

Abstract
Wiskott–Aldrich Syndrome protein (WASp) regulates the cytoskeleton in hematopoietic cells and mutations in its gene cause the Wiskott–Aldrich Syndrome (WAS), a primary immunodeficiency with microthrombocytopenia, eczema and a higher susceptibility to develop tumors. Autoimmune manifestations, frequently observed in WAS patients, are associated with an increased risk of mortality and still represent an unsolved aspect of the disease. B cells play a crucial role both in immune competence and self-tolerance and defects in their development and function result in immunodeficiency and/or autoimmunity. We performed a phenotypical and molecular analysis of central and peripheral B-cell compartments in WAS pediatric patients. We found a decreased proportion of immature B cells in the bone marrow correlating with an increased presence of transitional B cells in the periphery. These results could be explained by the defective migratory response of WAS B cells to SDF-1α, essential for the retention of immature B cells in the BM. In the periphery, we observed an unusual expansion of CD21low B-cell population and increased plasma BAFF levels that may contribute to the high susceptibility to develop autoimmune manifestations in WAS patients. WAS memory B cells were characterized by a reduced in vivo proliferation, decreased somatic hypermutation and preferential usage of IGHV4-34, an immunoglobulin gene commonly found in autoreactive B cells.
In conclusion, our findings demonstrate that WASp-deficiency perturbs B-cell homeostasis thus adding a new layer of immune dysregulation concurring to the increased susceptibility to develop autoimmunity in WAS patients.
Keywords: Wiskott–Aldrich Syndrome, Primary immunodeficiency, Autoimmunity, B lymphocyte, B-cell activating factor (BAFF), Immunoglobulin repertoire
Highlights
-
•
WASp-deficiency affects both central and peripheral B-cell development.
-
•
An early egress of immature B cells leads to an increase of transitional B cells in periphery.
-
•
Reduced maturation status of WAS memory B cells.
-
•
Altered selection of both protective and autoreactive Ig gene families in WAS patients.
-
•
Potentially autoreactive CD21low B cells are expanded in WAS patients.
1. Introduction
Wiskott–Aldrich Syndrome (WAS) is a monogenic X-linked primary immunodeficiency characterized by the classic triad of microthrombocytopenia, eczema and recurrent infections. Multiple autoimmune manifestations and tumors are serious complications and the life expectancy of WAS patients is severely reduced, unless they are successfully cured by hematopoietic stem cell transplantation [1].
WAS is caused by mutations in the WAS gene [2,3] impairing the expression and/or function of the WAS protein (WASp), a hematopoietic-specific regulator of cytoskeletal reorganization also involved in signal transduction of cells [4]. The absence or reduction of WASp compromises multiple processes of different immune cell types involved in innate and adaptive responses, resulting in a variable and progressive immunodeficiency. Autoimmune complications affect 22–72% of WAS patients and are associated with poor prognosis [5,6]. Susceptibility of WAS patients to develop autoimmune diseases has been mainly attributed to the breakdown of self tolerance sustained by dysfunction of both natural T regulatory and effector T cells (reviewed in Ref. [7]). However, B-cell intrinsic defects have been demonstrated to critically contribute to WAS-associated autoimmunity in Was−/− mouse model [8,9]. In humans, the contribution of B-cell defects in the pathogenesis of WAS has been partially investigated. B cells from patients exhibit lower motility, migratory and adhesive capacities [10], likely due to defective F-actin nucleation [11]. In contrast, despite the role of WASp in B-cell receptor (BCR) signaling [7,12], abnormalities in B-cell activation still remain controversial [13,14]. A skewed distribution of serum immunoglobulin (Ig) classes [5] and the inability to mount a proper antibody response, particularly to T-cell independent (TI) antigens [15], suggest defects in B-cell effector function.
Previous findings in WAS patients [16,17] show phenotypical B-cell perturbations in the periphery. In order to evaluate whether an abnormal B-cell development might generate a B-cell repertoire unable to unsure full protection against pathogens and tolerance against self-antigens, we have further studied the B-cell compartment in WAS patients. To this end, we have combined a detailed phenotypical analysis of B-cell maturation stages, from the bone marrow (BM) to the periphery, with a molecular study of Ig repertoire and in vivo B-cell maturation processes in a large cohort of WAS pediatric patients. Our data show that WASp-deficiency affects critical stages of central and peripheral B-cell differentiation contributing to abnormalities in humoral immunity and B-cell tolerance in humans.
2. Material and methods
2.1. Patients
The diagnoses were clinically defined and confirmed by genetic analysis. A description of all patients is reported in Supplementary Table 1. Human samples were obtained according to The Code of Ethics of the World Medical Association (Declaration of Helsinki) with the approval of the local Medical Ethical Committees of the Erasmus MC and the San Raffaele Scientific Institute Internal Review Board (TIGET02). All legal representatives gave written informed consent. All results obtained from samples of WAS patients were compared to age and sex matched healthy donors (HDs).
