

Abstract
Stress early in life is a known risk factor for the development of affective disorders later in life. Epigenetic mechanisms, such as DNA methylation, may have an important role in mediating that risk. Recent epigenetic research reported on the long-term relationship between traumatic stress in childhood and DNA methylation in adulthood. In this study, we examined the impact of various types of stress (perinatal stress, stressful life events (SLEs) and traumatic youth experiences) on methylation of the glucocorticoid receptor gene (NR3C1) in the blood of a population sample of 468 adolescents (50.4% female, mean age 16.1 years). Second, we determined whether stress at different ages was associated with higher NR3C1 methylation. NR3C1 methylation rates were higher after exposure to SLEs and after exposure to traumatic youth experiences. NR3C1 methylation in adolescence was not higher after exposure to perinatal stress. Experience of SLEs in adolescence was associated with a higher NR3C1 methylation, independently of childhood SLEs. We demonstrate that not only traumatic youth experiences but also (more common) SLEs are associated with higher NR3C1 methylation. In addition, our findings underline the relevance of adolescent stress for epigenetic changes in the NR3C1 gene.
Introduction
Severe maltreatment or neglect in childhood are known risk factors for development of affective disorders and have been associated with altered programming of the hypothalamic-pituitary-adrenal (HPA) axis.1, 2, 3, 4 Epigenetic modifications are thought to link early-life stress to later susceptibility to affective disorders, such as anxiety or depression, through interference with the development and functioning of the HPA axis early in life.5 More recently, the notion that stress can have a direct effect on epigenetic modifications across the life span has been proposed, which in turn affects brain plasticity and may lead to anxiety.6 The epigenetic process of DNA methylation involves the addition of methyl groups on cytosine–guanine dinucleotides (CpGs) in gene promoters and regulatory regions, which regulate gene transcription.7 The presence of these methyl groups is associated with reduced gene expression by reducing access to the DNA. Methyl groups on CpGs in regulatory regions for transcription factors can directly interfere with the binding of transcription factors to their recognition elements. Methylated regions can also repress transcription indirectly by attracting methylated DNA-binding proteins, which can alter the chromatin formation, disabling access to DNA for transcription.8, 9, 10, 11, 12 Whereas much research focuses on the function of epigenetic modifications, less is known about how they are environmentally induced.
Whereas the genome is fixed, the epigenome is considered to be dynamic—that is, under the influence of environmental factors.13,14 In rats, exposure to early-life stress, measured as reduced levels of maternal licking and grooming (LG) behavior towards their offspring, led to increased methylation in the glucocorticoid receptor (GR) gene (Nr3c1) of their offspring. The GRs regulate the release of glucocorticoids through a negative feedback mechanism in the HPA axis. Increased methylation in offspring and reduced expression of GRs in the hippocampus by low maternal LG behavior resulted in a diminished feedback sensitivity of the HPA axis.15 Differences in methylation between offspring from high-LG and low-LG mothers persisted into adulthood, illustrating long-lasting effects of early programming on the epigenome.
These animal findings were translated to humans by McGowan et al.16 They reported increased levels of methylation and decreased levels of GR expression in post-mortem hippocampal tissue of suicide completers who were abused during childhood, compared with non-abused suicide completers and non-abused controls.16 Other studies using peripheral DNA, from blood of infants, adolescents or adults, have shown increased levels of NR3C1 methylation in response to perinatal stress17, 18, 19 and abuse or neglect during childhood.20,21 Most studies thus far reported on DNA methylation in adults after enduring stress or traumatic events such as abuse or neglect.16,20, 21, 22 Fewer studies have investigated whether NR3C1 methylation in humans can be induced by other, more common, stressful life events (SLEs—for example, parental divorce, loss of a family member) as well.21,23,24
Besides types of stress, the role of timing of stress on methylation is understudied. Humans are subjected to a high variety of stressors throughout life. The perinatal period and childhood years are regarded as sensitive periods for the developing brain in which the organism could be particularly susceptible to epigenetic modifications that influence HPA axis development.25 However, some brain regions keep developing at least until early adulthood;26 hence, there is a possibility that epigenetic modifications are not restricted to childhood.27, 28, 29 Whereas there is preliminary evidence for epigenetic modification of NR3C1 due to stressors in childhood,16,20,21 the adolescent period, in spite of its obvious importance as a period of increased susceptibility to stress-related mental disorders,30,31 has had no examination of the impact of stress on NR3C1 methylation independent of stress experienced in childhood.6
We studied the effects of stress on NR3C1 methylation in a large prospective population study of adolescents in two ways. First, we examined the impact of various types of stress (for example, perinatal stress, SLEs and traumatic youth experiences) on NR3C1 methylation. Second, we determined whether stress at different ages was associated with higher NR3C1 methylation. On the basis of previous findings15, 16, 17, 18, 19, 20, 21, 22,32 we hypothesized that perinatal stress, many SLEs and traumatic youth experiences would relate to higher NR3C1 methylation in adolescence. We further expected that SLEs experienced in childhood would, independently of later adolescent stress, relate to increased NR3C1 methylation in adolescence.
