

Abstract
Integrins are important therapeutic targets. However, current RGD-based anti-integrin drugs are also partial agonists, inducing conformational changes that trigger potentially fatal immune reactions and paradoxical cell adhesion. Here we describe the first crystal structure of αVβ3 bound to a physiologic ligand: the 10th type III RGD-domain of wild-type fibronectin (wtFN10), or to a high affinity mutant (hFN10) that acts as a pure antagonist. Comparison of these structures revealed a central π - π interaction between Trp1496 in the RGD-containing loop of hFN10 and Tyr122 of the β3-subunit that blocked conformational changes triggered by wtFN10, and trapped hFN10-bound αVβ3 in an inactive conformation. Removing the Trp1496 or Tyr122 side-chains, or reorienting Trp1496 away from Tyr122, converted hFN10 into a partial agonist. The findings offer new insights on the mechanism of integrin activation and a basis for design of RGD-based pure antagonists.
Introduction
Integrins are α/β heterodimeric cell adhesion receptors which consist of a bilobular head and two legs that span the plasma membrane1–2. Integrins are unusual receptors, as they normally exist on the cell surface in an inactive state, unable to engage physiologic ligand. This is critical for integrin biology as it allows, for example, patrolling blood platelets and immune cells to circulate with minimal aggregation or interaction with vessel walls. Physiologic stimuli (e.g. chemokines), acting through the short integrin cytoplasmic tails, induce allosteric changes in the ectodomain required for extracellular ligand binding (“inside-out” activation)3. Binding of physiologic ligands induces “outside-in” signaling by initiating additional structural rearrangements in the ectodomain4, which induce conformational epitopes (Ligand-induced binding sites, LIBS) such as those recognized by monoclonal antibodies AP5 (ref. 5), LIBS-1 and LIBS-6 (ref. 6). These ligand-induced structural rearrangements trigger cell spreading7 via connections between the integrin cytoplasmic tails and filamentous actin8. Disruption of these processes contributes to the pathogenesis of many diseases9–11.
Despite the clinical efficacy of cyclic RGD-like molecules that target platelet integrin αIIbβ3 in preventing thrombosis, parenteral ligand-mimetic antagonists of αIIbβ3, such as the cyclic heptapeptide eptifibatide, can induce severe thrombocytopenia—a major life-threatening complication—in up to 2% of treated patients12. In addition, during oral therapy of acute coronary syndromes, RGD-mimetic drugs paradoxically induced thrombosis and mortality, contributing to the failure of clinical trials9.
Crystal structures of integrin ectodomains in complex with small RGD-based peptidomimetics13,14 show the RGD motif binding the integrin head: Arg contacts the Propeller domain of the α-subunit, and Asp binds the βA domain of the β-subunit at a metal-ion-dependent-adhesion-site (MIDAS) via Mg2+ (or Mn2+). Two regulatory Ca2+ cations at the ligand-associated metal binding site (LIMBS or synergistic, SyMBS) and at adjacent to MIDAS (ADMIDAS), flank the MIDAS metal ion13. The Ca2+ at ADMIDAS links the N- and C-terminal helices, stabilizing the inactive conformation15. Binding of ligand-mimetic compounds is mechanically coupled to tertiary changes in the βA domain involving inward movement of the N-terminal α1 helix towards the MIDAS, forcing reorganization of the C-terminal F-strand/α7 loop, a one-turn displacement of helix α7 and a Hybrid domain swing-out (reviewed in ref. 3). These movements may persist after dissociation of these compounds, contributing to immune thrombocytopenia16, and facilitating physiologic ligand binding to a “primed” integrin, an effect that may have contributed to the increased mortality in patients treated with RGD-mimetic compounds targeting platelet αIIbβ3 (ref. 17). RGD-containing αVβ3 antagonists (e.g. cilengitide18) can paradoxically stimulate model tumor growth and angiogenesis19, possibly through partial agonism of αVβ3. A recent attempt to address the issue of partial agonism utilized a non-RGD small molecule that acts competitively to destabilize Mg2+ at MIDAS in integrin αIIbβ3, an effect reversed at higher Mg2+ concentrations20. To date however no RGD-based pure antagonists have been identified.
In the modular matrix protein fibronectin (FN), the wild-type 10 kDa 10th type III domain (wtFN10) is necessary and sufficient for binding to αVβ3 (ref. 21). Here we report the first crystal structure of an integrin αVβ3 ectodomain bound to a physiologically relevant ligand: the FN10 domain. We show that in contrast wtFN10, a high affinity form of this ligand (hFN10) acts as a pure antagonist for αVβ3. To clarify the structural basis for this activity, we also determined the crystal structure of the hFN10–αVβ3. Comparisons of the structures revealed a novel mechanism underlying pure antagonism by an RGD containing ligand that suggests a path to the generation of RGD-based drugs that can act as pure antagonists.
RESULTS
Binding of hFN10 to resting and activated cellular αVβ3
We compared the integrin binding properties of wtFN10 to those of a high affinity form of FN10 (hFN10) that was selected for specific αVβ3 binding from a FN10 phage display library, where five residues N- and C-terminal to the RGD motif were randomized22. In hFN10, the sequence 1492PRGDWNEG replaces 1492GRGDSPAS of wtFN10. Interestingly, RGDWN is also the core sequence in the disintegrin barbourin (on which the drug eptifibatide was based23), excepting an R-to-K substitution, which enhances specificity of barbourin for platelet αIIbβ3 over αVβ3 (ref. 14).
The binding of fluoresceinated fluid-phase wtFN10 to stably expressed wild-type αVβ3 in K562 cells (K562-αVβ3) was a low background in physiological concentrations of Ca2+ and Mg2+ (Ca2+-Mg2+, 1mM each) (Fig. 1a), as expected, since soluble physiologic ligands do not bind to the inactive integrin in physiological Ca2+ /Mg2+ containing buffers. Binding of soluble wtFN10 was increased 10-fold in presence of the integrin activator Mn2+, a mimic of inside-out activation24 (Fig. 1a), or 6-fold by an N339S mutation in the βA domain, which constitutively activates αVβ3 both in vitro and in vivo25)(Fig. 1b). By contrast, strong binding, 6-fold background, of soluble fluoresceinated hFN10 to K562-αVβ3 occurred in Ca2+-Mg2+ and was increased a further ~1.5 fold by Mn2+ (Fig. 1a). Binding of hFN10 was similar both on wild type (WT) αVβ3 and to αVβ3(N339S), each transiently-expressed on HEK293T cells (Fig. 1b).
Figure 1. Binding properties of hFN10 and wtFN10 to αVβ3.

Binding of fluoresceinated wtFN10 (a) and hFN10 (b) or LIBS mAbs (alone (c) or in presence of wtFN10 and hFN10 (d) to αVβ3+ cells. In (c), mAb-binding was assessed using K562-αVβ3 and M21 cells. MFI, mean fluorescence intensity. Histograms in a-d represent mean+SD, n=3 independent experiments. (e) Hydrodynamic analyses of unliganded αVβ3 and αVβ3-FN10 complexes in presence of Ca2+, Ca2+-Mg2+ or Mn2+. Stokes radii (in nm) are shown in parentheses. AU: absorbance unit. (f–h) Mn2+-induced spreading of K562-αVβ3 on wtFN10 (f), hFN10 (g) (mean+SD, n=3 independent experiments), or on full-length FN (f) (two independent experiments are shown). Spreading under all conditions was eliminated by mAb LM609 against αVβ3 (not shown). (g,h) Representative phase contrast images of K562-αVβ3 spreading on wtFN10 (g) and hFN10 (h). Bar = 20µM.