2.2. Flow cytometry and purification of B-cell subsets
The composition of the precursor B-cell compartment was analyzed by flow cytometric immunophenotyping as described in the Supplementary Material. For the analysis of replication history and somatic hypermutation, four B-cell subsets were isolated from thawed peripheral blood mononuclear cells (PBMCs) using a FACS DiVa cell sorter (BD Biosciences) [18]. Gating on CD19+ cells, transitional (CD27−CD24highCD38high), mature naïve (CD27−IgD+CD24dimCD38dim), natural effector (CD27+IgD+) and memory (CD27+IgD−) B-cell subsets were sorted with a purity of >95% for all fractions. For intracytoplasmic detection of human WASp, cells were fixed and permeabilized using a Cytofix/Cytoperm kit (BD Pharmingen, Oregon, USA). The anti-WASp antibody 503 (a kind gift from Prof H. D. Ochs, Seattle, WA, and L. D. Notarangelo, Boston, MA) was used, followed by detection with Pacific Blue-labeled anti-rabbit IgG secondary antibody (Invitrogen, San Diego, USA). Samples were acquired on a FACSCanto cytometer.
2.3. Chemotaxis assay
CD20 positive cells were purified from PBMCs of pediatric WAS patients and age-matched HDs by immunomagnetic beads (Miltenyi Biotec, Germany) or FACS sorting. The purity of the isolated cells were analyzed by FACS and ranged from 84% to 98%. After isolation, cells were left overnight at 37 °C in culture medium composed of RPMI-1640, 10% FBS, 2 mM glutamine, 100 IU/mL penicillin and 100 μg/mL streptomycin (Lonza, Basel, Switzerland). In vitro chemotaxis assay was performed using 5 μM pore-size Transwell inserts (Costar Corporation, Corning, NY, US) in 24-well plates. Filters were prewet 30 min at 37 °C in presence of 600 μL of medium supplemented with 250 ng/mL of recombinant human stromal cell-derived factor (SDF)-1α (CXCL12; Peprotech, Rocky Hill, US). Fifty thousand CD20 positive or negative cells were resuspended in 100 μL of culture medium, seeded in the upper chamber and incubated at 37 °C for 3 h. Transmigrated cells, collected in the lower chamber, were counted for viable cells and stained with anti-CD19, anti-CD24, anti-CD38, anti-CD27 and anti-CD3 (BD Biosciences) for the phenotypical analysis by FACS. Migration frequency was estimated as the [(cell n° at the lower chamber/the initial B cell input in the upper chamber) × 100].
2.4. ELISA assay
Levels of B-cell activating factor (BAFF) were measured in duplicate in plasma samples of WAS patients and HDs using a Quantikine Human BAFF/BLyS/TNFSF13B Immunoassay kit (R&D Systems, Minneapolis, USA). The assay was performed according to manufacturer's instructions and the OD was determined using a microplate reader set to 450 nm.
2.5. Sequence analysis of Igκ and IGH gene rearrangements
RNA was isolated from PBMCs and reverse-transcribed using random hexamers. Igκ and IGH gene rearrangements were amplified and analyzed as described in the Supplementary Material.
2.6. Analysis of replication history by KREC assay
DNA was extracted from sorted B-cell subsets with the GenElute Mammalian Total DNA Miniprep kit (Sigma–Aldrich) and used to perform the Igκ-deleting recombination excision circles (KREC) assay [19]. The amounts of coding and signal joints of the Igκ-deleting rearrangement were measured by real-time quantitative PCR on an ABI Prism 7000 sequence detection system (Applied Biosystems).
2.7. Analysis of somatic hypermutation by IgκREHMA
Igκ restriction enzyme-based hot-spot mutation assay (IgκREHMA) was performed on genomic DNA isolated from sorted B-cell subsets [19]. Briefly, Vκ3-20-Jκ rearrangements were PCR-amplified and the PCR products (∼500 bp) were first digested using the restriction enzyme Fnu4HI and then KpnI. The unmutated gene products can be visualized as 244 or 247 bp HEX-coupled fragments and the mutated as 262 bp HEX-coupled fragment.
2.8. Statistical analyses
All results are expressed as mean and SD if not stated otherwise. Statistical significance was assessed using a two-tailed Mann Whitney test. Comparisons between proportions were calculated by using the chi-square test (χ2 test) (with continuity correction). P values of less than 0.05 were considered significant.
3. Results
3.1. Altered distribution of the precursor B-cell compartment in the bone marrow of WAS patients
Until now, the composition of the B-cell compartment in the BM of WAS patients has not been reported. To evaluate the effect of WASp-deficiency on B-cell development in the BM, we characterized five stages of B-cell differentiation [20] in three WAS patients. We observed a decreased frequency of immature B lymphocytes in WAS patients (Fig. 1A). This observation was further assessed in four additional patients, focusing on late stages of B-cell differentiation based on the expression of CD10 and CD20 [21] (gating strategy is shown in Suppl. Fig. 1). We confirmed a significant decrease in the frequency of immature B cells compared with age-matched HDs (Fig. 1B). In addition, at the earliest differentiation stages, we observed a significant increase of small Pre-B-II cells in WAS patients; while no statistical difference was detected in large Pre-B-II cell frequency (Fig. 1C). These findings might reflect a partial block in the BM B-cell differentiation between the small Pre-B-II and immature B-cell stages in WAS, or an early egress of immature B cells into the periphery.
Fig. 1.

Reduced frequency of immature B cells in WAS BM samples. A. The precursor B-cell differentiation pattern in the BM was analyzed by flow cytometry (corrected for blood contamination) and compared with the average values of age-matched HDs (y = years). B–C. Frequencies of Immature B cells (CD10intCD20+) (B), Pre-B-II small (CD10intCD20−) and Pre-B-II large (CD10intCD20int) cells (C) were determined on CD34−CD19+ cells and calculated on total BM precursor B cells excluding recirculating mature B cells (CD10−CD20+). The graphs show data points for all examined donors and patients. ***P < 0.0001; **P = 0.0029. (HD, n = 27; WAS, n = 7).