Materials and methods
Sample selection
Data from the TRAILS (TRacking Adolescents' Individual Lives Survey) study were used. TRAILS is a prospective population study of Dutch adolescents (N=2230) who are being followed from pre-adolescence into adulthood. Assessment waves are conducted biennially or triennially, and four assessment waves have been completed so far. Written consent was obtained from each subject and their parents at every assessment wave. The present study involves data collected during the first assessment wave (T1, 2001–2002, mean age 11.1 years, s.d.=0.55), second (T2, 2003–2004, mean age 13.6 years, s.d.=0.53), third (T3, 2005–2007, mean age 16.3 years, s.d.=0.71) and fourth (T4, 2008–2010, mean age 19.1 years, s.d.=0.60) assessment waves. At T3, 715 TRAILS subjects (focus sample) participated in more extensive experimental data collection. Adolescents with an increased risk of mental health problems had a greater chance of being selected for the experimental session. Increased risk was defined as having at least one of the following risk factors: child temperament (high frustration and fearfulness, low effortful control), parental psychopathology (depression, anxiety, addiction, psychoses or antisocial behavior) and environmental risk (living in a single-parent family), all measured at T1. Although high-risk adolescents were slightly oversampled (66% of the focus sample, the remaining 34% of the focus sample were selected at random from the low-risk TRAILS participants), the sample included the total range of mental health problems present in a community population of adolescents. The study was approved by the Dutch Central Medical Ethics Committee and all subjects received compensation for their participation. A detailed description of sampling and methods can be found in Huisman et al.33 and Ormel et al.34 DNA had been isolated from blood for 654 of these subjects. Initial selection for methylation analyses (N=475) was obtained by excluding subjects with non-Dutch ethnicity (N=58), unknown or insufficient DNA concentration (N=116), and randomly excluding one of each sibling pair (N=5). Following drop-out after DNA methylation analyses, 468 subjects were eligible for analysis. Our subsample subjects did not differ significantly (P>0.05) from the TRAILS focus sample with regard to sex, socioeconomic status (T1), age (T3), internalizing problems (T3) and externalizing problems (T3).
Stress measures
Perinatal stress was operationalized as the sum of maternal psychological problems during pregnancy or the 3 months after delivery, preterm delivery (⩽33 weeks), low birth weight (⩽2500 g), hospitalization of mother or child within 1 month after delivery and maternal alcohol use or smoking during pregnancy. For birth weight and gestational age, we used records of the Preventive Child Healthcare services.35 The other stressors were measured in a detailed interview with the parents at T1.
SLEs experienced between ages 0 and 15 years were assessed for the age categories of 0–5, 6–11, 12–13 and 14–15 years, as described by Bosch et al.4 Information on SLEs in early childhood (0–5 years) and middle childhood (6–11 years) was collected during a detailed interview with the parents at T1 and included the number of times the child had experienced parental divorce, hospitalization, the death of a family member or friend, out-of-home placement, parental addiction or parental mental health problems.4 The total number of SLEs experienced in early adolescence (12–13 years) was assessed with a self-report questionnaire at T2.4 The 25 SLEs included illness or injury of the participant, a family member or a friend; parental divorce; death of a family member or friend; changes in family composition; parental unemployment; conflicts with family or friends; and being bullied. SLEs in middle adolescence (14–15 years) were assessed at T3 in an Event History Calendar Interview36 with the adolescent. The list of possible events consisted of conflicts, physical or sexual intimidation, victim of bullying/gossiping, loss or lack of friends, psychological/addiction problems of family or friends, out-of-home placement, running away from home, death/illness of family member, hospitalization of participant and parental divorce. On the basis of the above-mentioned event measures, we calculated a measure of total SLEs experienced between ages 0 and 15 years by standardizing the sum score of the number of events for each age category and summing the standardized scores. This procedure was chosen to account for differences in the number of possible SLEs by age group. The resulting total sum score (SLEs 0–15 years) was standardized for analyses.
Traumatic youth experiences
At T4, information on sexual, physical and other traumatic experiences before the age of 16 years was obtained with a 16-item self-report questionnaire. To determine sexual abuse, the participants were asked whether an adult family member, friend of the family or stranger had ever, before the participant was 16, showed his/her genitals or masturbated in front of them; had sexually assaulted them; had forced them to touch him/her in a sexual manner; had attempted to have intercourse or had actually had intercourse with them. To determine physical abuse, participants were asked whether a parent or caretaker had ever, before the participant was 16, hit them with a belt, brush, stick or other hard object; had hit them with a fist or kicked them very hard; had shaken or pinched them; had beaten them up (that is, hit them in succession) or had threatened them with a knife or other weapon. To determine other trauma, participants were asked whether, before the age of 16, they had been involved in a life-threatening accident; had witnessed severe injury or death; had been a victim of physical violence or assault; had been threatened with a weapon; had been held captive or abducted or had been involved in a fire, flood or other (natural) disasters. Answers were coded into a single exposure or multiple exposures to traumatic youth experiences. We had no data on the timing of traumatic life stress. We cross-tabbed the three SLE profiles with traumatic experiences (Supplementary Table 1) and found that traumatic experiences were not strongly overrepresented in the childhood SLE group or the adolescence SLE group.
