

Abstract
Frequently, proteomic LC-MS/MS data may contain sets of modifications that evade identification during standard database search. For many laboratories, the standard technique to seek posttranslational modifications (PTMs) adds a short list of specified mass shifts to database search configuration. This technique provides information for only the specified PTMs, takes substantial time to run, and drives false discoveries upward through an exponential expansion of search space. This protocol describes a more structured approach to blind PTM discovery through reducing protein lists, targeting attention to a data-driven list of mass shifts, and seeking the resulting short list of modifications through targeted search.
Keywords: Shotgun proteomics, Proteome informatics, Posttranslational modification, Protein identification, Sequence tagging, Database search, Software
1 Introduction
Algorithms to identify peptides from LC-MS/MS were introduced in 1994 (1), and the ability to identify PTMs by these algorithms was added only 1 year later (2). In essence, the database search strategy trawls a protein sequence database for candidate peptides of the approximate mass of the precursor ion for an MS/MS. These candidates are then compared to the observed MS/MS by predicting what fragment ions should be observed for each sequence and comparing to the observed set. Configuring these tools to find PTMs enables them to convolute these peptide sequences with a small, defined set of mass shifts associated with particular residues (such as potentially adding 16 Da to methionine residues), creating an exponential expansion of the search space. To curtail the time required to seek a wide variety of PTMs, Craig and Beavis introduced the “refined search” technique in 2003 (3). This approach first determines the set of unmodified peptides found in a mixture, produces a reduced protein set, and then seeks a wide variety of modifications for those proteins. In practice, this technique mixes two different kinds of search results in a single result set, complicating the determination of which spectra have been identified successfully.
Although initially published in the same year as database search, the sequence tagging strategy took longer to fully automate than database search (4). The approach infers a partial sequence from each tandem mass spectrum (5) and then uses these inferred tags as filters to reduce the set of peptide sequences compared to each spectrum. Where database search employs only the observed precursor mass to select compared peptides, tagging is able to use the set of inferred partial sequences as well as the masses flanking this tag to select comparisons. Mass shifts can then be introduced in the flanking mass regions to reflect PTMs. The InsPecT algorithm was the first to automate all elements of this process (6). Tsur et al. leveraged the ability to infer sequences from spectra in a “blind search” for modifications of unknown mass and specificity (7). This protocol emulates a blind search technique based in sequence tagging that was described by Dasari in the context of toxicological proteomics (8).
This implementation integrates the MyriMatch database search engine (9), the DirecTag sequence inference engine (10), the TagRecon tag matcher (11), and the IDPicker protein assembly environment (12, 13). The use of the first three is coordinated through the BumberDash user interface (14), and IDPicker features its own graphical user interface. This protocol deploys this strategy in the context of formalin-fixed, paraffin-embedded tissues rather than the toxicological data set described earlier, but the principal features are the same: (1) build a reduced set of proteins that can be confidently identified from unmodified peptides, (2) infer a reduced set of PTMs for which substantial MS/MS evidence can be found, and (3) conduct a limited search for PTMs employing sequence tag information.
2 Materials
This protocol describes an informatics method for detecting protein posttranslational modifications from shotgun proteomics data sets. For this, readers will need the BumberDash search software suite, MS/MS data files, a protein sequence database, and IDPicker (version 3.0) software for results filtering and protein assembly.
2.1 BumberDash Search Software Suite
The BumberDash suite incorporates the MyriMatch and DirecTag-TagRecon software. MyriMatch is a database search engine designed for shotgun protein identification. DirecTag-TagRecon is a sequence tagging-based search engine optimized for finding unexpected modifications in peptides using shotgun proteomics data. Both these tools accept two types of inputs: a raw MS/MS data file and a protein sequence database. The software accepts MS/MS data in a variety of instrument native and derived formats (see Note 1 for a full list of acceptable formats). Protein sequences are read from a FASTA-formatted file.
MyriMatch derives peptide sequences from the protein database, predicts fragmentation spectra, and compares them to the experimental MS/MS. The quality of each peptide-spectrum match (PSM) is assessed using the intensity and mass error associated with matched fragments. Finally, the software records the top five matches for each MS/MS to an identification file in either pep-XML or mzIdentML format.
DirecTag-TagRecon software starts by generating short amino acid tags for each MS/MS in the data file. The software matches the tags to candidate peptide sequences from the protein database. Mass mismatches between the tag and candidate sequence are interpreted as modifications. Theoretical tandem spectra are predicted for candidate peptides and compared to the experimental MS/MS. PSMs are scored, and identifications are recorded to standard formats following the same method employed by MyriMatch.
2.2 IDPicker Software
IDPicker (version 3.0) filters raw identifications from the search software to a confident set using the target-decoy strategy. The software reads the identifications from pepXML or mzIdentML files. IDPicker computes false discovery rates (FDR) of all identifications and filters the results to meet a user-defined target FDR. Finally, the software assembles a minimal protein list that encompasses all filtered peptides using the rules of parsimony. IDPicker writes the results to a SQL database and presents them to the user in a flexible graphical user interface. The software allows the user to arrange the results into an experimental hierarchy (biological replicates, technical replicates, controls, experiments, et cetera). The software can also create protein, peptide, and spectral identification reports in text format. These reports are useful for downstream analysis like spectral counting-based protein differentiation.
BumberDash and IDPicker (version 3.0) software are available for download free of charge from the Internet Web site http://fenchurch.mc.vanderbilt.edu. To employ this protocol, readers should download and install the software on a local computer.
2.3 LC-MS/MS Data Sets
We demonstrate this protocol with a shotgun proteomic data set generated from formalin-fixed paraffin-embedded (FFPE) colon tissues (15). Tissues were stored in FFPE for 1, 3, 5, or 10 years. Independently, tissues were fixed in formalin for 0, 1, 2, or 4 days. Proteins in each fixation were reduced with dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. Resulting peptide mixtures were analyzed via LC-MS/MS in replicates on a Thermo LTQ mass spectrometer. All the raw data files can be downloaded from the Proteome Commons Tranche Web site https://proteomecommons.org/dataset.jsp?i=77376. Readers can select subsets of the data for analysis to save computation time.
3 Methods
Figure 1 illustrates the three-stage workflow for detecting unexpected PTMs from shotgun proteomics data sets. First, we characterize the protein content of the sample with a simple MyriMatch database search (protein identification). Resulting PSMs are filtered with IDPicker software at 2 % FDR. Proteins with at least two unique peptide identifications are included in a subset FASTA database. Next, we query the proteins in the subset database for unanticipated PTMs using DirecTag-TagRecon software (blind PTM search). Peptide and PTM identifications are filtered with IDPicker software at a stringent 2 % FDR. Confident PTMs are identified using IDPicker software (see Subheading 3.3). Finally, we refine the PTM identification results with a directed PTM search. In this step, we re-query the subset FASTA with DirecTag-TagRecon software configured to look for only the confident PTMs. This (optional) refinement step is designed to improve the sensitivity and specificity of the PTM search. Identifications from the directed PTM search are filtered with IDPicker software at 2 % FDR.
Fig. 1.