3.2. Perturbed peripheral B-cell maturation in WAS patients
Next we examined the distribution of B-cell subsets in the peripheral blood of WAS patients and age-matched HDs. When available, we evaluated the absolute count of total B cells, which was found significantly reduced in patients less than three years old (<3y), while no differences were observed in the older WAS group (Fig. 2A) confirming previously published results [16]. Four B-cell subsets were identified according to the expression of CD24, CD38, IgD and CD27: transitional, mature naïve, memory and plasmablasts (Fig. 2B). Transitional B cells (CD24hiCD38hi) are the most immature peripheral B-cell population that were significantly increased in both WAS age groups as frequency (Fig. 2C) and absolute number (Suppl. Fig. 2). The next maturation stage, represented by mature naïve B cells (CD27−IgD+CD24dimCD38dim), was significantly decreased only in WAS patients less than 3 years old both in frequency (Fig. 2C) and absolute number (Suppl. Fig. 2). Finally, the frequency of plasmablasts (CD24−CD38hi) was significantly increased in the older group of patients (Fig. 2C), while the absolute number of both memory B cells (CD27+IgD−CD24dimCD38dim) and plasmablasts were significantly decreased in the younger group of WAS patients (Suppl. Fig. 2).
Fig. 2.

Perturbed peripheral B-cell maturation in WAS patients. A. Absolute counts of CD19+ cells of WAS patients (black dots) and age-matched HDs (white dots) were analyzed when available. B. Representative plots of flow cytometric analysis of blood B-cell subsets of a HD and a WAS patient (age range: <3y) are shown. C. Graphs display frequencies of the four main B-cell subsets in the blood of WAS patients and age-matched HDs. Individual data points are displayed with mean and SD. Groups are compared to controls and significant values are indicated: ***P ≤ 0.0005; **P < 0.005; *P < 0.05. (HD < 3y, n = 29; HD 3–12y, n = 27).
Altogether, these findings demonstrate that WAS patients have a perturbed peripheral B-cell distribution.
3.3. B lymphocytes from WAS patients display a reduced migratory ability in response to SDF-1α
The overrepresentation of transitional B cells in the blood of WAS patients led us to hypothesize that an early egress of immature B cells from BM could occur. CXCR4, the chemokine receptor for SDF-1α, has an important role in retaining B-cell precursors within the BM in order to prevent premature migration to the periphery [22–24]. We tested the chemotactic response to SDF-1α of B lymphocytes of WAS patients and HDs by transwell migration assay. As shown in Fig. 3, both transitional and mature peripheral B cells were less responsive to SDF-1α in the absence of WASp (Fig. 3A–C). This defect was specific for B lymphocytes since CD3 positive cells of WAS patients showed a normal migratory capacity to SDF-1α (Suppl. Fig. 3). Thus, we hypothesize that a diminished retention signal mediated by CXCR4/SDF-1α could explain the premature egress of immature B cells from the BM leading to an increase of transitional B cells in periphery.
Fig. 3.
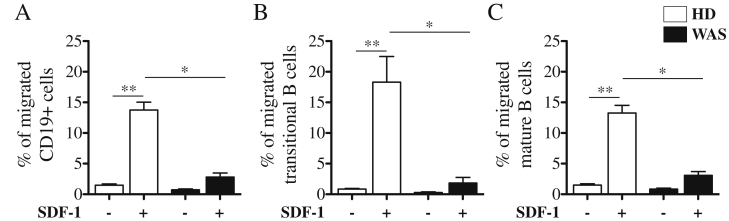
Reduced in vitro migration to SDF-1α of B cells from WAS patients. CD20 positive cells were isolated from PBMCs of WAS patients (black bars; n = 4) and age matched HDs (white bars; n = 5) and subjected to the chemotaxis assay in the presence of 250 ng/mL of SDF-1α or medium alone. The percentage of migrated CD19+ cells (A), CD19+CD24hiCD38hi transitional (B) and CD19+CD24dimCD38dim mature B cells (C) were determined by flow cytometric analysis. The graphs summarize three independent experiments and represented as bars with mean and SEM. WAS Groups are compared to HDs and statistically significant values are indicated: **P < 0.005; *P < 0.05.
3.4. WAS patients display alterations in potentially autoreactive B-cell subsets and elevated B-cell activating factor plasma levels
To further characterize the peripheral B-cell distribution, we focused our analysis on B-cell subsets found expanded in autoimmune diseases and potentially autoreactive [25–28]. We found a significant increase of CD19+CD21−CD35− subset in both WAS age groups (Fig. 4A), confirming data previously reported by Park and colleagues [17]. We also analyzed the frequency of CD19+CD21−CD38− cells (referred to as CD21low) described to be expanded in autoimmune diseases and immunodeficiencies [26,27] [29]. Interestingly, we observed that the frequency of this unusual population was markedly increased in WAS patients, both in the younger and older groups (Fig. 4B). In addition, higher levels of soluble BAFF were found in the plasma of WAS patients as compared to pediatric HDs (Fig. 4C). This finding was also associated with a decreased expression of its receptor, BAFFR, in transitional B cells of WAS patients (Fig. 4D). Because of the role of BAFF in B-cell homeostasis and peripheral B-cell tolerance [30,31], high BAFF levels may affect the stringency of peripheral B-cell selection thus favoring the survival of B-cell subsets containing potentially autoreactive clones.
Fig. 4.