Effects hFN10 binding to αVβ3
The integrin activator Mn2+ induces expression of LIBS mAbs AP5 (ref. 5), LIBS-1 and LIBS-2 (ref. 6), each recognizing distinct epitopes in the β3-subunit. mAb AP5 recognizes an N-terminal sequence in the PSI (plexin-semaphorin-integrin) domain5, mAb LIBS-1 recognizes a different epitope from AP5, and LIBS-6 binds the C-terminal membrane proximal βTD domain. LIBS expression is further increased upon binding of physiologic ligand5.
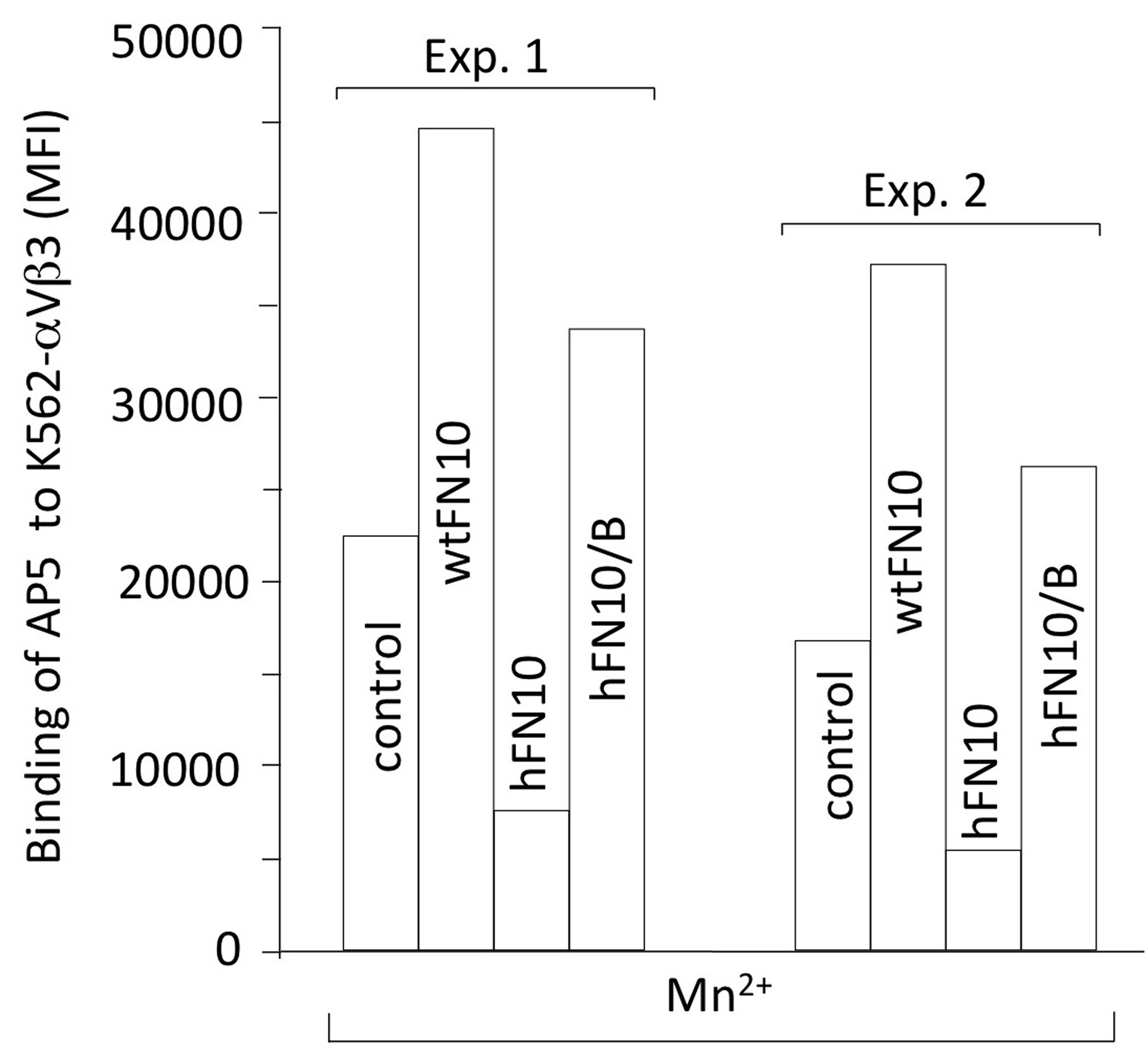
As expected, Mn2+-driven binding of wtFN10 to K562-αVβ3 increased expression of LIBS mAbs AP5, LIBS-1 and LIBS-6 above that induced by Mn2+ alone (Fig. 1c). In contrast, binding of hFN10 not only did not induce LIBS expression, but also significantly decreased LIBS expression induced by Mn2+ alone (Fig. 1c). We saw this effect whether αVβ3 was expressed artificially (on K562 cells) or constitutively (on melanoma M21 cells). And while binding of wtFN10 to the constitutively active αVβ3(N339S) integrin, increased AP5 epitope expression by ~2-fold, binding of hFN10 did not (Fig. 1d).
It is known that Mn2+ or the binding of soluble RGD-based ligands induces conformational changes in αVβ3 ectodomain, detected by changes in the hydrodynamic radius of the ectodomain4,26,27. We therefore examined the effects of binding of hFN10 to the αVβ3 ectodomain in solution by comparing the hydrodynamic radii of αVβ3-hFN10 and αVβ3-wtFN10 complexes by molecular sieve chromatography4,27. Preformed αVβ3-hFN10 and αVβ3-wtFN10 complexes (Supplementary Fig. 1a) were chromatographed on a molecular sieve column equilibrated in the relevant metal ion-containing buffer, and Stokes’ radii were derived as described previously4. The complexes displayed dramatically different profiles (Fig. 1e and Supplementary Fig. 2b): wtFN10 increased the Stokes’ radius (Rs) of αVβ3 in Mn2+ (6.6nm), compared to the integrin size in Mn2+ alone (Rs 6.3 nm), as expected. However, hFN10 had little effect on the Rs of αVβ3 in Mn2+ (6.3 nm) or in Ca2+/Mg2+ (6.0 nm vs. 5.9 nm in the absence of hFN10).
Cell spreading is a reporter of ligand-induced outside-in signaling28. To determine the effect of hFN10 on spreading, we compared spreading of αVβ3-expressing cells on surfaces coated with native full-length FN (positive control) (Fig.1f), wtFN10 (Fig.1f, g) or hFN10 (Fig. 1f, h). After 2h, approximately 90% of attached cells spread on native FN and 60% on wtFN10. In contrast, less than 20% of attached cells spread on hFN10. Cell attachment under all conditions was eliminated when assays were carried out in presence of the function-blocking LM609 mAb against αVβ3 (not shown).
