 1 d after injection in both KA- and NMDA-injected brains. In the penumbra region, all three functional markers disappeared in the KA-injected brain beginning after 3 h and/or 1 day, but were unchanged [EAAT2, white arrows in (A)] or slightly increased [Kir4.1 and GS, white arrowheads in (B, C)] in the NMDA-injected brain. Scale bars = 1 mm (upper panels) and 20 μm (lower panels). Values are means ± SEMs of more than three samples (*p < 0.01; **p < 0.001). n.s, not significant.
We obtained similar results using S100β, another marker of astrocytes. S100β+ astrocytes disappeared in the KA-injected hemisphere at 1–7 days (Fig. 3B). Interestingly, S100β immunoreactivity rapidly decreased prior to GFAP immunoreactivity (Fig. 3A, KA, 3 h vs. Fig. 3B, KA, 3 h). Double labeling using anti-GAF and anti-S100β antibodies revealed that intact astrocytes co-expressed GFAP and S100β (white arrows in Fig. 3C), whereas GFAP+ reactive astrocytes in the KA-injected cortex lost S100β immunoreactivity at 3 h (white arrowheads in Fig. 3C). At 1 day, neither GFAP nor S100β immunoreactivity was detectable (Fig. 3C). In the NMDA-injected cortex, S100β+ astrocytes disappeared in the injection site (Fig. 3B, region ‘a’), but remained in the peripheral region (black arrows in Fig. 3B, region ‘b’). In the PBS-injected brain, GFAP+ astrocytes exhibited a reactive morphology in the injection site, but showed no evidence of death (Supplementary Fig. 2). Most importantly, in the KA-injected cortex, the GFAP+ astrocyte-absent area exactly matched with that of delayed neuronal death (Fig. 3D). These results suggest that loss of astrocyte support may be closely related to delayed neuronal death in the KA-injected cortex.
We further examined several functional markers of astrocytes in both NMDA- and KA-injected cortexes. EAAT2 (GLT-1) and Kir4.1 are known to be mainly expressed in astrocytes and to regulate extracellular glutamate and potassium concentration (Olsen et al., 2006; Rothstein et al., 1996). GS is also expressed in astrocytes and catalyzes the formation of glutamine from glutamate and ammonia (Hoshi et al., 2006). Similar to GFAP+ astrocytes, EAAT2+, Kir4.1+ and GS+ astrocytes disappeared in almost the entire hemisphere 1 day after KA injection, but not after NMDA injection (Fig. 4).
A more detailed analysis showed that EAAT2 immunoreactivity was present in the astrocyte processes around neuronal cell bodies in the intact brain (white arrows in Fig. 4A). In the KA-injected cortex, EAAT2+ astrocytes were maintained at 3 h (white arrows in Fig. 4A, regions ‘a, b’), but subsequently disappeared at 1–7 days (Fig. 4A). In the NMDA-injected cortex, EAAT2+ astrocytes began to disappear at 1 day in the damage core (Fig. 4A, region ‘a’), but remained for up to 7 d in the penumbra (Fig. 4A, region ‘b’).
Kir4.1 immunoreactivity was detectable in the intact brain (white arrows in Fig. 4B). In the KA-injected cortex, Kir4.1 immunoreactivity was increased slightly at 3 h in the damage core and penumbra (white arrowheads in Fig 4B), but eventually disappeared beginning at 1 day (Fig 4B). In the NMDA-injected core, Kir4.1+ astrocytes disappeared at 3 h and thereafter (Fig. 4B, region ‘a’), but Kir4.1 immunoreactivity increased in the penumbra region at 3 h and 7 days (white arrowheads in Fig. 4B, region ‘b’).