Amplicon selection
Three amplicons (genomic regions) within the NR3C1 cytosine-guanine dinucleotide (CpG) island in the promoter region were selected for analyses (Figure 1). CpG-island position was determined using criteria from UCSC genome browser—that is, a genomic region with a CpG content of 50% or greater, length greater than 200 bp and a ratio greater than 0.6 of observed number of CG dinucleotides to the expected number on the basis of the number of Gs and Cs in the segment (UCSC human February 2009 assembly GRCh37/hg19, (http://genome.ucsc.edu). Two primer sets were designed using the software EpiDesigner by Sequenom (www.epidesigner.com), covering the edges of the NR3C1 CpG island. In addition, we analyzed a region of the CpG island using the primer set previously used by McGowan et al.16 This genomic region encompasses exon 1F that corresponds to the rat exon 17. Sequence information and primer properties can be found in Supplementary Figure 1 and Supplementary Table 2.
Figure 1.
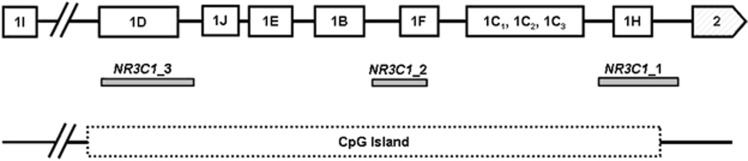
Schematic representation of the glucocorticoid receptor gene (NR3C1). Amplicons (NR3C1_1, NR3C1_2 and NR3C1_3, shown in gray) are shown in relation to the NR3C1 CpG island (chr5:142782072–142785071, dotted box) and untranslated first exons (line boxes) upstream of exon 2 (striped box). Image based on Labonte et al.37 and Turner et al.38
DNA methylation
Analysis
DNA was extracted from whole-blood samples using a manual salting-out procedure as described by Miller et al.39 DNA-methylation rates were analyzed using bisulfite-treated DNA, PCR, reverse transcription, base-specific cleavage of in vitro transcribed RNA product and mass spectrometry (Sequenom EpiTYPER, San Diego, CA, USA). Bisulfite conversion of DNA was performed using EZ-96 DNA Methylation Kit (Shallow; Zymo Research, CA, USA). Bisulfite treatment was performed according to the manufacturers' protocol. It must be noted that bisulfite conversion does not differentiate between the different types of cytosine methylation (for example, hydroxymethylation). PCR, reverse transcription, cleavage and mass spectrometry were performed in triplicate according to the EpiTYPER protocol. Amplification conditions can be found in Supplementary Table 3. The mass signal patterns generated are translated to quantitative methylation rates for different CpG units by the MassARRAY EpiTYPER analyzer software from Sequenom. (v1.0, build1.0.6.88 Sequenom). Fragments with CpG dinucleotides are referred to as CpG units. One CpG unit can contain one or more CpG dinucleotide.
Data cleaning
All samples were analyzed in triplicate, and samples with a s.d. of ⩾10% between replicates were removed for analysis. CpG units with ⩾25% missing values were not included in the analyses. For each CpG unit, methylation scores of the triplicates were averaged. We accounted for mass change in CpG units by single nucleotide length polymorphisms (only when minor allele frequency >5%) by removing CpG units from analyses containing the single nucleotide length polymorphism and removing units with overlapping mass caused by single nucleotide length polymorphisms in non-CpG units. Amplicon 1 consisted of 11 eligible CpG units, amplicon 2 contained 10 and amplicon 3 contained nine eligible CpG units.
Statistical analyses
Main analyses
Linear regression analyses were conducted to examine the effects of type and timing of stress on NR3C1 methylation in adolescence. Amplicons were analyzed separately.40 For each CpG unit, methylation was mean-centered (resulting in mean methylation of 0, with original s.d.), to account for high methylation in a small number of CpG units. As individuals could have drop-out in one or more CpG units, an average methylation score for each amplicon was calculated by taking the average of the mean-centered methylation scores of the CpG units within an amplicon. Separate models were run for perinatal stress, SLEs (0–15 years), the three categories of traumatic youth experiences (sexual, physical and other trauma). Smoking, gender and age at T3 were considered as potential confounders; however, including them in the models appeared not to change the regression coefficients of stress variables with more than 10%. Therefore, these confounders were not included in the final models. To test our hypothesis on the timing of stress and NR3C1 methylation, we ran a linear regression model with the two SLE variables (0–11 and 12–15 years).