PTM identification workflow
3.1 Protein Identification
Launch the BumberDash software.
Click on the bottommost row or go to “File->New Job” to add a new job. This opens an Add Job dialogue (see Fig. 2).
In the Add Job dialogue, select “MyriMatch- Database Search” as the “Type of Search” (default choice in BumberDash).
Name the search job and check the box if results should be stored as a subfolder in the output directory. This step may be omitted if desired (see Note 2).
Select the “Input Files” using the “Browse” button. BumberDash accepts MS/MS data files in mzXML, mzML, mgf, mz5, or any vendor raw format. The user can hold down ctrl to select multiple data files.
Change the “Output Directory” to the desired location. By default, BumberDash writes the output files to the folder containing the “Input Files.”
Select a “FASTA database” containing the protein sequences for the search. BumberDash accepts the protein databases in FASTA format.
If you have a premade MyriMatch configuration file, select it with the “Browse” button. Otherwise, create a new configuration file using the “New” button next to the configuration drop-down (see Note 3).
Clicking the “New” button will start the MyriMatch configuration editor (Fig. 3). Make changes to the desired parameters. You can either save the changes either temporarily or permanently. Click the “Use Once” button if you want to use the configuration for the current job only. Click “Save As New” and supply a filename if you want to retain the configuration for future searches. Return to the Add Job dialogue.
Append the job to the search queue with the “Add” button. Figure 4 shows an example of BumberDash job queue. Users can track the status of jobs in the queue. Jobs can also be cancelled (click the “X” button) from the queue (see Note 4).
Wait for the MyriMatch job to finish. Launch IDPicker (version 3.0) software to process the database search results. If IDPicker 3 is already installed on the computer it can be launched from BumberDash’s File->Run menu.
Import the resulting identification files (pepXML) into IDPicker. The pepXML files are found in the “Output Directory” of the corresponding BumberDash job. To import the files, launch the IDPicker results navigator with “File->Import Files” option. IDPicker results navigator has two panels. The left panel navigates the folder structure containing the results files. The right panel shows the pepXML files found in a folder selected in the left panel. To import files, browse to the output folder containing the pepXML files in the left panel and click the “>” button to see the files in the right panel. Make sure that the desired files are checked and click “Open.”
After this step, IDPicker will display an “Import Settings” dialogue (see Fig. 5). These settings are used by the software to filter the PSMs present in the pepXML files. Set the “Database” to the FASTA file used for the database search. Change “Max FDR” setting to “0.02,” “Max Rank” to “1,” and “Qonverter Settings” to “MyriMatch Optimized.” Click “OK” to start creating the IDPicker report and wait for it to finish.
The filtered report contains information about the proteins, peptides, and PSMs identified in the data set. We need to create a small subset FASTA database containing sequences of proteins identified in the data set. This subset FASTA is used for subsequent PTM searches (Fig. 1). To accomplish this, go to File->Export->Subset FASTA, instruct IDPicker to add decoy sequences to the generated database, and save the database to a location of your choice.
Fig. 2.