Presence of B-cell populations potentially autoreactive with increased BAFF levels in WAS patients. A-B. Representative plots (left panels) and relative graph (right panel) show the decreased expression of CD21 and CD35 (A) and the frequency of CD21low B cells (B) gated on CD19+ cells in WAS patients. Individual data points are displayed in the graphs. C. Bar graph, with mean and SD, shows BAFF levels in plasma of WAS patients and HDs with an age range of 0–5 year old. D. Histogram shows the expression of BAFFR in transitional B cells of representative WAS (Pt20, gray line) and HD (black line) subjects. Solid gray curve is the unstained control. Bar graph shows the mean fluorescence intensity (MFI) of BAFFR. Significant values compared to normal controls are indicated: ***P ≤ 0.0005; **P < 0.005; *P < 0.05.
3.5. WASp-deficiency affects antigen-dependent B-cell differentiation and maturation
In order to evaluate the effect of WASp-deficiency in an antigen dependent context, we analyzed in detail the memory B-cell compartment of WAS patients. We identified six distinct memory B-cell subsets [32] (gating strategy is shown in Suppl. Fig. 4): CD27+IgM+IgD− (also named “IgM-only”) and CD27−IgG+ deriving from primary germinal center (GC) responses; IgG+ CD27+ and IgA+ CD27+ switched memory B cells originating from secondary GC responses; CD27+IgD+ (named “natural effector”) and CD27−IgA+ generated in a TI manner in the gastrointestinal tract or in the splenic marginal zone area, respectively [33]. We did not observe differences in memory B-cell populations deriving from both primary and secondary GC responses between WAS patients and HDs (Fig. 5A–B). A marked reduction in natural effector B cells was found in both age groups of WAS patients both as frequency (Fig. 5C) and absolute number (Suppl. Fig. 5). The frequency of CD27−IgA+ B cells was decreased in 3–12 years old patients compared with HDs (Fig. 5C). In conclusion, the absence of WASp mainly affects TI responses of B cells, whereas only mildly impairs the generation of early memory B cells in GC without influencing the class-switched memory B-cell compartment.
Fig. 5.

Alterations in the memory B-cell compartment of WAS patients. Graphs indicate the frequencies of: IgM-only B cells (left) and CD27−IgG+ B cells (right), representing memory B cells firstly involved in a GC response (A); switched memory B cells identified as CD27+IgG+ (left) and CD27+IgA+ (right) B cells (B); GC-independent memory B cells (C). Significant values compared to normal controls are indicated. ***P ≤ 0.0005; **P < 0.005; *P < 0.05.
We determined the in vivo B-cell proliferation history by KREC assay and in parallel SHM by IgκREHMA in sorted transitional, mature naïve, natural effector and memory B cells. As expected, transitional B lymphocytes did not undergo cell divisions and mature naïve B cells had a limited number of cell divisions in HDs (Fig. 6A). In HDs both B-cell subsets were characterized by the absence of SHM (Fig. 6B) and we did not find differences in transitional and mature naïve B cells of WAS subjects (Fig. 6A–B). In contrast, the memory compartment showed less proliferation in WAS memory B cells in association with a reduced SHM level (Fig. 6A–B). Interestingly, the defect in maturation was even more pronounced in natural effector WAS B lymphocytes. To study SHM also in the Ig heavy chain (IGH), we analyzed the frequency of mutated nucleotides in rearranged Variable region of IGH (IGHV) genes by cloning and sequencing the most frequent IGHV subgroups, the IGHV3 and IGHV4 gene families [34], of both γ- and α-chain of Constant regions (Cγ and Cα). The mutational frequency of WAS B cells was significantly lower for both Cγ and Cα transcripts in all domains of V region (Fig. 6C and Suppl. Fig. 6). Thus, the decreased in vivo proliferation and SHM levels of WAS memory B cells could be responsible of a lower protection against infections and persistence of pathogens that finally lead to autoimmunity.
Fig. 6.
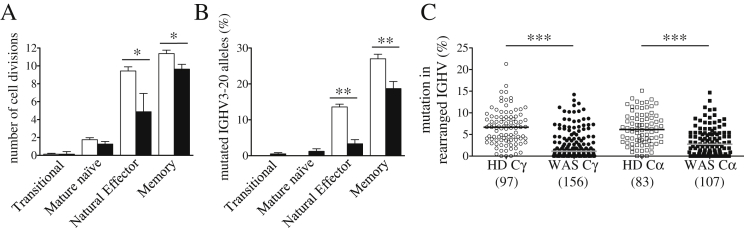
Defective in vivo B-cell maturation processes in WAS. A. Number of cell divisions was determined by KREC assay performed on DNA samples isolated from sorted peripheral B-cell subsets of 5 WAS patients (black bars) and 10 HDs (white bars). B. Frequency of somatic hypermutation, expressed as percentage of mutated hotspot in any rearranged IGκV3-20 gene segment, was determined by IgκREHMA on the same DNA samples. Bars represent mean values ± SEM. Significant values are indicated. ***P ≤ 0.0005; **P < 0.005; *P < 0.05. C. Frequencies of mutated nucleotides in rearranged IGHV genes subdivided for γ- or α-chain constant regions (Cγ or Cα) are shown. All individual data points are shown as white (HD) or black (WAS) dots and total number of analyzed sequences is reported in brackets. Differences between each WAS group compared with age-matched HDs (***P < 0.0001).