As a post hoc robustness check, we repeated analyses on type and timing of stress on methylation of amplicon 2 on an additional random sample of 454 TRAILS subjects, who were not part of the T3 focus sample. Information on the other two amplicons was not available for this sample. Amplicon 2 was chosen a priori for its correspondence with the study by McGowan et al.16 and the known presence of the nerve-growth-factor-inducible-protein-A (NGFI-A)-binding site.
Exploratory analyses
In addition to the analyses of the three above-mentioned amplicons, we performed exploratory linear regression analyses of type and timing of stress on mean-centered methylation scores of three individual CpG units, selected based on their relatively high methylation rates and s.d.'s (Supplementary Table 4).
The tests were conducted using SPSS (IBM SPSS, v.20.0., IBM Corp, Armonk, NY, USA), whereas the latent profile analysis (LPA) was performed in Mplus 5 (Muthén & Muthén, Los Angeles, CA, USA).
Results
Approximately half of the sample was female (50.4%) and the mean age of the adolescents was 16.1 years at the time of the DNA collection, with a range from 14 to 18 years. Methylation was not correlated with age. Table 1 presents the descriptive statistics of perinatal stress, SLEs and traumatic youth experiences (uncentered, unstandardized).
Table 1. Descriptives of perinatal stress, SLEs and traumatic youth experiences.
| Median (min–max) | N (%) | |
|---|---|---|
| Perinatal stress | 1 (0–6) | |
| SLEs (0–15 years) | ||
| 0–15 years | 6 (0–23) | |
| 0–5 years | 1 (0–10) | |
| 6–11 years | 1 (0–8) | |
| 12–13 years | 2 (0–12) | |
| 14–15 years | 1 (0–11) | |
| Traumatic youth experiences (0–16 years) | ||
| Sexual abuse | ||
| None | 403 (84.8%) | |
| Single exposure | 22 (4.6%) | |
| Repeated exposure | 12 (2.5%) | |
| Physical abuse | ||
| None | 253 (53.2%) | |
| Single exposure | 170 (35.8%) | |
| Repeated exposure | 14 (2.9%) | |
| Other trauma | ||
| None | 319 (67.2%) | |
| Single exposure | 90 (18.9%) | |
| Repeated exposure | 28 (5.9%) | |
Abbreviation: SLE, stressful life event.
Life stress and methylation
The results of the linear regression models with stress variables as predictors of NR3C1 methylation are presented in Table 2. Exposure to SLEs 0–15 years and to traumatic youth experiences significantly predicted higher methylation rates in amplicon 1. In amplicon 2, only single exposure to sexual abuse predicted higher methylation rates (B=0.44, P<0.001). For amplicon 3, repeated exposure to other traumatic youth experiences was associated with lower methylation rates (B=−0.26, P<0.01).
Table 2. Life stress and NR3C1 methylation scores by amplicons.
|
NR3C1_1 |
NR3C1_2 |
NR3C1_3 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| B | s.e. | P | B | s.e. | P | B | s.e. | P | |
| Perinatal stress | 0.03 | 0.02 | 0.14 | −0.01 | 0.02 | 0.78 | 0.01 | 0.02 | 0.53 |
| Traumatic youth experiencesa | |||||||||
| Sexual abuse | |||||||||
| Single exposure | 0.37 | 0.09 | <0.0001 | 0.44 | 0.12 | <0.001 | −0.08 | 0.10 | 0.40 |
| Repeated exposure | 0.50 | 0.12 | <0.0001 | 0.13 | 0.17 | 0.45 | −0.26 | 0.13 | 0.05 |
| Physical abuse | |||||||||
| Single exposure | 0.01 | 0.04 | 0.73 | 0.02 | 0.06 | 0.68 | −0.02 | 0.05 | 0.66 |
| Repeated exposure | 0.49 | 0.12 | <0.0001 | −0.09 | 0.15 | 0.56 | −0.18 | 0.12 | 0.15 |
| Other trauma | |||||||||
| Single exposure | 0.09 | 0.05 | 0.06 | −0.04 | 0.07 | 0.52 | −0.10 | 0.06 | 0.08 |
| Repeated exposure | 0.53 | 0.08 | <0.0001 | 0.07 | 0.11 | 0.56 | −0.26 | 0.09 | <0.01 |
| SLEsb | |||||||||
| Total (0–15 years) | 0.05 | 0.02 | 0.02 | 0.03 | 0.03 | 0.25 | −0.03 | 0.02 | 0.20 |
| Childhood (0–11 years) | 0.01 | 0.02 | 0.57 | 0.00 | 0.03 | 0.97 | −0.03 | 0.02 | 0.22 |
| Adolescence (12–15 years) | 0.05 | 0.02 | <0.01 | 0.04 | 0.03 | 0.13 | −0.01 | 0.02 | 0.62 |
Abbreviations: B, regression coefficient; SLEs, stressful life events.