GS immunoreactivity was also detectable in intact astrocytes (white arrows in Fig. 4C). In the KA-injected cortex, GS+ astrocytes disappeared from 3 h to 7 days in both core and penumbra regions (Fig. 4C, region ‘a, b’). In the NMDA-injected brain, however, GS+ astrocytes disappeared only in the core, but GS immunoreactivity increased and/or maintained in astrocytes in the penumbra region at 3 h and thereafter (Fig. 4C, region ‘a, b’). EAAT2+, Kir4.1+, and GS+ astrocytes in the PBS-injected brain were similar to those in the intact brain (data not shown). Taken together, these results also show that loss of astrocyte function occurred in the KA model where delayed neuronal GFAP and NeuN antibodies showed that GFAP+ astrocytes and NeuN+ neurons were healthy in intact and PBS-injected brains (Fig. 5A). However, in the KA-injected cortex, the intensity of dark purple-colored astrocytes decreased slightly at 3 h, making brown-colored neurons more obvious (Fig. 5A). At 1 day, dark purple-colored astrocytes had almost disappeared, but neurons were still present, suggesting that disappearance of astrocytes precedes neuronal death in the KA-injected cortex (arrowheads in Fig. 5A). We further examined whether the remaining neurons where astrocytes disappeared (black arrowheads in Fig. 5A) were healthy. Intact NeuN+ neurons exhibited round cell bodies and homogenously stained DAPI+ nuclei (white arrows in Fig. 5B). However, at 3 h, cell bodies and nuclei of most NeuN+ neurons in the core region had shrunk and/or showed DNA condensation (white arrowheads in Fig. 5B, region ‘a’). In the penumbra region, most NeuN+ neurons still had intact nuclei at 3 h (white arrows in Fig. 5B, region ‘b’), but at 1 day, the remaining faintly stained NeuN+ neurons were shrunken and showed DNA condensation, as in the core region (white arrowheads in Fig. 5B, regions ‘a, b’). Double labeling using NeuN and GS antibodies also showed that the functional loss of astrocytes preceded neuronal death. In the intact brain, NeuN+ neurons and GS+ astrocytes coexisted (Fig. 5C). However, at 3 h after KA injection, GS+ astrocytes rapidly disappeared in ‘a’ and ‘b’ regions whereas NeuN+ neurons remained (Fig. 5C). Taken together, these results suggest that loss of neuron-supportive astrocytes eventually led to delayed neuronal death.
Fig. 5.
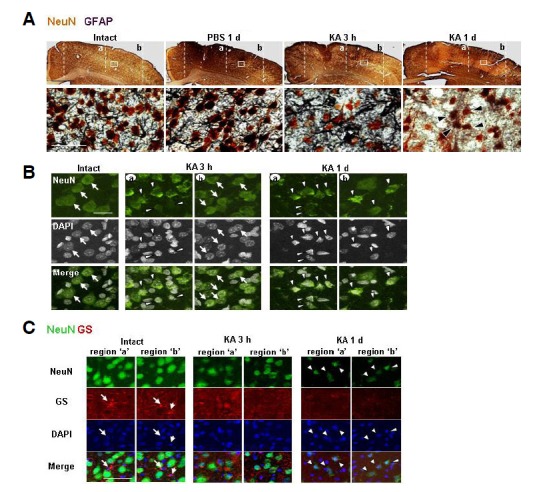
Astrocyte loss preceded delayed neuronal death in the KA-injected cortex. (A) Brain sections were double labeled with primary antibodies against NeuN and GFAP, and visualized using peroxidaseconjugated secondary antibodies. Color reactions used DAB for NeuN (brown) and DAB/nickel sulfate for GFAP (dark purple). In intact and PBS-injected brains, GFAP+ astrocytes and NeuN+ neurons were labeled. At 3 h GFAP immunoreactivity slightly decreased in the KA-injected brain, although both types of cells were detected. However, at 1 day, most GFAP+ astrocytes disappeared whereas NeuN+ neurons were still present (black arrowheads). (B, C) Brain sections obtained at the indicated times after KA injection were labeled with antibodies against NeuN (B), NeuN and GS (C), and visualized using Alexa Fluor 488- and/or 555-conjugated antibodies. Nuclei were labeled with DAPI. (B) Intact NeuN+ neurons exhibited round cell bodies and homogenously stained DAPI+ nuclei (black arrows). Shrunken and/or condensed neurons (white arrows) were detected in the core region at 3 h, and in both core and penumbra regions at 1 day. (C) At 3 h after injection, GS+ astrocytes (white arrows) disappeared, whereas NeuN+ neurons were still present in both ‘a’ and ‘b’ regions. At 1 day, GS+ astrocytes had completely disappeared, and shrunken NeuN+ neurons (white arrowheads) were detectable. Scale bars = 1 mm [upper panels in (A)], 50 μm [lower panels in (A), and (C)], and 20 μm (B).