3.6. Altered selection of antibody repertoire in B cells from WAS patients
Next we studied the IGHV gene repertoire within the IGHV3 and IGHV4 subgroups. WAS B cells showed an increased frequency of IGHV3-30 in both Ig classes (Fig. 7A and Suppl. Fig. 7A) and the absence of IGHV3-48 genes in Cα sequences (Suppl. Fig. 7A). In IGHV4 transcripts, we noticed a dominant usage of the IGHV4-34 genes in WAS Cγ (Fig. 7B) and Cα transcripts (Suppl. Fig. 7B), also characterized by lower mutation frequency (Fig. 7C) [35]. Moreover, the antibody repertoire of WAS B cells was devoid of IGHV4-59, a gene commonly used in B cells from HDs [36]. The distribution of D families was only slightly altered in WAS patients (Suppl. Fig. 7C–D).
Fig. 7.

Skewed Ig gene usage of WAS B cells. A-B. Graphs show the frequency of IGHV3 (A) and IGHV4 (B) genes used by IgG of HDs (panel A, n = 7; panel B, n = 5) and WAS patients (panel A–B, n = 12). Total number of analyzed sequences is reported in brackets. C. Graph indicates the percentage of mutated (W7, A23, Y25 positions) or not mutated IGHV4-34 sequences within the FR1 region. D. Schematic representation of the constant region of the human IGH locus. E–F. Percentage distribution of IgG (E) and IgA (F) receptor subclasses used in IGH rearrangements of B cells are represented in the pie charts reflecting their order in the human IGH locus. Total number of analyzed sequences is indicated in the center of each pie. Differences were statistically analyzed by χ2 test (*P < 0.05).
Finally, we evaluated the CSR process in total B cells by analyzing the frequency of Ig subclasses in sequenced IGH transcripts. A preferential usage of γ-chain C region 3 (Cγ3) and Cγ1, IGH-proximal genes (the IGH locus is schematized in Fig. 7D), accompanied by a reduction in the expression of distal Cγ2 and Cγ4, were detected in WAS patients (Fig. 7E). The analysis of IgA transcripts showed no significant difference in the subclass usage (Fig. 7E). In conclusion WAS patients show a skewed Ig gene usage suggesting an altered selection of the antibody repertoire during B-cell development.
4. Discussion and conclusions
We reported herein that WASp-deficiency affects many aspects of B-cell development, first in the BM and then in the periphery. Indeed, precursor B-cell as well as memory B-cell development and selection appear affected by the absence of functional WASp.
In the BM we have observed a decreased frequency of immature B cells, while in the periphery an overrepresentation of transitional B cells likely due to a reduced retention in the BM and/or a decreased migration to peripheral lymphoid tissues. The interaction of the chemokine receptor CXCR4 with its ligand SDF-1α is required for the retention of developing B cells in the BM [22,24]. WASp is actively involved in CXCR4 signaling [37–39] and Was−/− murine B cells show defective migratory response to SDF-1α [10]. Consistently, here we demonstrate in WAS patients that B cells migrate less to SDF-1α. Thus, WAS immature B cells could be unable to properly sense retention signals derived from CXCR4 in the BM leading to a premature release in the periphery. The increase of transitional B cells could also reflect an altered migration to the peripheral lymphoid organs resulting in a prolonged persistence in the circulation. Consistently, our data allow us to exclude that the expansion of transitional B cells is due to homeostatic proliferation since we demonstrated the absence of replication history in this subset.
Transitional B cells are found expanded in immunodeficient conditions [40] or autoimmune diseases [41] and represent a reservoir of autoreactive B cells [42]. The cohort of patients analyzed here did not show any overt sign of autoimmunity, probably due to their young age. However, the analysis of serum autoantibodies performed in four patients showed positivity for anti-nuclear antibodies in three patients. One of them also showed the presence of anti-platelet antibodies (data not shown). Importantly, an increased frequency of CD21low B cells was observed. These cells are enriched in anergic autoreactive clones [26] and expanded in systemic lupus erythematosus (SLE), rheumatoid arthritis and in common variable immunodeficiency groupIa patients developing autoimmune syndromes [26,28]. BAFF levels and signals through BAFFR coordinate the maintenance of the primary B-cell pool and the fate of self-reactive B cells [30]. Herein, we report for the first time increased BAFF levels and decreased BAFFR expression in WAS patients. Elevated BAFF serum levels are often present in immunodeficiencies [43], autoimmune diseases [44,45] and viral infections [46] and may lower the thresholds for the survival of autoreactive B cell clones [30]. The enrichment of CD21low B cells and the alterations in BAFF levels and expression of its receptor suggest that the mechanisms of B-cell selection could be altered in WAS patients. Our data also suggest the presence of a defective selection of WAS B cells producing high-affinity antibodies. Indeed, we noticed a restricted or null presence of IGHV3-48 and IGHV4-59 gene families and a preferential usage of IGHV3-30 and IGHV4-34. In particular, IGHV3-48 gene is selectively used against polysaccharide antigens [47] and its decrease in WAS could account for an inefficient antibody response. In contrast, IGHV3-30 is highly represented among anti-platelet autoantibodies from patients with idiopathic thrombocytopenic purpura [48] and in SLE patients [49]. Additionally, IGHV4-34 – encoded antibodies are intrinsically autoreactive when unmutated [35,50], as observed in our patients. This peculiar antibody repertoire reflects an altered selection of both protective and autoreactive Ig gene families. The perturbation of B-cell homeostasis present in WAS patients supports the administration of anti-CD20 mAb in the conditioning regimen of gene therapy to deplete B cells [51].