Linear regression of early-life stress and multivariate regression analyses of SLEs in two age categories on NR3C1 methylation scores. Bold numbers indicate significant results (P<0.05).
No exposure is the reference category.
Z-scores.
Secondly, we analyzed timing of SLEs. We had no specific hypotheses for early versus late adolescence or for early versus late childhood stress.4 As these variables are correlated more strongly within childhood and adolescence age groups than between (see Supplementary Table 5), we considered creating a childhood SLE variable and an adolescence SLE variable. We used LPA to explore profiles of SLE in our sample. We found three distinct profiles (see Supplementary Table 6 and Supplementary Figure 2): a ‘low stress group' at all ages, a ‘high childhood stress group (0–5 years and 6–11 years)' and a ‘high adolescent stress group (12–13 years and 14–15 years)'. Sample size was too small to use the three profiles as predictor of NR3C1 methylation; however, the LPA analysis provided the support to distinguish between SLEs in childhood (0–11 years) and SLEs in adolescence (12–15 years). The variables were constructed by summing standardized scores for each age period. The SLE scores (0–11 years and 12–15 years) were standardized before analyses.
Experience of SLEs in adolescence was associated with a higher methylation scores independently of childhood SLEs in amplicon 1 (B=0.05, P<0.01) but not in amplicons 2 and 3 (Table 2).
Exploratory analyses
Exploratory analyses on CpG unit-specific methylation (Table 3) showed higher methylation rates with more exposure to SLEs 0–15 years and to traumatic youth experiences in all three CpG units. Perinatal stress was not related to CpG unit-specific methylation. In addition, the CpG unit-specific analyses show that SLEs in adolescence predicted higher NR3C1 methylation in NR3C1_1CpGU12 and NR3C1_2CpGU13, whereas childhood SLEs predicted higher NR3C1 methylation in NR3C1_2CpGU14.
Table 3. Life stress and NR3C1 methylation scores of the three highest methylated CpG units.
|
NR3C1_1CpGU12 |
NR3C1_2CpGU13 |
NR3C1_2CpGU14 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| B | s.e. | P | B | s.e. | P | B | s.e. | P | |
| Perinatal stress | 0.05 | 0.11 | 0.64 | 0.05 | 0.20 | 0.81 | −0.05 | 0.09 | 0.58 |
| Traumatic youth experiencesa | |||||||||
| Sexual abuse | |||||||||
| Single exposure | 1.95 | 0.52 | <0.001 | 5.06 | 0.95 | <0.0001 | 0.91 | 0.43 | 0.04 |
| Repeated exposure | 3.07 | 0.70 | <0.0001 | 4.21 | 1.29 | <0.01 | 0.50 | 0.59 | 0.40 |
| Physical abuse | |||||||||
| Single exposure | 0.07 | 0.24 | 0.78 | 0.34 | 0.45 | 0.46 | 0.15 | 0.20 | 0.46 |
| Repeated exposure | 2.79 | 0.67 | <0.0001 | 2.68 | 1.20 | 0.03 | −0.34 | 0.54 | 0.52 |
| Other trauma | |||||||||
| Single exposure | 0.36 | 0.28 | 0.20 | −0.02 | 0.53 | 0.98 | 0.07 | 0.24 | 0.78 |
| Repeated exposure | 3.32 | 0.46 | <0.0001 | 3.86 | 0.83 | <0.0001 | 0.22 | 0.39 | 0.57 |
| SLEsb | |||||||||
| Total (0–15 years) | 0.38 | 0.12 | <0.01 | 0.45 | 0.21 | 0.03 | 0.24 | 0.09 | 0.01 |
| Childhood (0–11 years) | 0.10 | 0.12 | 0.41 | −0.05 | 0.21 | 0.82 | 0.21 | 0.09 | 0.02 |
| Adolescence (12–15 years) | 0.41 | 0.12 | <0.001 | 0.68 | 0.21 | <0.01 | 0.10 | 0.09 | 0.26 |
Abbreviations: B, regression coefficient; CpG, cytosine–guanine dinucleotide; SLE, stressful life events. Linear regression of early life stress and multivariate regression analyses of SLEs in two age categories on NR3C1 methylation scores. Bold numbers indicate significant results (P<0.05).
No exposure is the reference category.
Z-scores.
In the post hoc robustness check (Supplementary Table 7), we repeated the analyses for amplicon 2 exclusively. The significant association with single exposure to sexual abuse in the original sample failed to replicate in the additional sample.
Discussion
In this study, experience of multiple SLEs and exposure to traumatic experiences between birth and adolescence were associated with higher NR3C1 methylation rates in adolescents. In contrast with our initial expectation, we found that not perinatal or childhood stress, but rather SLEs in adolescence, were associated with higher NR3C1 methylation. To the best of our knowledge no other comparable studies have focused on the timing of stressful events when investigating DNA methylation. Our results on traumatic youth experiences are consistent with prior studies,16,20, 21, 22 with higher methylation rates in individuals who have experienced traumatic youth experiences.