Astrocyte functional loss preceded delayed neuronal death in the KA-injected cortex
Previously, we reported that delayed neuronal death in contusion-induced spinal cord injury is spatially and temporally correlated with astrocyte death, suggesting that loss of astrocyte function may be critical for delayed neuronal death (Min et al., 2012). In this study, we investigated a possible correlation between progressive degeneration of astrocytes and delayed neuronal cell death in the KA model. Double labeling using GFAP and NeuN antibodies showed that GFAP+ astrocytes and NeuN+ neurons were healthy in intact and PBS-injected brains (Fig. 5A). However, in the KA-injected cortex, the intensity of dark purple-colored astrocytes decreased slightly at 3 h, making brown-colored neurons more obvious (Fig. 5A). At 1 day, dark purple-colored astrocytes had almost disappeared, but neurons were still present, suggesting that disappearance of astrocytes precedes neuronal death in the KA-injected cortex (arrowheads in Fig. 5A). We further examined whether the remaining neurons where astrocytes disappeared (black arrowheads in Fig. 5A) were healthy. Intact NeuN+ neurons exhibited round cell bodies and homogenously stained DAPI+ nuclei (white arrows in Fig. 5B). However, at 3 h, cell bodies and nuclei of most NeuN+ neurons in the core region had shrunk and/or showed DNA condensation (white arrowheads in Fig. 5B, region ‘a’). In the penumbra region, most NeuN+ neurons still had intact nuclei at 3 h (white arrows in Fig. 5B, region ‘b’), but at 1 day, the remaining faintly stained NeuN+ neurons were shrunken and showed DNA condensation, as in the core region (white arrowheads in Fig. 5B, regions ‘a, b’). Double labeling using NeuN and GS antibodies also showed that the functional loss of astrocytes preceded neuronal death. In the intact brain, NeuN+ neurons and GS+ astrocytes coexisted (Fig. 5C). However, at 3 h after KA injection, GS+ astrocytes rapidly disappeared in ‘a’ and ‘b’ regions whereas NeuN+ neurons remained (Fig. 5C). Taken together, these results suggest that loss of neuron-supportive astrocytes eventually led to delayed neuronal death.
Astrogliosis appeared to protect neurons from further damage in the NMDA-injected cortex
Finally, we examined whether the absence of delayed neuronal death in the NMDA-injected brain was due to survival of astrocytes. As shown above (Figs. 1 and 3), both astrocytes and neurons died in the core at 3 h after injection. In further analysis of behavior of astrocytes and neurons, we found that at 3 h, atrocyte-absent areas were much smaller than neuron-death areas (red lines in Figs. 6A and 6B), and astrocytes outside of the dead areas underwent gliosis, as confirmed by an increase in GFAP expression levels (Fig. 6A). Astrocyte gliosis continued to progress at 7 days, and neurons were healthy in areas where gliosis occurred (red lines in Figs. 6A, 6B, and 6C). Taken together with our demonstration that surviving astrocytes in the NMDA-injected brain highly expressed not only GFAP but several functional markers, including EAAT1, Kir4.1 and GS (Fig. 3), these results suggest that astrogliosis may be a mechanism for protecting neurons in the injured brain. Therefore, neuronal death could be caused by the death and/or loss of function of astrocytes required to support neuronal survival.
Fig. 6.