Crystal structures of αVβ3-wtFN10 and αVβ3-hFN10 complexes
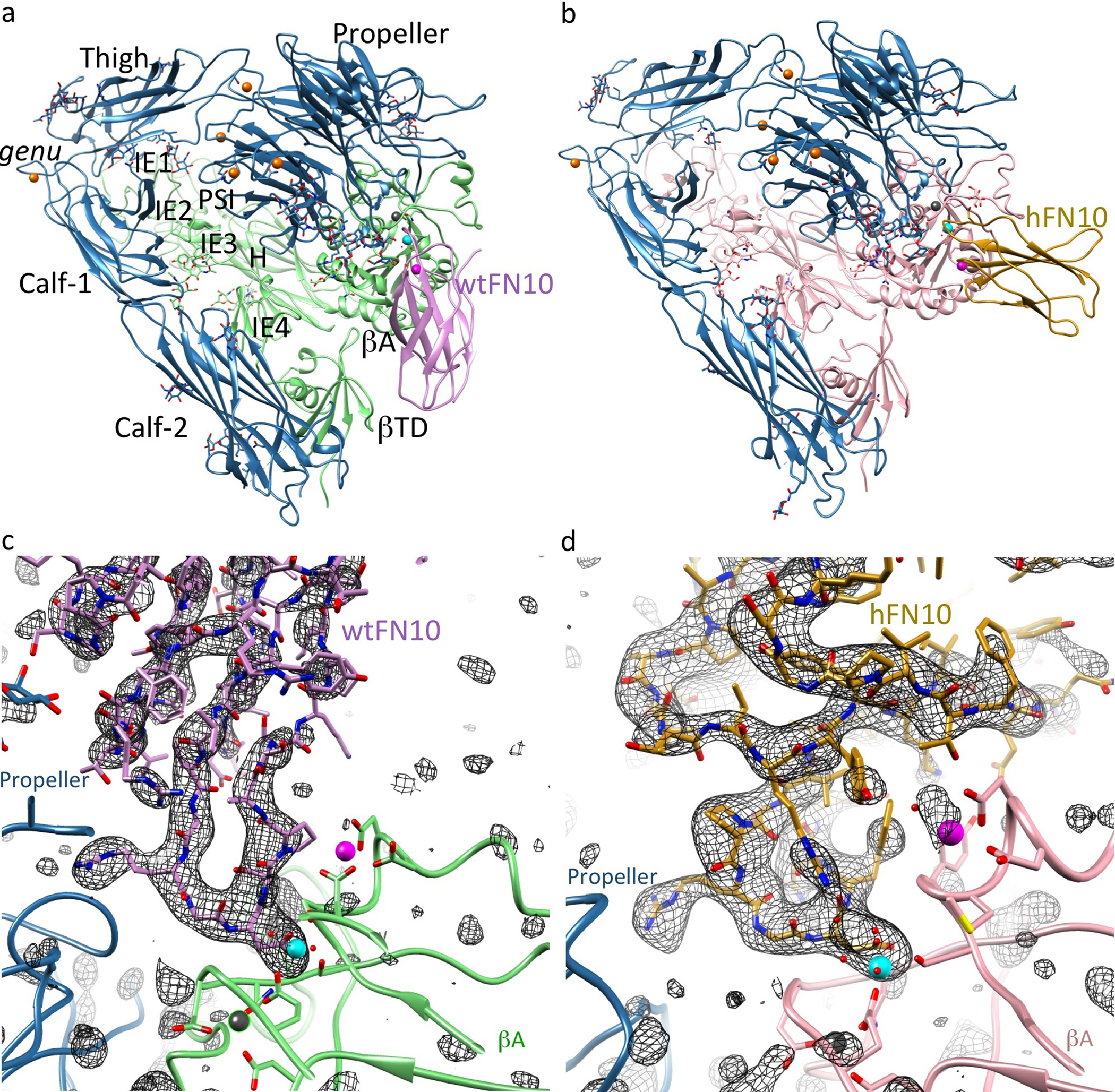
To clarify the structural basis for the inhibitory effects of bound hFN10 on conformational changes and function of αVβ3, we soaked the macromolecular ligands hFN10 or wtFN10 into crystals of the αVβ3 ectodomain4 in 2mM MnCl2, and determined the crystal structures of the resulting αVβ3-hFN10 and αVβ3-wtFN10 complexes (Fig. 2a, b, Supplementary Fig. 2, and Table 1). hFN10- or wtFN10-bound αVβ3 remained genuflected, with each ligand bound at the integrin head, as expected. However, orientation of FN10 relative to the βA domain differed dramatically between the two complexes, with a ~60° rotation around the RGD-loop (Fig. 2c). Fo-Fc omit maps (generated after omitting the FN10 ligand), revealed clear positive densities (Supplementary Fig. 2c, d), reflecting stable engagement of the integrin head by ligand. The omit maps showed clear density for the complete hFN10 domain, but for only ~60% of wtFN10, that facing the integrin, with the wtFN10 segment farthest away from the integrin showing minimal density, consistent with its low affinity and the likely flexibility of this region in the crystal.
Figure 2. Structures of αVβ3 bound to FN10.
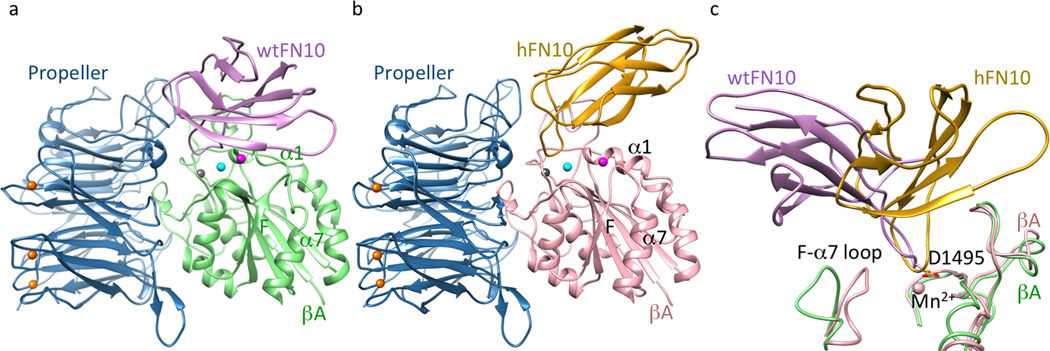
Ribbon diagrams of αVβ3 head bound to wtFN10 (a) or hFN10 (b). Orientation of the integrin head in (a) and (b) is identical. Mn2+ ions at LIMBS (gray), MIDAS (cyan) and ADMIDAS (magenta) are shown as spheres (also in Figs. 3a–b, 4c). (c) Orientation of bound FN10 relative to the superimposed βA domains (chain colors as in a, b). Mn2+ at MIDAS the ligand Asp (D1495) and the F-α7 loop are shown.
Table 1.