The absence of a significant relationship between NR3C1 methylation and perinatal stress in our sample appears to contrast with previous human studies that showed higher NR3C1 methylation after exposure to perinatal stress.17, 18, 19 NR3C1 methylation in newborns was positively associated with depressed maternal mood in the third trimester of pregnancy.17 In addition, war stress during pregnancy affected newborn NR3C1-methylation rates.19 As these studies involve newborns, no statement could be made on the effect of stressful experiences on NR3C1 methylation later in life. Radtke18 reported higher NR3C1 methylation in 25 adolescents with maternal exposure to intimate partner violence during pregnancy. However, they did not consider the possible influences of more recent stressful experiences on NR3C1 methylation of the adolescents.
Our timing analysis on SLEs indicates that NR3C1 methylation was independently associated with SLEs in adolescence. Although this may not be very surprising, given that adolescence is a significant neurodevelopmental stage, this is the first study to show that adolescent stress actually co-occurs with higher NR3C1 methylation. Our study did not support the notion of a sensitive period to SLEs in childhood for NR3C1 methylation in adolescents, despite suggestive findings in animal studies15,32,41 and a study on long-term effects of adversities on cortisol stress response in our TRAILS participants.4 However, in line with our results, a study in rodents did not report alterations in NR3C1 methylation following early-life stress,42 and others reported changes in NR3C1 methylation following chronic and acute stress in adult rats.43 Possibly, some epigenetic modifications by stress exposure may be short-term effects, which may allow for a more adaptive stress regulation. This may also explain the discrepancy between our study and those investigating methylation in newborns following maternal stress. Recent evidence on active removal of methyl groups44,45 gives rise to the possibility of dynamic regulation and reversibility of DNA methylation. Reversal of NR3C1 methylation in adult rats was proven possible through pharmacological manipulation;46 however, it is currently unknown whether active demethylation can be triggered by environmental factors—for example, positive events following a stressful early life.
We expected to find associations with stress in amplicon 2 in particular. This amplicon is identical to the one studied by McGowan et al.16 and covers the exon 1F (analog to first exon 17 in rats47) promoter containing the transcription factor NGFI-A binding site. In rats and humans, DNA methylation inhibited binding of NGFI-A to its binding site, causing a reduction in transcriptional activity.16,48 However, most of the associations with stress measures were in amplicon 1, at the edge of the CpG island (Figure 1). It is likely that the environment exerts its influence on other alternative first exon promoters spanning the CpG island as well, as methylation is highly variable between individuals in these promoters.49 Further, the presence of other CpG-rich transcription factor-binding sites may also have a role.49 Recently, childhood abuse was related to methylation of other first exon promoters than the exon 1F promoter in human post-mortem brain tissue.37
The exploratory analyses on individual CpG units with relatively high methylation scores showed more pronounced relations between stress and methylation than the main analyses. In addition, these analyses also showed higher methylation in a single CpG unit after childhood stress exposure. Differences between CpG units emphasize the need to understand which regions have a regulatory function and may be more responsive to environmental stimuli. In our post hoc robustness check on amplicon 2, the significant association with sexual abuse in the original sample failed to replicate. This may be due to the fewer individuals that have experienced a single exposure to sexual abuse in the replication sample or the initial association may have been due to chance.
It has to be noted that our assessment of SLEs differed between time periods because we aimed to measure age-appropriate SLEs for the different developmental stages of childhood and adolescence and used different informants to optimize the reliability of event recall. As a consequence, some SLEs that were assessed in adolescence were not measured in childhood, including conflicts with family and friends, being bullied, sexual intimidation and loss or lack of friends. These added events in adolescence may have contributed to the difference in methylation rates we found for childhood and adolescence SLEs. As children lack the capacity to remember early childhood experiences, childhood SLEs were based on parent reports. SLEs in adolescence were based on self-report because the parents may no longer be aware of all aspects of their children's lives as they grow towards independence. Considering the nature of our study design and our desire to include as many relevant SLEs as possible, we could not fully overcome these dissimilarities but we do acknowledge that they warrant caution when interpreting the results. It is possible, for instance, that self-reported SLEs more closely reflect the actual stress levels experienced than parent-reported events. Furthermore, some SLEs that were assessed only in adolescence may be more chronic than the SLEs measured in childhood (for example, sexual intimidation or lack/loss of friends) and thus have a more lasting influence on stress levels. Hence, although our findings suggest that SLEs in adolescence are independently associated with NR3C1 methylation, we cannot completely exclude the possibility that this can be ascribed to specific SLEs measured in adolescence but not in childhood, or to informant differences.