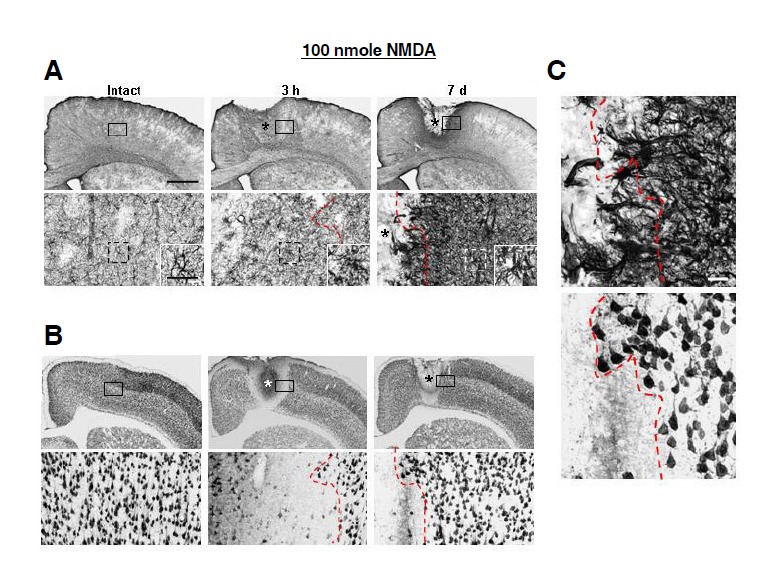
Neurons were healthy in the NMDA-injected cortex where astrogliosis occurred. (A, B) Locations of GFAP-labeled astrocytes and NeuN-labeled neurons were analyzed in brain sections obtained at 3 h and 7 days after NMDA injection. Lower panels are higher magnification of boxed areas in upper panels. *, drug injection sites. Insets in lower panels in (A) indicate higher magnification of dotted boxes. At 3 h after NMDA injection, GFAP expression significantly increased compared to the intact brain, and processes became thicker. At 7 days, GFAP expression further increased. (C) Notably, neurons were healthy in areas where astrogliosis occurred. Red dotted lines in (A, B, and C) indicate the borders of dead- and live-neuron areas. Scale bars = 1 mm [upper panels in (A, B)], 100 μm [lower panels in (A, B)], and 20 μm [inset in lower panels in (A), and (C)].
DISCUSSION
In this study, we showed that 1) KA injection, but not NMDA injection, induced delayed neuronal death, but there was little difference in inflammatory responses between KA- and NMDA-injected cortexes; 2) extensive astrocyte loss occurred in the KA-injected cortex; 3) the astrocyte loss territory correlated with the delayed neuronal damage area in the KA-injected cortex; and 4) the disappearance of astrocyte markers preceded delayed neuronal death in the KA-injected cortex. On the basis of these findings, we suggest that loss of astrocyte support is a critical cause of delayed neuronal death in the injured brain.
Many studies have reported that brain inflammation is neurotoxic and aggravates progressive neuronal death in neurode-generative diseases (Chao et al., 1992; de Bock et al., 1998; Giulian et al., 1993; Kaushal and Schlichter, 2008; Ryu et al., 2004; Scali et al., 2000). However, we and others have reported that brain inflammation is not neurotoxic but is instead neuroprotective and functions to repair the injured brain (Batchelor et al., 1999; Elkabes et al., 1996; Howe and Barres, 2012; Jeong et al., 2010; 2013a; 2013b; Kim et al., 2010; Koch and Szecsey, 2000; Lehrmann et al., 1998; Min et al., 2012; Reines et al., 2004; Scharf et al., 1999; van Gool et al., 2003; Vinet et al., 2012; Yang et al., 2007). In this study, we demonstrated that inflammatory responses were similar in both KA-and NMDA-injected cortexes. Regardless of the presence or absence of delayed neuronal death, resident microglia died at 3 h and monocytes infiltrated at 7 days after KA or NMDA injection (Fig. 2). Quantitative mRNA expression analyses showed that TNF-α and IL-1β significantly increased at 12 h in both NMDA- and KA-injected cortexes (Fig. 2B) compared to cortexes injected with PBS, although TNF-α and IL-1β levels were slightly elevated in the NMDA- and KA-injected cortex, respectively (Fig. 2). In addition, infiltration of neurotoxic iNOS-expressing neutrophils was very low in both NMDA- and KA-injected brains, and iNOS levels were similarly increased at 12 h to 1 day in both NMDA- and KA-injected cortexes (Fig. 2). Therefore, inflammatory responses appeared to have little effect on delayed neuronal death in the KA-injected brain.