Data collection and refinement statistics (molecular replacement)
| αVβ3–hFN10 | αVβ3–wtFN10 | αVβ3–hFN10/B | |
|---|---|---|---|
| Data collection | |||
| Space group | P3221 | P3221 | P3221 |
| Cell dimensions | |||
| a, b, c (Å) | 129.8, 129.8, 307.6 | 129.7, 129.7, 305.8 | 130.0, 130.0, 308.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 50-3.1 (3.21–3.1)* | 75.49-3.32 (3.5–3.32) | 50-3.17 (3.28–3.17) |
| Rsym | 7.9 (79.7) | 10.9 (67.2) | 10.4 (89.4) |
| I / σ I | 27.1 (2.3) | 13.9 (3.3) | 12.6 (2.2) |
| Completeness (%) | 99.9 (100) | 88.0 (88.0) | 99.7 (99.7) |
| Redundancy | 6.1 (6.1) | 6.2 (6.4) | 5.3 (5.1) |
| Refinement | |||
| Resolution (Å) | 49.3–3.1 | 42.5–3.32 | 49.4–3.18 |
| No. reflections | 55,243 | 39,536 | 51,260 |
| Rwork / Rfree | 20.5/ 25.5 | 21.1/ 25.8 | 20.5/ 23.9 |
| No. atoms | 13,501 | 13,626 | 13,000 |
| Protein | 12,794 | 12,922 | 12,922 |
| Ligand/Ion | |||
| FN10 | 690 | 694 | 65 |
| Mn2+ | 8 | 8 | 8 |
| Water | 9 | 2 | 5 |
| B factors | |||
| All atoms (Å2) | 116.7 | 102.8 | 75.8 |
| Protein | 114.2 | 98.5 | 75.7 |
| Ligand/Ion | |||
| FN10 | 163.2 | 181.8 | 83.7 |
| Mn2+ | 135.9 | 102.9 | 80.5 |
| Water | 95.2 | 67.7 | 54.8 |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.004 | 0.003 | 0.005 |
| Bond angles(°) | 0.89 | 0.9 | 0.98 |
Values in parenthesis are for highest-resolution shell. One crystal was used for each dataset.
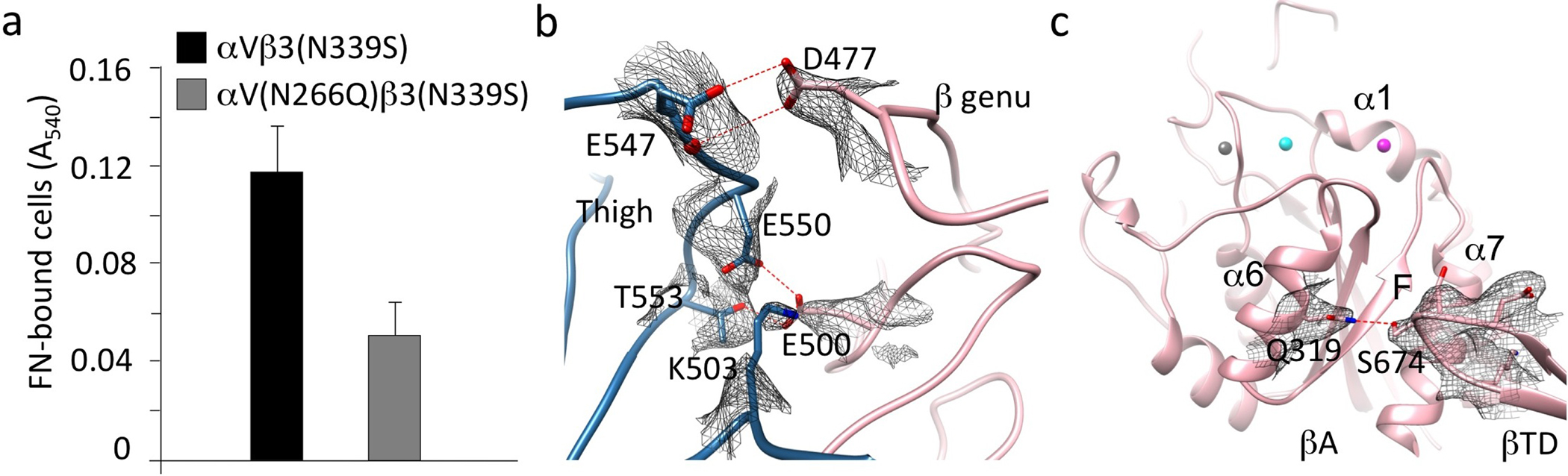
The RGD motif of each ligand bound the αVβ3 head in an identical manner (Fig. 3a, b), and as shown previously for the RGD-containing pentapeptide, cilengitide13: RGD inserted into the crevice between the Propeller and βA domains, and contacted both. The αVβ3-wtFN10 interface was modestly larger than the αVβ3-cilengitide interface, mainly due to contacts wtFN10 made with the glycan at the propeller residue Asn266, which included H-bonds with mannose 2271 (MAN2271) (Fig. 3a). An N266Q substitution in cellular αVβ3 did not impair heterodimer formation (as judged by binding of the heterodimer-specific mAb LM609, not shown) but reduced adhesion of HEK293T cells expressing the constitutively active mutant integrin αV(N266Q)β3(N339S) to immobilized full-length FN by 56% vs. adhesion mediated by αVβ3(N339S) in Ca2+-Mg2+ buffer (p=0.003, n=3 independent experiments)(Supplementary Fig.3a).
Figure 3. αVβ3-FN10 interfaces, conformational changes and structure validation.

Ribbon diagrams showing key electrostatic and H-bond interactions and metal ion coordinations in αVβ3-wtFN10 (a) and αVβ3-hFN10 (b) structures. Chain colors are as in Fig. 2. Inset in b, enlarged view of σA weighted 2Fo-Fc map contoured at 1.0σ of Trp1496 and Tyr122 side-chains in αVβ3-hFN10 complex. Inward movement (blue arrow) of Tyr122 (in light green) in wtFN10-bound βA would clash with Trp1496 side chain. (c) Left panel, βA domain from αVβ3-hFN10 (in pink) superimposed on that of αVβ3-wtFN10 (in light green) and on βA domain (in dark green) from unliganded αVβ3 (pdb 3ije) (right panel). Blue arrow in left panel in (c) indicates direction of wtFN10-induced inward movement of α1 helix (and ADMIDAS ion) towards MIDAS. Spheres representing the three metal ions bear the color of respective βA. The major tertiary change observed in the F-α7 loop of wtFN10-bound βA (c, left panel) was not translated into a one-turn displacement of α7, possibly the result of crystal contacts when the complete ectodomain is used in crystallization. Except for ligand-occupancy and resulting changes in SDL loop, structures of unliganded- and hFN10-bound βA domains are identical (c, right panel) (LIMBS and MIDAS are not occupied by metal in unliganded βA). (d) Left panel, binding of fluoresceinated AP5 to M21 cells in absence (control) or presence of unlabeled wtFN10, hFN10 or hFN10W/S. Right panel, binding of fluoresceinated AP5 to αVβ3+ or or αVβ3(Y122A) + HEK293T in presence of unlabeled wt- or hFN10. Histograms represent mean±SD, n=3 independent experiments.