Our study has several strengths: The TRAILS study provides data on NR3C1 methylation in a large population-based sample. Furthermore, we had a detailed account of SLEs between birth and adolescence. This study is the first to explore the effect of SLEs during childhood and adolescence separately. A limitation of the study was that blood was collected at T3 only, preventing analyses of changes in methylation. For this reason, we could not establish any causal links between SLEs or trauma and methylation. In addition, our sample overrepresents adolescents with an increased risk of mental health problems. However, the use of sampling weights to reproduce the distribution in the total TRAILS sample50 did not affect our results. In addition, unlike for the SLE measure, no timing data were available for traumatic youth experiences. Another limitation is that our robustness check was only possible for amplicon 2.
Together, our findings add to the existing literature by showing that both SLEs and traumatic stress affect NR3C1 methylation in adolescents. In addition, it is the first study to show epigenetic effects of stress experienced in adolescence.
Acknowledgments
This research is part of the TRacking Adolescents' Individual Lives Survey (TRAILS). Participating centers of TRAILS include various departments of the University Medical Center and University of Groningen, the Erasmus University Medical Center Rotterdam, the University of Utrecht, the Radboud Medical Center Nijmegen and the Parnassia Bavo group, all in the Netherlands. Van der Knaap and the epigenetic assays are supported by the Sophia Children's Hospital Foundation, project number 634. TRAILS has been financially supported by various grants from the Netherlands Organization for Scientific Research (NWO), ZonMW, GB-MaGW, the Sophia Foundation for Medical Research, the Dutch Ministry of Justice, the European Science Foundation, BBMRI-NL and the participating universities. We are grateful to all adolescents, their parents and teachers who participated in this research, and to everyone who worked on this project and made it possible.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Translational Psychiatry website (http://www.nature.com/tp)
Supplementary Material
References
- Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33:693–710. doi: 10.1016/j.psyneuen.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Heim C, Newport DJ, Wagner D, Wilcox MM, Miller AH, Nemeroff CB. The role of early adverse experience and adulthood stress in the prediction of neuroendocrine stress reactivity in women: a multiple regression analysis. Depress Anxiety. 2002;15:117–125. doi: 10.1002/da.10015. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Bosch NM, Riese H, Reijneveld SA, Bakker MP, Verhulst FC, Ormel J, et al. Timing matters: long term effects of adversities from prenatal period up to adolescence on adolescents' cortisol stress response. The TRAILS study. Psychoneuroendocrinology. 2012;37:1439–1447. doi: 10.1016/j.psyneuen.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Xiong F, Zhang L. Role of the hypothalamic-pituitary-adrenal axis in developmental programming of health and disease. Front Neuroendocrinol. 2013;34:27–46. doi: 10.1016/j.yfrne.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RG, McEwen BS. Stress and anxiety across the lifespan: structural plasticity and epigenetic regulation. Epigenomics. 2013;5:177–194. doi: 10.2217/epi.13.8. [DOI] [PubMed] [Google Scholar]
- Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev. 1991;55:451–458. doi: 10.1128/mr.55.3.451-458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J. 1998;17:4905–4908. doi: 10.1093/emboj/17.17.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- Bird AP, Wolffe AP. Methylation-induced repression—belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Szyf M. The dynamic epigenome and its implications in toxicology. Toxicol Sci. 2007;100:7–23. doi: 10.1093/toxsci/kfm177. [DOI] [PubMed] [Google Scholar]
- Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–797. doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyf M. The early life environment and the epigenome. Biochim Biophys Acta. 2009;1790:878–885. doi: 10.1016/j.bbagen.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, et al. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry. 2011;1:e21. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan CJ, D'Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroud N, Paoloni-Giacobino A, Prada P, Olie E, Salzmann A, Nicastro R, et al. Increased methylation of glucocorticoid receptor gene (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Transl Psychiatry. 2011;1:e59. doi: 10.1038/tp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PLoS One. 2012;7:e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroud N, Dayer A, Piguet C, Nallet A, Favre S, Malafosse A, et al. Childhood maltreatment and methylation of the glucocorticoid receptor gene NR3C1 in bipolar disorder. Br J Psychiatry. 2013;204:30–35. doi: 10.1192/bjp.bp.112.120055. [DOI] [PubMed] [Google Scholar]