Add Job dialogue of BumberDash
Fig. 3.

BumberDash configuration editor
Fig. 4.
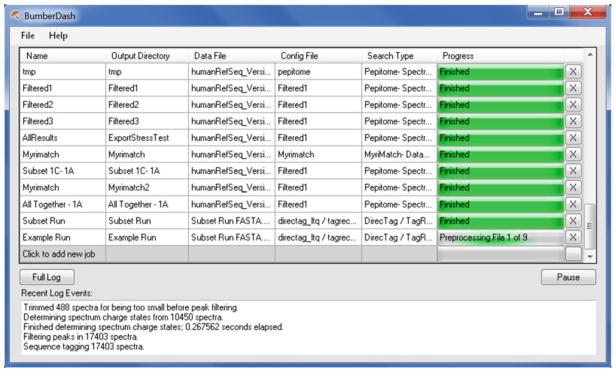
BumberDash job queue
Fig. 5.

IDPicker (version 3) “Import Settings” dialogue
3.2 Blind PTM Search for Finding Unexpected Sequence Modifications
Load BumberDash and bring up the Add Job dialogue (see Subheading 3.1, step 2).
Select “DirecTag/TagRecon- Sequence Tagging” as the “Type of Search.”
Name the job, select the input MS/MS data files, and specify an output directory (see Subheading 3.1, steps 4–6). The input files selected for this search should be identical to those selected for the protein identification step (see Subheading 3.1).
Select the subset FASTA produced at the end of Subheading 3.1 as input “FASTA database.”
Next, we need to configure DirecTag and TagRecon separately. Both these configurations are shown as separate drop-down boxes in the Add Job dialogue (Fig. 2).
If you want to use a premade DirecTag configuration file click on the “Browse” button next to the DirecTag configuration drop-down box. Otherwise, click the “New” button to start the configuration editor (Fig. 3). Make changes to the desired parameters. You can save the changes either temporarily or permanently. Click the “Use Once” button if you want to use the configuration for the current job only. Click “Save As New” and supply a filename if you want to retain the configuration for future searches. Return to the Add Job dialogue (see Note 5).
Repeat the previous step to configure TagRecon. Ensure that the “Explain Unknown Mass Shifts As” option is set to “blind-ptms.” Return to the Add Job dialogue.
Add the job to the search queue (see Subheading 3.1, step 10) and wait for completion (see Note 6).
Launch IDPicker (version 3.0) software to process “Blind PTM” search results (see Subheading 3.1, step 11). Add the search pepXML files to IDPicker (see Subheading 3.1, step 12).
Configure the “Import Settings” of IDPicker as described in Subheading 3.1, step 12 with two important changes. Set the “Database” to the subset FASTA file used in the search and change the “Qonverter Settings” to “TagRecon Optimized.” Start the IDPicker results filtering using the “OK” button.
The “Blind PTM” IDPicker report will contain a summary of proteins, peptides, PSMs, and PTMs present in the data set. The PTM summary (Fig. 6) is located in the modification form (by default located in the lower right quarter of the IDPicker GUI). We need to use this form to identify a list of confident PTMs present in the sample (see Subheading 3.3). These confident PTMs are used in the final “Directed PTM” search (Fig. 1) (see Note 7).
Fig. 6.