One structural feature which coincided with the higher affinity of hFN10 compared to wtFN10 was the more extensive αVβ3-hFN10 interface, largely due to the additional and distinct contacts hFN10 made mainly with the MIDAS face and with the specificity-determining loop (SDL) of the βA domain (Fig. 3b). These contributed to the different orientation of hFN10 on αVβ3 and the coordination patterns of the Mn2+ ion at MIDAS and ADMIDAS (Fig. 3b). In contrast to the αVβ3-wtFN10 interface, hFN10 made no contacts with the Propeller glycan at Asn266. At the center of the hFN10-βA contacts was a π - π edge-to-face interaction of the mutant Trp1496 in hFN10 with Tyr122 in the α1 helix of βA (Fig. 3b). Tyr122 also H-bonded to Glu1462 of hFN10, which also formed a salt bridge with Lys125 of βA. The hydroxyl oxygen of hFN10-Tyr1446 also coordinated the Mn2+ at ADMIDAS via a water molecule. The outcome of these interactions was that the tertiary changes induced by the physiologic ligand wtFN10 (i.e. inward movement of the N-terminal α1 helix towards MIDAS, and reorganization of the C-terminal F-strand/α7 loop) were absent in the hFN10-bound βA domain (Fig. 3c). Indeed, the structure of hFN10-bound βA was superimposable on that of the unliganded βA domain (Fig. 3c). That is to say: the hFN10-bound βA domain assumed the same conformation as the unliganded βA domain. The hFN10-bound αVβ3 structure also displayed other features of an inactive state4 that were absent from the αVβ3-wtFN10 structure. These included electrostatic interactions between the two subunits at the β-genu (Supplementary Fig. 3b), and between the βA and βTD within the β3 subunit (Supplementary Fig. 3c). Together these findings suggested that when bound to hFN10, αVβ3 was at or close to an inactive ground state that would not transduce outside-in signals, despite ligand occupancy.
The Trp1496-Tyr122 π - π interaction was crucial for blocking conformational changes induced by binding of ligand as shown in mutational studies. Removing Trp1496 side chain, through Trp1496 to Ser substitution (hFN10W/S) converted hFN10 into a partial agonist that could induce binding of the LIBS mAb AP5 to cellular αVβ3 (Fig. 3d)29. Similarly, when the Tyr122 side chain was removed, through a Tyr to Ala mutation in the βA domain, hFN10-bound αVβ3(Y122A) now also expressed LIBS (Fig. 3d).
Reorienting Trp1496 converts hFN10 into a partial agonist
The core RGDWN sequence in hFN10 matches that in barbourin, a partial agonist of αIIbβ3. Yet, superposition of the RGD-containing loops in hFN10, barbourin (pdb id 1q7j) and the drug eptifibatide (pdb id 2vdn14) revealed drastically different orientations of the Trp side-chain in hFN10 compared to that in barbourin/eptifibatide. In contrast to Trp1496 side-chain in hFN10, this points inwards towards the center of the RGD loop in barbourin/eptifibatide structures, away from the critical Tyr122 side chain of the βA domain (Fig. 4a). And binding of either barbourin or eptifibatide induces expression of AP5 LIBS14. We mutated three loop residues flanking the RGDWN sequence in hFN10 (TPRGDWNE) so as to match the barbourin sequence (IAKGDWND), and purified this hFN10/B domain. The crystal structure of the αVβ3-hFN10/B complex (Supplementary Fig. 4) showed that the main-chain and side-chain structure of the RGD-loop residues IARGDWN were clear in the Fo-Fc omit map (Supplementary Fig. 4), but the rest of hFN10/B domain was not visible, likely reflecting flexibility at the new N- and C-termini of the RGD-containing loop. It was clear however, that the main-chain structure of this loop changed to position the Cα and Cβ of Trp1496 in the αVβ3-hFN10/B complex as in barbourin or eptifibatide (Fig. 4b), with the Trp1496 side-chain in hFN10/B repositioned so that it no longer faces the Tyr122 side-chain of βA (Fig. 4c). This reorientation was associated with the unhindered inward movement of Tyr122 in the α1 helix, as in the eptifibatide-bound integrin14. The binding of hFN10/B to cellular αVβ3 consistently induced expression of mAb AP5 LIBS similar to that induced by wtFN10 (Fig. 4d and Supplementary Fig. 5).
Figure 4. RGD-containing loop structures in wild type and modified FN10.

(a) Superimposed R/KGD-containing loops of hFN10, eptifibatide (pdb id 2vdn) and barbourin (pdb id 1q7j, model 2). Residues R/KGDWN common to hFN10 and barbourin are labeled in black and the three flanking residues are in the respective loop color. (b) Superimposed structures of RGD-containing loops of αVβ3-hFN10, αVβ3-wtFN10, barbourin and αVβ3-hFN10/B. The position the Cα and Cβ of Trp1496 in the αVβ3-hFN10/B complex is as that in barbourin or eptifibatide. (c) Main ionic interactions at the αVβ3-hFN10/B interface involving the RGD-containing loop (in dark cyan). (d) Binding (mean±SD, n=3 independent experiments) of fluoresceinated AP5 mAb to M21 cells in absence (control) and presence of wtFN10, hFN10 or hFN10/B.
DISCUSSION
In this study we report the crystal structures of integrin αVβ3 in complex with an RGD-bearing domain of the physiologically relevant macromolecular ligand fibronectin (wtFN10), and with a high affinity form of this domain of fibronectin (hFN10) carrying substitutions adjacent to the RGD sequence. Our major finding is that hFN10 unexpectedly acts as a pure antagonist of αVβ3, based on biochemical, biophysical and cell-biological data, and lacks the partial agonism of the native ligands also observed in other RGD-like ligands. The structural basis for antagonism was revealed by the crystal structures of αVβ3–hFN10 and αVβ3–hFN10.
When it bound to cellular αVβ3, hFN10, unlike wtFN10, did not induce activation-specific conformational LIBS mAb epitopes in the integrin N- and C-terminal domains. And it reduced LIBS expression induced both by the activating cation Mn2+ and by constitutive (mutational) activation of αVβ3. Second, hFN10 bound to, but did not dramatically alter the hydrodynamic behavior of the soluble αVβ3 ectodomain, unlike wtFN10 or cyclic RGD-based peptides4. Third, cell spreading on immobilized hFN10 was significantly reduced compared with wtFN10 or native FN. Cell spreading reports outside-in signaling by ligand-occupied integrins28.
The αVβ3-hFN10 structure defines for the first time the interface an integrin makes with a macromolecular physiologic ligand. Interestingly, this interface was surprisingly modest even relative to αVβ3-cilengitide interface13 and was distinguished by contacts with the glycan at Asn266 of the α-subunit Propeller domain. These contacts significantly contributed to the adhesion function of cellular αVβ3. The glycan at Asn266 is conserved in the fibronectin receptor α5β1 (ref. 30), and mutation of the equivalent residue in α5 (N275Q) impaired α5β1-mediated cell adhesion31, suggesting that the glycan contact may also be in the α5β1/FN interface. This interface is also expected to be more robust than the αVβ3/FN interface, due to an interaction of FN-type III domain 9- with the α-subunit Propeller, an interaction that is not used by αVβ3 (ref. 21). This may explain the greater susceptibility of the smaller αVβ3-FN interface to force-induced binding/unbinding events, which may make it more suitable than the more extensive α5β1-FN interface for mediating dynamic outside-in signal transduction32. Thus, the wtFN10–αVβ3 structure provides a molecular explanation for the distinct cellular responses seen when different integrins bind to the same ligand-domain.