In the memory B-cell compartment, the frequency of isotype-switched IgG+ B cells was found normal in WAS patients, despite a deficit of CD27+ B cells [17]. We observed a normal frequency of memory B cells and of switched CD27+IgG+ and CD27+IgA+ B cells in our cohort of WAS patients. This suggests that T-cell dependent antigen response is induced normally in WAS patients. In contrast, the frequency and the SHM level of TI memory B-cell subsets were reduced, likely due to their reduced in vivo proliferation. The marked reduction in the natural effector B-cell subset, resembling marginal zone B cells in the spleen, mirrors the decreased number of splenic marginal zone B cells and the histological defects in the marginal zone area of the spleen already described in Was−/− mice [9,52,53] and patients [54]. In addition, both the reduction in natural effector and IgA+CD27− B cells, which are generated during TI responses, provide evidence of a defect in B-cell function independent from the cross-talk with T lymphocytes.
The mutational status of both heavy and light Ig chains is reduced in WAS in presence of a diminished rate of proliferation in the total memory B-cell pool. In addition, the analysis of Ig gene selection showed a preferential use of IGH-proximal genes (IGHG1 and IGHG3) accompanied by reduced SHM suggesting that WAS B cells have undergone less GC reactions [33]. Reduced IgG2 switching in WAS patients could account for the poor response to TI antigens which is typical of the syndrome [15]. The cytokine profile of WAS patients shows an impairment in the production of Th1 cytokines [7] that could explain their reduced switching to IgG2 subclass. Moreover, the isotype-switching outcome is also influenced by two additional factors: cell division [55] and antibody affinity to antigens [56]. The defective proliferation of memory and natural effector B cells and the reduced affinity maturation found in WAS might contribute to these alterations in class switching.
Our results in WAS patients show an early egress of immature B cells from BM leading to an overrepresentation of transitional B cells in the periphery. The memory compartment is characterized by a reduced maturation status that could affect the B-cell effector functions contributing to a lower clearance of pathogens and leading to chronic inflammation that can break tolerance.
In conclusion, our results add novel immunological features of WAS B-cell phenotype that can complement the evaluation of the efficacy of various treatment approaches.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgments
We thank H. Ijspeert (Department of Immunology, University Medical Center Rotterdam, Rotterdam, The Netherlands) and Dr L. Devine (Yale University School of Medicine, New Haven, CT, USA) for help in the experiments and C. Brombin (San Raffaele University, Milan, Italy) in statistical analyses.
This work was supported by Telethon (A.V.), Ministero della Salute (RF 2007-2008-2009 Giovani Ricercatori Grant) (M.B.), fellowship from European Society of Immunodeficiencies (M.C.C.) and by NIH-NIAID (Grants AI061093, AI071087, AI082713 and AI095848) (E.M.). M.v.d.B. is supported by Dutch Scientific Organization (ZonMW Vidi grant 016.126.323) and M.C.v.Z. by an Erasmus University Rotterdam (EUR-) Fellowship. M.C.C. conducted this study as partial fulfillment of her PhD in Molecular Medicine, Program in Basic and Applied Immunology, San Raffaele University, Milan, Italy.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Appendix A. Supplementary data
References
- 1.Bosticardo M., Marangoni F., Aiuti A., Villa A., Grazia Roncarolo M. Recent advances in understanding the pathophysiology of Wiskott–Aldrich syndrome. Blood. 2009;113:6288–6295. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- 2.Derry J.M., Ochs H.D., Francke U. Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- 3.Villa A., Notarangelo L., Macchi P., Mantuano E., Cavagni G., Brugnoni D. X-linked thrombocytopenia and Wiskott–Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat Genet. 1995;9:414–417. doi: 10.1038/ng0495-414. [DOI] [PubMed] [Google Scholar]
- 4.Thrasher A.J., Burns S.O. WASP: a key immunological multitasker. Nat Rev Immunol. 2010;10:182–192. doi: 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- 5.Dupuis-Girod S., Medioni J., Haddad E., Quartier P., Cavazzana-Calvo M., Le Deist F. Autoimmunity in Wiskott–Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111:e622–e627. doi: 10.1542/peds.111.5.e622. [DOI] [PubMed] [Google Scholar]
- 6.Imai K., Morio T., Zhu Y., Jin Y., Itoh S., Kajiwara M. Clinical course of patients with WASP gene mutations. Blood. 2004;103:456–464. doi: 10.1182/blood-2003-05-1480. [DOI] [PubMed] [Google Scholar]
- 7.Catucci M., Castiello M.C., Pala F., Bosticardo M., Villa A. Autoimmunity in Wiskott–Aldrich syndrome: an unsolved Enigma. Front Immunol. 2012;3:209. doi: 10.3389/fimmu.2012.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker-Herman S., Meyer-Bahlburg A., Schwartz M.A., Jackson S.W., Hudkins K.L., Liu C. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. 2011;208:2033–2042. doi: 10.1084/jem.20110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Recher M., Burns S.O., de la Fuente M.A., Volpi S., Dahlberg C., Walter J.E. B cell-intrinsic deficiency of the Wiskott–Aldrich syndrome protein causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. 2012;119:2819–2828. doi: 10.1182/blood-2011-09-379412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westerberg L., Larsson M., Hardy S.J., Fernandez C., Thrasher A.J., Severinson E. Wiskott–Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood. 2005;105:1144–1152. doi: 10.1182/blood-2004-03-1003. [DOI] [PubMed] [Google Scholar]