It is well known that astrocytes regulate the brain microenvironment and support neurons by supplying glucose and neurotrophic factors, regulating oxidative stress and excitotoxicity through glutathione (GSH) production and uptake of glutamate and potassium, and regulating extracellular water content (Badaut et al., 2002; Chih and Roberts Jr., 2003; Müller and Seifert, 1982; Olsen et al., 2006; Raps et al., 1989; Rothstein et al., 1996; Simard and Nedergaard, 2004; Tsacopoulos and Magistretti, 1996). Thus, astrocyte support is required for neuronal survival (Jeong et al., 2013c; Min et al., 2012). A loss of EAAT2/GLT-1 and decreased glutamate transport have been identified in human ALS patients (Rothstein et al., 1992; 1995). In addition, astrocyte functional loss precedes neuronal death in spinal cord injury (Min et al., 2012), and transplantation of human astrocytes facilitates functional recovery after the injury (Ermakova et al., 2005; Myer et al., 2006). In the previous study, we showed that in ATP-injected brain, astrogliosis occurs and delayed neuronal death is absent (Jeong et al., 2013c). The results presented in the current study also showed that neuronal death is closely linked to astrocyte loss. Notably, astrocytes gradually disappeared throughout the entire hemisphere at 7 days in the KA-injected cortex, whereas astrocyte disappearance was confined to the injection core in the NMDA-injected cortex at 7 days (Figs. 1 and 3). Moreover, only the KA-injected cortex showed an extensive disappearance of proteins important for astrocyte function, such as EAAT2, Kir4.1 and GS, throughout the hemisphere (Fig. 4). Accordingly, GFAP+ as well as S100β+ and GS+ astrocytes disappeared at 1 day, before the death of NeuN+ neurons (Fig. 5 and Supplementary Fig. 3). Another important feature of the NMDA-injected brain was astrogliosis (reactive astrocytes) in the penumbra region (Fig. 6) where neurons were healthy (Fig. 1). Therefore, the territories of loss of astrocyte support correlated with the neuron-damaged area in both NMDA- and KA-injected cortexes (Figs. 1 and 3), indicating that reactive astrocytes protect neurons in the injured brain as previously reported in several brain injury models (Ermakova et al., 2005; Haj-Yasein et al., 2012; Myer et al., 2006; Rothstein et al., 1992; 1995; Shi et al., 2012; Zeng et al., 2012).
The discrepancy between astrocyte behavior in the NMDA-and KA-injected brain may reflect differences in the expression levels of NMDA receptors and KA receptors in astrocytes. Astrocytes rarely express NMDA receptors (Kettenmann and Schachner, 1985). Thus, NMDA does not induce astrocyte cell death. On the other hand, astrocytes are vulnerable to KA (David et al., 1996). Therefore, differences in the vulnerability of astrocytes to NMDA and KA appear to determine the absence and presence of delayed neuronal death.
In conclusion, astrocyte functional loss may be a critical cause of delayed neuronal death in the injured brain. Our results highlight the physiological importance of astrocytes in protecting neurons from a harmful microenvironment. Therefore, preservation of astrocytes and/or astrocyte functional support could be a therapeutic strategy for preventing delayed neuronal death in the injured brain.
Supplementary Data
Acknowledgments
This work was supported by a KOSEF NRL Program grant (no. 2-2008025-0) funded by the Korean government (MEST) and a grant (NRF-2012R1A5A2048183) from KOSEF through the Chronic Inflammatory Disease Research Center (CIDRC) at Ajou University.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Badaut J., Lasbennes F., Magistretti P.J., Regli L. Aquaporins in brain: distribution, physiology, and pathophysiology. J. Cereb. Blood Flow Metab. 2002;22:367–378. doi: 10.1097/00004647-200204000-00001. [DOI] [PubMed] [Google Scholar]
- Batchelor P.E., Liberatore G.T., Wong J.Y., Porritt M.J., Frerichs F., Donnan G.A., Howells D.W. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J. Neurosci. 1999;19:1708–1716. doi: 10.1523/JNEUROSCI.19-05-01708.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C.C., Hu S., Molitor T.W., Shaskan E.G., Peterson P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- Chih C.P., Roberts E.L., Jr Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 2003;23:1263–1281. doi: 10.1097/01.WCB.0000081369.51727.6F. [DOI] [PubMed] [Google Scholar]