Structure of the αVβ3–hFN10 complex identified a basis for the unexpected action of hFN10 as a pure antagonist despite its prototypical RGD motif. Structural and mutational studies support a critical role for the novel Trp1496-Tyr122 π - π interaction in “freezing’ the integrin in an inactive conformation. First, removing the Trp1496 side chain from hFN10 resulted in a domain that acted as wtFN10, as reported by LIBS expression. LIBS were also induced by hFN10 binding to cellular αVβ3 lacking the Tyr122 side chain. Second, changing the orientation of the Trp1496 side chain in hFN10 such that it no longer faced Tyr122 (as seen in the structure of αVβ3–hFN10/B complex) also lead to induction of LIBS when cellular αVβ3 bound hFN10/B. These data strongly argue that blocking the inward movement of the α1 helix towards MIDAS is sufficient to halt associated tertiary changes in the βA domain leading to outside-in signaling. Thus, altering side-chain orientation of Trp1496 by design or selection of its local environment can dramatically affect the tertiary or quaternary changes induced by binding of RGD-based ligands. This could perhaps be replicated. For example: in a cyclized form of the RGD-containing loop of hFN10; by changing orientation of Trp side chain in eptifibatide; or by replacing D-Phe with D-Trp in a modified cilengitide. The critical βA-Tyr122 is also conserved in α5β1 (ref. 30) and β2 integrins33, which like αVβ3 are drug targets34. Our surprising results thus clearly suggest a path to a structure-based drug design of a new generation of ligand-mimetic integrin inhibitors that can act as pure antagonists.
Online Methods
Reagents and antibodies
Restriction and modification enzymes were obtained from New England Biolabs Inc. (Beverly, MA). Cell culture reagents were purchased from Invitrogen Corp (San Diego, CA) or Fisher Scientific (Hampton, NH). Human plasma fibronectin was obtained from Sigma-Aldrich (St Louis, MO). The non-inhibitory mouse monoclonal antibody (mAb) AP3 (American Type Culture Collection, ATCC) detects the β3-subunit in all conformations. Mouse mAb AP5, kindly provided by T.J. Kunicki (Blood Centre, Madison, WI), detects residues 1–6 in the PSI domain of the in β3-subunit only in high-affinity/ligand-bound states. Mouse mAbs LIBS-1 and LIBS-6 to the human β3-subunit6 were kindly provided by Dr. M. Ginsberg (University of California, San Diego). LIBS-1 binds a neoepitope distinct from that of AP5, and LIBS-6 binds the βTD (residues 602–690) (ref. 5). The Fab fragment of AP5 was prepared by papain digestion followed by anion exchange and size-exclusion chromatography. The function-blocking and heterodimer-specific mAb LM609 to αVβ3 (ref. 35) was from Millipore (Danvers, MA) and APC-labeled goat anti-mouse Fc-specific antibody was from Jackson ImmunoResearch (West Grove, PA).
Plasmids, mutagenesis, protein expression and purification
Human αVβ3 ectodomain was expressed in insect cells and purified as described36. The activating N339S mutation in the β3 subunit was generated as described37. Expression plasmids encoding wild-type human N-terminally His-tagged FN10 (S1417-T1509) were generated by PCR from a plasmid containing human FN7-10 kindly provided by H.P. Erickson (Duke University Medical Center, Durham, NC)38. Plasmid encoding His-tagged high affinity FN10 (hFN10) was PCR-generated by replacing the cDNA encoding the loop sequence 1492GRGDSPAS in wtFN10 with 1492PRGDWNEG. RGD-loop substitutions W1496S and TPRGDWNE to IARGDWND (substituted residues underlined) in hFN10 to produce hFN10W/S and hFN10/B, respectively, were generated using PCR-based mutagenesis with the Quick-change kit (Agilent Technologies), cloned into bacterial expression plasmid pET11a and verified by DNA sequencing. The double mutation N266/Q (in αV Propeller) plus N339S (in βA domain), and the Y122A mutation were generated by PCR in pcDNA3 expression plasmids and confirmed by DNA sequencing. FN10 forms were expressed in bacteria and purified by affinity chromatography on nickel columns followed by gel filtration. Thrombin-cleaved FN10 was further purified by gel filtration. Protein purity was confirmed by SDS PAGE.
Cell lines, cell culture and transfection
The human erythroleukemia cell line K562 stably expressing recombinant αVβ3 (K562-αVβ3)4 and the human melanoma cell line M21, which constitutively expresses αVβ3, have been previously described39. Cells were maintained in Iscove’s modified Dulbecco’s medium plus G418 (0.5–1.0 mg/ml) (K562-αVβ3) or RPMI1640 (M21), supplemented with 10% fetal calf serum, 2mM L-glutamine, penicillin and streptomycin. HEK293T (ATCC) cells cultured in Dulbecco modified Eagle’s medium supplemented with 10% fetal calf serum, 2mM L-glutamine, 1 mM sodium pyruvate, penicillin and streptomycin, were transiently co-transfected with pcDNA3 plasmids encoding full-length WT αVβ3, αVβ3(Y122A), αVβ3(N339S), or αV(N266Q)β3(N339S) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol.
Fluorescent labeling of FN10 and mAbs
FN10, and mAbs AP5 (Fab) and AP3 (IgG) were labeled respectively with N-hydroxy succinimidyl esters of Fluor 488 (Alexa488) and Alexa647, (Invitrogen) according to the manufacturer’s instructions. Excess dye was removed using Centri-Spin size-exclusion microcentrifuge columns (Princeton Separations, Adelphia, NJ). The final FN10, AP5 and AP3 concentrations and dye to protein molar ratios (F/P) were determined spectrophotometrically, giving F/P molar ratios of 1.2 (for FN10) and 3 (for AP5 and AP3).
Ligand binding and flow cytometry
In cellular and biochemical assays where calcium, magnesium or manganese ions were used, they were each at final concentrations of 1 mM. Cells stably (K562) or transiently (HEK293T) expressing WT or mutant forms of αVβ3 were harvested by incubating in 10 mM EDTA in PBS (5 min; 25°C), followed by washing three times in Hepes-buffered saline (20 mM Hepes, 150 mM NaCl, pH 7.4) containing bovine serum albumin (BSA) (0.1% w/v; washing buffer, WB). 1×106 cells were suspended in 100µl WB containing CaCl2 plus MgCl2 or MnCl2 (10 min; 37°C), and incubated first with Alexa488-labeled wt- or hFN10 (each at 3–10 µg/ml) (30 min; 25°C) in the dark, then with Alexa647-conjugated AP3 (10 µg/ml) for an additional 30 min on ice. Cells were washed in the respective metal ion-containing buffer, resuspended, fixed in 4% paraformaldehyde and analyzed using FACSCalibur or BD-LSRII flow cytometers (BD Biosciences). Binding of soluble FN10 to αVβ3+ cells was expressed as mean fluorescence intensity (MFI), as determined using FlowJo software. Binding of soluble FN10 to HEK293T was normalized by dividing its MFI by the MFI of Alexa647-conjugated AP3 to the same cells and multiplying by 100. Mean and SD from independent experiments were calculated, and compared using Student’s t test.