- 11.Facchetti F., Blanzuoli L., Vermi W., Notarangelo L.D., Giliani S., Fiorini M. Defective actin polymerization in EBV-transformed B-cell lines from patients with the Wiskott–Aldrich syndrome. J Pathol. 1998;185:99–107. doi: 10.1002/(SICI)1096-9896(199805)185:1<99::AID-PATH48>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 12.Liu C., Miller H., Hui K.L., Grooman B., Bolland S., Upadhyaya A. A balance of Bruton's tyrosine kinase and SHIP activation regulates B cell receptor cluster formation by controlling actin remodeling. J Immunol. 2011;187:230–239. doi: 10.4049/jimmunol.1100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon H.U., Mills G.B., Hashimoto S., Siminovitch K.A. Evidence for defective transmembrane signaling in B cells from patients with Wiskott–Aldrich syndrome. J Clin Invest. 1992;90:1396–1405. doi: 10.1172/JCI116006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henriquez N.V., Rijkers G.T., Zegers B.J. Antigen receptor-mediated transmembrane signaling in Wiskott–Aldrich syndrome. J Immunol. 1994;153:395–399. [PubMed] [Google Scholar]
- 15.Ochs H.D., Thrasher A.J. The Wiskott–Aldrich syndrome. J Allergy Clin Immunol. 2006;117:725–738. doi: 10.1016/j.jaci.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Park J.Y., Kob M., Prodeus A.P., Rosen F.S., Shcherbina A., Remold-O'Donnell E. Early deficit of lymphocytes in Wiskott–Aldrich syndrome: possible role of WASP in human lymphocyte maturation. Clin Exp Immunol. 2004;136:104–110. doi: 10.1111/j.1365-2249.2004.02409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J.Y., Shcherbina A., Rosen F.S., Prodeus A.P., Remold-O'Donnell E. Phenotypic perturbation of B cells in the Wiskott–Aldrich syndrome. Clin Exp Immunol. 2005;139:297–305. doi: 10.1111/j.1365-2249.2005.02693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Zelm M.C., Reisli I., van der Burg M., Castano D., van Noesel C.J., van Tol M.J. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 19.van Zelm M.C., Szczepanski T., van der Burg M., van Dongen J.J. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007;204:645–655. doi: 10.1084/jem.20060964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noordzij J.G., de Bruin-Versteeg S., Verkaik N.S., Vossen J.M., de Groot R., Bernatowska E. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood. 2002;100:2145–2152. [PubMed] [Google Scholar]
- 21.van Lochem E.G., van der Velden V.H., Wind H.K., te Marvelde J.G., Westerdaal N.A., van Dongen J.J. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age-related changes and disease-induced shifts. Cytometry B Clin Cytom. 2004;60:1–13. doi: 10.1002/cyto.b.20008. [DOI] [PubMed] [Google Scholar]
- 22.Ma Q., Jones D., Springer T.A. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463–471. doi: 10.1016/s1074-7613(00)80046-1. [DOI] [PubMed] [Google Scholar]
- 23.Nie Y., Waite J., Brewer F., Sunshine M.J., Littman D.R., Zou Y.R. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. 2004;200:1145–1156. doi: 10.1084/jem.20041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira J.P., An J., Xu Y., Huang Y., Cyster J.G. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol. 2009;10:403–411. doi: 10.1038/ni.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdei A., Isaak A., Torok K., Sandor N., Kremlitzka M., Prechl J. Expression and role of CR1 and CR2 on B and T lymphocytes under physiological and autoimmune conditions. Mol Immunol. 2009;46:2767–2773. doi: 10.1016/j.molimm.2009.05.181. [DOI] [PubMed] [Google Scholar]
- 26.Isnardi I., Ng Y.S., Menard L., Meyers G., Saadoun D., Srdanovic I. Complement receptor 2/CD21-human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wehr C., Eibel H., Masilamani M., Illges H., Schlesier M., Peter H.H. A new CD21low B cell population in the peripheral blood of patients with SLE. Clin Immunol. 2004;113:161–171. doi: 10.1016/j.clim.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Warnatz K., Wehr C., Drager R., Schmidt S., Eibel H., Schlesier M. Expansion of CD19(hi)CD21(lo/neg) B cells in common variable immunodeficiency (CVID) patients with autoimmune cytopenia. Immunobiology. 2002;206:502–513. doi: 10.1078/0171-2985-00198. [DOI] [PubMed] [Google Scholar]
- 29.Rakhmanov M., Keller B., Gutenberger S., Foerster C., Hoenig M., Driessen G. Circulating CD21low B cells in common variable immunodeficiency resemble tissue homing, innate-like B cells. Proc Natl Acad Sci U S A. 2009;106:13451–13456. doi: 10.1073/pnas.0901984106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancro M.P. Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol. 2009;9:657–661. doi: 10.1038/nri2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Ann N Y Acad Sci. 2011;1246:1–10. doi: 10.1111/j.1749-6632.2011.06347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Driessen G.J., van Zelm M.C., van Hagen P.M., Hartwig N.G., Trip M., Warris A. B-cell replication history and somatic hypermutation status identify distinct pathophysiological backgrounds in common variable immunodeficiency. Blood. 2011;118:6814–6823. doi: 10.1182/blood-2011-06-361881. [DOI] [PubMed] [Google Scholar]