- de Rooij SR, Costello PM, Veenendaal MV, Lillycrop KA, Gluckman PD, Hanson MA, et al. Associations between DNA methylation of a glucocorticoid receptor promoter and acute stress responses in a large healthy adult population are largely explained by lifestyle and educational differences. Psychoneuroendocrinology. 2012;37:782–788. doi: 10.1016/j.psyneuen.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Essex MJ, Boyce WT, Hertzman C, Lam LL, Armstrong JM, Neumann SM, et al. Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 2013;84:58–75. doi: 10.1111/j.1467-8624.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgatroyd C, Spengler D. Epigenetics of early child development. Front Psychiatry. 2011;2:16. doi: 10.3389/fpsyt.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talens RP, Christensen K, Putter H, Willemsen G, Christiansen L, Kremer D, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11:694–703. doi: 10.1111/j.1474-9726.2012.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CC, Caspi A, Williams B, Craig IW, Houts R, Ambler A, et al. A longitudinal study of epigenetic variation in twins. Epigenetics. 2010;5:516–526. doi: 10.4161/epi.5.6.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenema AH. Early life stress, the development of aggression and neuroendocrine and neurobiological correlates: what can we learn from animal models. Front Neuroendocrinol. 2009;30:497–518. doi: 10.1016/j.yfrne.2009.03.003. [DOI] [PubMed] [Google Scholar]
- McGorry PD, Purcell R, Goldstone S, Amminger GP. Age of onset and timing of treatment for mental and substance use disorders: implications for preventive intervention strategies and models of care. Curr Opin Psychiatry. 2011;24:301–306. doi: 10.1097/YCO.0b013e3283477a09. [DOI] [PubMed] [Google Scholar]
- Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmuhl Y, Fischer D, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12:1559–1566. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- Huisman M, Oldehinkel AJ, de Winter A, Minderaa RB, de Bildt A, Huizink AC, et al. Cohort profile: the Dutch 'TRacking Adolescents' Individual Lives' Survey' TRAILS. Int J Epidemiol. 2008;37:1227–1235. doi: 10.1093/ije/dym273. [DOI] [PubMed] [Google Scholar]
- Ormel J, Oldehinkel AJ, Sijtsema J, van Oort F, Raven D, Veenstra R, et al. The TRacking Adolescents' Individual Lives Survey (TRAILS): design, current status, and selected findings. J Am Acad Child Adolesc Psychiatry. 2012;51:1020–1036. doi: 10.1016/j.jaac.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Reijneveld SA, Brugman E, Verhulst FC, Verloove-Vanhorick SP. Identification and management of psychosocial problems among toddlers in Dutch preventive child health care. Arch Pediatr Adolesc Med. 2004;158:811–817. doi: 10.1001/archpedi.158.8.811. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Thornton A, Freedman D, Amell JW, Harrington H, et al. The life history calendar: A research and clinical assessment method for collecting retrospective event-history data. Int J Methods Psychiatr Res. 1996;6:101–114. [Google Scholar]
- Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, et al. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biol Psychiatry. 2012;72:41–48. doi: 10.1016/j.biopsych.2012.01.034. [DOI] [PubMed] [Google Scholar]
- Turner JD, Alt SR, Cao L, Vernocchi S, Trifonova S, Battello N, et al. Transcriptional control of the glucocorticoid receptor: CpG islands, epigenetics and more. Biochem Pharmacol. 2010;80:1860–1868. doi: 10.1016/j.bcp.2010.06.037. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke PM, Stirzaker C, Melki JR, Millar DS, Paul CL, Clark SJ. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997;25:4422–4426. doi: 10.1093/nar/25.21.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. Genes learn from stress: how infantile trauma programs us for depression. Epigenetics. 2010;5:194–199. doi: 10.4161/epi.5.3.11375. [DOI] [PubMed] [Google Scholar]
- Daniels WM, Fairbairn LR, van Tilburg G, McEvoy CR, Zigmond MJ, Russell VA, et al. Maternal separation alters nerve growth factor and corticosterone levels but not the DNA methylation status of the exon 1(7) glucocorticoid receptor promoter region. Metab Brain Dis. 2009;24:615–627. doi: 10.1007/s11011-009-9163-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzmann SR, Turner JD, Meriaux SB, Meijer OC, Muller CP. Epigenetic regulation of the glucocorticoid receptor promoter 1(7) in adult rats. Epigenetics. 2012;7:1290–1301. doi: 10.4161/epi.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2012;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Champagne FA, Brown SE, Dymov S, Sharma S, Meaney MJ, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25:11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JD, Schote AB, Macedo JA, Pelascini LP, Muller CP. Tissue specific glucocorticoid receptor expression, a role for alternative first exon usage. Biochem Pharmacol. 2006;72:1529–1537. doi: 10.1016/j.bcp.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Weaver IC, D'Alessio AC, Brown SE, Hellstrom IC, Dymov S, Sharma S, et al. The transcription factor nerve growth factor-inducible protein a mediates epigenetic programming: altering epigenetic marks by immediate-early genes. J Neurosci. 2007;27:1756–1768. doi: 10.1523/JNEUROSCI.4164-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JD, Pelascini LP, Macedo JA, Muller CP. Highly individual methylation patterns of alternative glucocorticoid receptor promoters suggest individualized epigenetic regulatory mechanisms. Nucleic Acids Res. 2008;36:7207–7218. doi: 10.1093/nar/gkn897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldehinkel AJ, Ormel J, Bosch NM, Bouma EM, Van Roon AM, Rosmalen JG, et al. Stressed out? Associations between perceived and physiological stress responses in adolescents: the TRAILS study. Psychophysiology. 2011;48:441–452. doi: 10.1111/j.1469-8986.2010.01118.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.