LIBS epitope expression
K562-αVβ3 cells, transiently transfected HEK293T or αVβ3+ M21 cells (0.5×106 in 100 µl WB) were incubated in the absence or presence of unlabeled soluble FN10 (5µg) in Ca2+/Mg2+ or in Mn2+ (30 min; at 25°C). Alexa647-labeled AP5 Fab, unlabeled anti-LIBS-1 (each to 10 µg/ml) or LIBS-6 ascites (to 1:50 dilution) were added, and the cells were incubated for an additional 30 min before washing. Alexa647-labeled AP3 was used with HEK293T cells in a separate set of tubes for normalization. APC-labeled goat anti-mouse Fc-specific antibody was added to anti-LIBS-1- or anti-LIBS-6-bound M21 cells for an additional 30 min at 4°C. Cells were then washed, resuspended, fixed in 4% paraformaldehyde and analyzed by flow cytometry. LIBS epitope expression was measured and expressed as MFI (in case of K562-αVβ3 or M21 cells) and normalized (in case of HEK293T cells) as above.
Hydrodynamic shift assays
αVβ3 ectodomain was incubated alone or with FN10 (at 2:1 FN10/integrin molar ratio) in 145 mM NaCl and 25 mM Tris-HCl, pH 7.4 (TBS) containing Ca2+, Ca2+/Mg2+ or Mn2+ (20°C; 1.5 h). Aliquots were then taken and chromatographed at room temperature on a precalibrated Superdex S-200 GL column equilibrated in buffer having the same metal ion composition used during the incubation, and Stokes radii were derived as described previously4. The elution profiles resolved by molecular sieve chromatography were monitored in-line by UV adsorption at 280 nm. Unliganded ± FN10-treated αVβ3 species were resolved as single discrete symmetrical peaks. Excess FN10 served as an internal standard. Identity of resolved peaks was formally confirmed by SDS-PAGE.
Cell adhesion assays
wtFN10, hFN10 or full-length FN (each at a 100µg/ml in PBS) was adsorbed to demarcated areas in Maxisorp Nunc Omni Tray plates (Sigma-Aldrich, St Louis, MO) overnight at 25° C. The various FN-coated surfaces were washed with PBS, and blocked with bovine serum albumin (5% w/v in PBS; 1h; 25°C). K562-αVβ3 cells (5×104) were added (in TBS; 1mM Mn2+), in the absence or presence of LM609 mAb (10 µg/ml, added 15 min prior to plating). Cells were incubated (2h; 37°C), washed three times in warm TBS and fixed with 4% formaldehyde (in PBS; 25°C; 10 min). Images were captured using an inverted phase microscope (Zeiss Axiovert 40CFL) fitted with a powershot G12 Cannon Camera. ImageJ 1.48a software (National Institutes of Health, USA) was used to quantify cell spreading of 300–400 random selected cells. Spread cells were clearly distinguishable from round, refringent non-spread cells. Representative phase contrast images were collected using Zeiss Axiovert 35 inverted microscope using a CCD camera and Spot software (Diagnostics Instruments). Adhesion of transiently transfected HEK293T expressing equivalent amounts of αVβ3(N339S) or αV(N266Q)β3(N339S) was done by adding cells (3×104) in TBS containing Ca2+-Mg2+ and 0.1% BSA (w/v) to FN-coated surfaces (45min; 25°C). Non-adherent cells were removed by washing and adherent cells were fixed (2% paraformaldehyde; 30 min; 25°C), stained (0.1% Crystal Violet; 30 min; 25°C), washed with water, air dried and then solubilized (1% SDS). Relative cell attachment was estimated by absorbance of the lysates at 540 nm measured using a SpectraMax M2E Microplate reader (Molecular Devices, Sunnyvale, CA).
Crystallography, structure determination, and refinement
αVβ3 ectodomain was crystallized at 4°C by vapor diffusion using the hanging drop method as previously described4. hFN10, wtFN10 or hFN10/B (at 1.5 mM) were soaked into αVβ3 crystals in the crystallization well solution containing 2 mM Mn2+ for 2–3 weeks. Crystals were harvested in 12% PEG 3500 (polyethylene glycol, molecular weight 3500), in 100 mM sodium acetate, pH 4.5, 800 mM NaCl plus 2 mM Mn2+ and FN10 (at 1.5 mM), cryoprotected by addition of glycerol in 2% increments up to 24% final concentration, and then flash-frozen in liquid nitrogen. Diffraction data from cryocooled crystals were collected on the ID19 beamline fitted with a CCD detector at the APS Facility (Chicago, IL). Data were indexed, integrated and scaled with HKL2000 program40 for αVβ3-hFN10, αVβ3-hFN10B, and with iMosflm41 for αVβ3-wtFN10, using the same Rfree set imported from pdb id 3ije structure factors. Phases were determined by molecular replacement using PHASER42, with structures of unliganded αVβ3 ectodomain (pdb 3ije) and FN10 domain (pdb 1fnf) used as search models. The resulting models were refined with 1.8.4 version of Phenix 1143 using simulated annealing, TLS, positional and individual temperature-factor refinement, and default restrains. Several cycles of refinement and model building using Coot44 were applied to refine the complex structures of αVβ3-hFN10, αVβ3-wtFN10 and αVβ3-hFN10/B (Table 1), with automatic optimization of X-ray and stereochemistry/ADP, and additional Ramachandran restrains in last cycles. Ramachandran statistics were as follows: αVβ3-hFN10 structure, 89% in most favored regions, 10.47% in additional allowed regions and 0.53% outliers; αVβ3-wtFN10 structure, 89% in most favored regions, 10.18% in additional allowed regions and 0.82% outliers; αVβ3-hFN10/B structure, 91% in most favored regions, 0.56% in additional allowed regions and 0.56% outliers. σA weighted Fo-Fc omit maps were generated by removing the FN10 ligand from the final complex models using phenix.maps. All structural illustrations were prepared with the Chimera software45.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Harold .P. Erickson (Duke University) for providing the FN7-10 plasmid, Dr. Thomas J. Kunicki (The Scripps Research Institute) for access to AP5 antibody, Dr. Mark Ginsberg (UC San Diego) for providing LIBS-1 and LIBS-6 mAbs, Dr. Gregory A. Petsko (Brandeis University) for helpful discussions and Zhiping Ding and Dirk Mueller-Pompalla for expert technical assistance. This work was supported by grants DK48549, DK096334 and DK007540 (M.A.A.) from NIDDK, National Institutes of Health.
Footnotes
Accession codes
The coordinates and structure factors of αVβ3–hFN10, αVβ3–wtFN10 and αVβ3–hFN10/B have been deposited in the Protein Data Bank under accession codes 4MMZ, 4MMX, and 4MMY, respectively.
Author Contributions
M.A.A. conceived and designed experiments. J.V.A., J-P.X. and S.L.G. made and purified proteins. JVA and JPX performed the crystallographic studies. J.L.A., X.R., J.V.A., M.A.A. and B.D.A. performed the biophysical, biochemical and cell-based assays. M.A.A., J.V.A., J-P.X. and J.L.A. interpreted data. M.A.A. wrote the manuscript with the assistance of S.L.G. and J-P.X.