- 33.Berkowska M.A., Driessen G.J., Bikos V., Grosserichter-Wagener C., Stamatopoulos K., Cerutti A. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood. 2011;118:2150–2158. doi: 10.1182/blood-2011-04-345579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brezinschek H.P., Brezinschek R.I., Lipsky P.E. Analysis of the heavy chain repertoire of human peripheral B cells using single-cell polymerase chain reaction. J Immunol. 1995;155:190–202. [PubMed] [Google Scholar]
- 35.Potter K.N., Hobby P., Klijn S., Stevenson F.K., Sutton B.J. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. 2002;169:3777–3782. doi: 10.4049/jimmunol.169.7.3777. [DOI] [PubMed] [Google Scholar]
- 36.Ng Y.S., Wardemann H., Chelnis J., Cunningham-Rundles C., Meffre E. Bruton's tyrosine kinase is essential for human B cell tolerance. J Exp Med. 2004;200:927–934. doi: 10.1084/jem.20040920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haddad E., Zugaza J.L., Louache F., Debili N., Crouin C., Schwarz K. The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis. Blood. 2001;97:33–38. doi: 10.1182/blood.v97.1.33. [DOI] [PubMed] [Google Scholar]
- 38.Okabe S., Fukuda S., Broxmeyer H.E. Activation of Wiskott–Aldrich syndrome protein and its association with other proteins by stromal cell-derived factor-1alpha is associated with cell migration in a T-lymphocyte line. Exp Hematol. 2002;30:761–766. doi: 10.1016/s0301-472x(02)00823-8. [DOI] [PubMed] [Google Scholar]
- 39.Stabile H., Carlino C., Mazza C., Giliani S., Morrone S., Notarangelo L.D. Impaired NK-cell migration in WAS/XLT patients: role of Cdc42/WASp pathway in the control of chemokine-induced beta2 integrin high-affinity state. Blood. 2010;115:2818–2826. doi: 10.1182/blood-2009-07-235804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avery D.T., Deenick E.K., Ma C.S., Suryani S., Simpson N., Chew G.Y. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207:155–171. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sims G.P., Ettinger R., Shirota Y., Yarboro C.H., Illei G.G., Lipsky P.E. Identification and characterization of circulating human transitional B cells. Blood. 2005;105:4390–4398. doi: 10.1182/blood-2004-11-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wardemann H., Yurasov S., Schaefer A., Young J.W., Meffre E., Nussenzweig M.C. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 43.Kreuzaler M., Rauch M., Salzer U., Birmelin J., Rizzi M., Grimbacher B. Soluble BAFF levels inversely correlate with peripheral B cell numbers and the expression of BAFF receptors. J Immunol. 2012;188:497–503. doi: 10.4049/jimmunol.1102321. [DOI] [PubMed] [Google Scholar]
- 44.Groom J., Kalled S.L., Cutler A.H., Olson C., Woodcock S.A., Schneider P. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J Clin Invest. 2002;109:59–68. doi: 10.1172/JCI14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker-Merok A., Nikolaisen C., Nossent H.C. B-lymphocyte activating factor in systemic lupus erythematosus and rheumatoid arthritis in relation to autoantibody levels, disease measures and time. Lupus. 2006;15:570–576. doi: 10.1177/0961203306071871. [DOI] [PubMed] [Google Scholar]
- 46.Fontaine J., Chagnon-Choquet J., Valcke H.S., Poudrier J., Roger M. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood. 2011;117:145–155. doi: 10.1182/blood-2010-08-301887. [DOI] [PubMed] [Google Scholar]
- 47.Baxendale H.E., Goldblatt D. Correlation of molecular characteristics, isotype, and in vitro functional activity of human antipneumococcal monoclonal antibodies. Infect Immun. 2006;74:1025–1031. doi: 10.1128/IAI.74.2.1025-1031.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roark J.H., Bussel J.B., Cines D.B., Siegel D.L. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. 2002;100:1388–1398. [PubMed] [Google Scholar]
- 49.Roben P., Barbas S.M., Sandoval L., Lecerf J.M., Stollar B.D., Solomon A. Repertoire cloning of lupus anti-DNA autoantibodies. J Clin Invest. 1996;98:2827–2837. doi: 10.1172/JCI119111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pugh-Bernard A.E., Silverman G.J., Cappione A.J., Villano M.E., Ryan D.H., Insel R.A. Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001;108:1061–1070. doi: 10.1172/JCI12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott–Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westerberg L.S., de la Fuente M.A., Wermeling F., Ochs H.D., Karlsson M.C., Snapper S.B. WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood. 2008;112:4139–4147. doi: 10.1182/blood-2008-02-140715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bosticardo M., Draghici E., Schena F., Sauer A.V., Fontana E., Castiello M.C. Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott–Aldrich syndrome. J Allergy Clin Immunol. 2011;127:1376–1384 e5. doi: 10.1016/j.jaci.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 54.Vermi W., Blanzuoli L., Kraus M.D., Grigolato P., Donato F., Loffredo G. The spleen in the Wiskott–Aldrich syndrome: histopathologic abnormalities of the white pulp correlate with the clinical phenotype of the disease. Am J Surg Pathol. 1999;23:182–191. doi: 10.1097/00000478-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Hodgkin P.D., Lee J.H., Lyons A.B. B cell differentiation and isotype switching is related to division cycle number. J Exp Med. 1996;184:277–281. doi: 10.1084/jem.184.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Turner M.L., Corcoran L.M., Brink R., Hodgkin P.D. High-affinity B cell receptor ligation by cognate antigen induces cytokine-independent isotype switching. J Immunol. 2010;184:6592–6599. doi: 10.4049/jimmunol.0903437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.