- David J.C., Yamada K.A., Bagwe M.R., Goldberg M.P. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J. Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bock F., Derijard B., Dornand J., Bockaert J., Rondouin G. The neuronal death induced by endotoxic shock but not that induced by excitatory amino acids requires TNF-alpha. Eur. J. Neurosci. 1998;10:3107–3114. doi: 10.1046/j.1460-9568.1998.00317.x. [DOI] [PubMed] [Google Scholar]
- Elkabes S., DiCicco-Bloom E.M., Black I.B. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J. Neurosci. 1996;16:2508–2521. doi: 10.1523/JNEUROSCI.16-08-02508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova I.V., Loseva E.V., Hodges H., Sinden J. Transplantation of cultured astrocytes attenuates degenerative changes in rats with kainic acid-induced brain damage. Bull. Exp. Biol. Med. 2005;140:677–681. doi: 10.1007/s10517-006-0052-0. [DOI] [PubMed] [Google Scholar]
- Giulian D., Corpuz M., Chapman S., Mansouri M., Robertson C. Reactive mononuclear phagocytes release neurotoxins after ischemic and traumatic injury to the central nervous system. J. Neurosci. Res. 1993;36:681–693. doi: 10.1002/jnr.490360609. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein N.N., Jensen V., Ostby I., Omholt S.W., Voipio J., Kaila K., Ottersen O.P., Hvalby O., Nagelhus E.A. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: a gene deletion study in mouse hippocampus. Glia. 2012;60:867–874. doi: 10.1002/glia.22319. [DOI] [PubMed] [Google Scholar]
- Hoshi A., Nakahara T., Kayama H., Yamamoto T. Ischemic tolerance in chemical preconditioning: possible role of astrocytic glutamine synthetase buffering glutamate-mediated neurotoxicity. J. Neurosci. Res. 2006;84:130–141. doi: 10.1002/jnr.20869. [DOI] [PubMed] [Google Scholar]
- Howe M.L., Barres B.A. A novel role for microglia in minimizing excitotoxicity. BMC Biol. 2012;10:7. doi: 10.1186/1741-7007-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.K., Ji K.M., Kim B., Kim J., Jou I., Joe E.H. Inflammatory responses are not sufficient to cause delayed neuronal death in ATP-induced acute brain injury. PLoS One. 2010;5:e13756. doi: 10.1371/journal.pone.0013756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.K., Ji K., Min K., Joe E.H. Brain inflammation and microglia: facts and misconceptions. Exp. Neurobiol. 2013a;22:59–67. doi: 10.5607/en.2013.22.2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.K., Ji K.M., Kim J., Jou I., Joe E.H. Repair of astrocytes, blood vessels, and myelin in the injured brain: possible roles of blood monocytes. Mol. Brain. 2013b;6:28. doi: 10.1186/1756-6606-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.K., Jou I., Joe E.H. Absence of delayed neuronal death in ATP-injected brain: possible roles of astrogliosis. Exp. Neurobiol. 2013c;22:308–314. doi: 10.5607/en.2013.22.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji K.A., Yang M.S., Jeong H.K., Min K.J., Kang S.H., Jou I., Joe E.H. Resident microglia die and infiltrated neutrophils and monocytes become major inflammatory cells in lipopolysaccharide-injected brain. Glia. 2007;55:1577–1588. doi: 10.1002/glia.20571. [DOI] [PubMed] [Google Scholar]
- Ji K.A., Eu M.Y., Kang S.H., Gwag B.J., Jou I., Joe E.H. Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. Glia. 2008;56:1039–1047. doi: 10.1002/glia.20677. [DOI] [PubMed] [Google Scholar]
- Kaul D.K., Liu X.D., Choong S., Belcher J.D., Vercellotti G.M., Hebbel R.P. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am. J. Physiol. Heart Circ. Physiol. 2004;287:H293–301. doi: 10.1152/ajpheart.01150.2003. [DOI] [PubMed] [Google Scholar]
- Kaushal V., Schlichter L.C. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J. Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H., Schachner M. Pharmacological properties of gamma-aminobutyric acid-, glutamate-, and aspartate-induced depolarizations in cultured astrocytes. J. Neurosci. 1985;5:3295–3301. doi: 10.1523/JNEUROSCI.05-12-03295.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Min K.J., Seol W., Jou I., Joe E.H. Astrocytes in injury states rapidly produce anti-inflammatory factors and attenuate microglial inflammatory responses. J. Neurochem. 2010;115:1161–1171. doi: 10.1111/j.1471-4159.2010.07004.x. [DOI] [PubMed] [Google Scholar]