Competing Financial Interests The authors declare no competing financial interests.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Xiong JP, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnaout MA, Goodman SL, Xiong JP. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19:495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong JP, et al. Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. The Journal of cell biology. 2009;186:589–600. doi: 10.1083/jcb.200905085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honda S, et al. Topography of ligand-induced binding sites, including a novel cation-sensitive epitope (AP5) at the amino terminus, of the human integrin beta 3 subunit. The Journal of biological chemistry. 1995;270:11947–11954. doi: 10.1074/jbc.270.20.11947. [DOI] [PubMed] [Google Scholar]
- 6.Frelinger AL, 3rd, Du XP, Plow EF, Ginsberg MH. Monoclonal antibodies to ligand-occupied conformers of integrin alpha IIb beta 3 (glycoprotein IIb-IIIa) alter receptor affinity, specificity, and function. The Journal of biological chemistry. 1991;266:17106–17111. [PubMed] [Google Scholar]
- 7.Raborn J, Wang W, Luo BH. Regulation of integrin alphaIIbbeta3 ligand binding and signaling by the metal ion binding sites in the beta I domain. Biochemistry. 2011;50:2084–2091. doi: 10.1021/bi2000092. [DOI] [PubMed] [Google Scholar]
- 8.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323:642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 9.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nature reviews. Drug discovery. 2010;9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 10.Gerber EE, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503:126–130. doi: 10.1038/nature12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maile LA, et al. A monoclonal antibody against alphaVbeta3 integrin inhibits development of atherosclerotic lesions in diabetic pigs. Science translational medicine. 2010;2:18ra11. doi: 10.1126/scitranslmed.3000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Journal of thrombosis and haemostasis : JTH. 2009;7:911–918. doi: 10.1111/j.1538-7836.2009.03360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong JP, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 14.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mould AP, Barton SJ, Askari JA, Craig SE, Humphries MJ. Role of ADMIDAS Cation-binding Site in Ligand Recognition by Integrin {alpha}5{beta}1. The Journal of biological chemistry. 2003;278:51622–51629. doi: 10.1074/jbc.M306655200. [DOI] [PubMed] [Google Scholar]
- 16.Gao C, et al. Eptifibatide-induced thrombocytopenia and thrombosis in humans require FcgammaRIIa and the integrin beta3 cytoplasmic domain. The Journal of clinical investigation. 2009;119:504–511. doi: 10.1172/JCI36745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bassler N, et al. A mechanistic model for paradoxical platelet activation by ligand-mimetic alphaIIb beta3 (GPIIb/IIIa) antagonists. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:e9–e15. doi: 10.1161/01.ATV.0000255307.65939.59. [DOI] [PubMed] [Google Scholar]
- 18.Alghisi GC, Ponsonnet L, Ruegg C. The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PloS one. 2009;4:e4449. doi: 10.1371/journal.pone.0004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reynolds AR, et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nature medicine. 2009;15:392–400. doi: 10.1038/nm.1941. [DOI] [PubMed] [Google Scholar]
- 20.Zhu J, et al. Structure-guided design of a high-affinity platelet integrin alphaIIbbeta3 receptor antagonist that disrupts Mg(2)(+) binding to the MIDAS. Science translational medicine. 2012;4:125ra132. doi: 10.1126/scitranslmed.3003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowditch RD, et al. Identification of a novel integrin binding site in fibronectin. Differential utilization by beta 3 integrins. Journal of Biological Chemistry. 1994;269:10856–10863. [PubMed] [Google Scholar]
- 22.Richards J, et al. Engineered fibronectin type III domain with a RGDWXE sequence binds with enhanced affinity and specificity to human alphavbeta3 integrin. Journal of molecular biology. 2003;326:1475–1488. doi: 10.1016/s0022-2836(03)00082-2. [DOI] [PubMed] [Google Scholar]
- 23.Scarborough RM. Development of eptifibatide. American heart journal. 1999;138:1093–1104. doi: 10.1016/s0002-8703(99)70075-x. [DOI] [PubMed] [Google Scholar]
- 24.Mould AP, et al. Integrin activation involves a conformational change in the alpha 1 helix of the beta subunit A-domain. The Journal of biological chemistry. 2002;277:19800–19805. doi: 10.1074/jbc.M201571200. [DOI] [PubMed] [Google Scholar]
- 25.Kendall T, Mukai L, Jannuzi AL, Bunch TA. Identification of integrin beta subunit mutations that alter affinity for extracellular matrix ligand. The Journal of biological chemistry. 2011;286:30981–30993. doi: 10.1074/jbc.M111.254797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 27.Adair BD, et al. Three-dimensional EM structure of the ectodomain of integrin {alpha}V{beta}3 in a complex with fibronectin. The Journal of cell biology. 2005;168:1109–1118. doi: 10.1083/jcb.200410068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cluzel C, et al. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. The Journal of cell biology. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Felding-Habermann B, et al. Involvement of tumor cell integrin alpha v beta 3 in hematogenous metastasis of human melanoma cells. Clinical & experimental metastasis. 2002;19:427–436. doi: 10.1023/a:1016377114119. [DOI] [PubMed] [Google Scholar]
- 30.Nagae M, et al. Crystal structure of alpha5beta1 integrin ectodomain: atomic details of the fibronectin receptor. The Journal of cell biology. 2012;197:131–140. doi: 10.1083/jcb.201111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato Y, et al. An N-glycosylation site on the beta-propeller domain of the integrin alpha5 subunit plays key roles in both its function and site-specific modification by beta1,4-N-acetylglucosaminyltransferase III. The Journal of biological chemistry. 2009;284:11873–11881. doi: 10.1074/jbc.M807660200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16245–16250. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie C, et al. Structure of an integrin with an alphaI domain, complement receptor type 4. The EMBO journal. 2010;29:666–679. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodman SL, Picard M. Integrins as therapeutic targets. Trends in pharmacological sciences. 2012;33:405–412. doi: 10.1016/j.tips.2012.04.002. [DOI] [PubMed] [Google Scholar]
References
- 35.Cheresh DA. Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:6471–6475. doi: 10.1073/pnas.84.18.6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehta RJ, et al. Transmembrane-truncated alphavbeta3 integrin retains high affinity for ligand binding: evidence for an 'inside-out' suppressor? Biochem J. 1998;330(Pt 2):861–869. doi: 10.1042/bj3300861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng M, et al. Mutation of a Conserved Asparagine in the I-like Domain Promotes Constitutively Active Integrins {alpha}Lbeta2 and {alpha}IIbbeta3. The Journal of biological chemistry. 2007;282:18225–18232. doi: 10.1074/jbc.M701386200. [DOI] [PubMed] [Google Scholar]
- 38.Leahy DJ, Aukhil I, Erickson HP. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 39.Mitjans F, et al. An anti-alpha v-integrin antibody that blocks integrin function inhibits the development of a human melanoma in nude mice. J Cell Sci. 1995;108(Pt 8):2825–2838. doi: 10.1242/jcs.108.8.2825. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Vol. 276. Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 41.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCoy AJ, et al. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.