- Koch H.J., Szecsey A. A randomized controlled trial of prednisone in Alzheimer’s disease. Neurology. 2000;55:1067. doi: 10.1212/wnl.55.7.1067. [DOI] [PubMed] [Google Scholar]
- Lehrmann E., Kiefer R., Christensen T., Toyka K.V., Zimmer J., Diemer N.H., Hartung H.P., Finsen B. Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia. 1998;24:437–448. doi: 10.1002/(sici)1098-1136(199812)24:4<437::aid-glia9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Min K.J., Jeong H.K., Kim B., Hwang D.H., Shin H.Y., Nguyen A.T., Kim J.H., Jou I., Kim B.G., Joe E.H. Spatial and temporal correlation in progressive degeneration of neurons and astrocytes in contusion-induced spinal cord injury. J. Neuroinflammation. 2012;9:100. doi: 10.1186/1742-2094-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H.W., Seifert W. A neurotrophic factor (NTF) released from primary glial cultures supports survival and fiber outgrowth of cultured hippocampal neurons. J. Neurosci. Res. 1982;8:195–204. doi: 10.1002/jnr.490080209. [DOI] [PubMed] [Google Scholar]
- Myer D.J., Gurkoff G.G., Lee S.M., Hovda D.A., Sofroniew M.V. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Olsen M.L., Higashimori H., Campbell S.L., Hablitz J.J., Sontheimer H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia. 2006;53:516–528. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raps S.P., Lai J.C., Hertz L., Cooper A.J. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989;493:398–401. doi: 10.1016/0006-8993(89)91178-5. [DOI] [PubMed] [Google Scholar]
- Reines S.A., Block G.A., Morris J.C., Liu G., Nessly M.L., Lines C.R., Norman B.A., Baranak C.C. Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- Rothstein J.D. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. Adv. Neurol. 1995;68:7–20. discussion 21–27. [PubMed] [Google Scholar]
- Rothstein J.D., Martin L.J., Kuncl R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl. J. Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein J.D., Van Kammen M., Levey A.I., Martin L.J., Kuncl R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Rothstein J.D., Dykes-Hoberg M., Pardo C.A., Bristol L.A., Jin L., Kuncl R.W., Kanai Y., Hediger M.A., Wang Y., Schielke J.P., et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Ryu J.K., Franciosi S., Sattayaprasert P., Kim S.U., McLarnon J.G. Minocycline inhibits neuronal death and glial activation induced by beta-amyloid peptide in rat hippocampus. Glia. 2004;48:85–90. doi: 10.1002/glia.20051. [DOI] [PubMed] [Google Scholar]
- Scali C., Prosperi C., Vannucchi M.G., Pepeu G., Casamenti F. Brain inflammatory reaction in an animal model of neuronal degeneration and its modulation by an anti-inflammatory drug: implication in Alzheimer’s disease. Eur. J. Neurosci. 2000;12:1900–1912. doi: 10.1046/j.1460-9568.2000.00075.x. [DOI] [PubMed] [Google Scholar]
- Scharf S., Mander A., Ugoni A., Vajda F., Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- Shi W.Z., Qi L.L., Fang S.H., Lu Y.B., Zhang W.P., Wei E.Q. Aggravated chronic brain injury after focal cerebral ischemia in aquaporin-4-deficient mice. Neurosci. Lett. 2012;520:121–125. doi: 10.1016/j.neulet.2012.05.052. [DOI] [PubMed] [Google Scholar]
- Simard M., Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- Streit W.J. Microglia and neuroprotection: implications for Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Tsacopoulos M., Magistretti P.J. Metabolic coupling between glia and neurons. J. Neurosci. 1996;16:877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool W.A., Aisen P.S., Eikelenboom P. Anti-inflammatory therapy in Alzheimer’s disease: is hope still alive? J. Neurol. 2003;250:788–792. doi: 10.1007/s00415-003-1146-5. [DOI] [PubMed] [Google Scholar]
- Vinet J., Weering H.R., Heinrich A., Kalin R.E., Wegner A., Brouwer N., Heppner F.L., Rooijen N., Boddeke H.W., Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.S., Min K.J., Joe E. Multiple mechanisms that prevent excessive brain inflammation. J. Neurosci. Res. 2007;85:2298–2305. doi: 10.1002/jnr.21254. [DOI] [PubMed] [Google Scholar]
- Zeng X.N., Xie L.L., Liang R., Sun X.L., Fan Y., Hu G. AQP4 knockout aggravates ischemia/reperfusion injury in mice. CNS Neurosci. Ther. 2012;18:388–394. doi: 10.1111/j.1755-5949.2012.00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.