IDPicker 3 modification detail view
3.3 Detecting Confident Mass Shifts Using IDPicker
From the modification grid, switch to detail view by clicking the button in the upper-right corner. This should show the list of possible modifications identified along with data associated with the modification.
Click the “Unimod Filter” button to bring up a list of all possible Unimod explanations for the data. If fewer modifications appear than expected it may be necessary to increase the “Round to Nearest” number (see Note 8).
Select the posttranslational checkbox and click outside the pop-up to show only modifications identified as possibly being posttranslational (Fig. 7). Generally modifications which are associated with many spectra and which contribute to at least two peptides are less likely to be erroneous results.
Choose three to eight modifications which are of greatest interest based on the data. Record the mass shifts and residue characters to use in the next step.
Fig. 7.

Unimod filter popup
3.4 Run-Directed Posttranslational Modification Search
Once more load BumberDash and start a “DirecTag/TagRecon- Sequence Tagging” job.
The input files, subset FASTA, and DirecTag configuration should remain the same. Select a new job name and output folder if desired.
IntheTagReconconfiguration,“ExplainUnknownMassShiftsAs” should be set to “preferredptms.” Modifications obtained from the previous step should be added to the configuration by entering the residue motif (or character), the modification delta mass, and the mod type set to “PreferredPTM.”
Run the job and load the results in IDPicker 3 (see Note 9).
Acknowledgments
The algorithms described in this protocol were developed through support to all three authors by R01 CA126218. In addition, J.D.H. was supported by U01 CA08402.
Footnotes
BumberDash supports mzML, mzXML, MGF, Agilent, Bruker FID/YEP/BAF, Thermo RAW, Waters RAW, MGF, MS2/CMS2/BMS2, and mzIdentML file formats.
Job naming is optional, if no name is given when files are selected a name will be generated automatically based on the top-level directory of the input files when they are selected. If the new folder box is checked then a new folder will be generated in the output directory in which to store the result files. On the main screen jobs which have not yet been run but are set to produce a subfolder have an output directory ending with a “+.” Jobs which have been or are in the process of being run will have a “*” at the end of the output directory indicating that it points to the subfolder that has been produced for the job. The full output directory can be viewed in the tooltip of the abbreviated output directory cell.
BumberDash comes preloaded with recommended settings for a few different instrument types, which can be loaded by selecting an option in the Instrument drop-down menu. Setting the specificity to semi-specific (or placing “MinTerminiCleavages = 1” in the configuration file) will yield a much better data set; however it will also greatly increase the required time compared to a fully specific (“MinTerminiCleavages = 2”) search. For the data from the Sprung article (15), search times on a Core 2 Quad CPU were approximately 5 h per file for fully specific searches and 35 h per file for semi-specific searches.
BumberDash runs at a “Below Normal” priority level, so it should not throttle the CPU usage of other programs running on the computer. To further allow BumberDash to run in the background it minimizes to the system tray, not taking up space in the task bar. To restore a minimized instance of BumberDash double-click the icon in the tray.
We recommend adding carbamidomethylation of cysteine as a static modification to DirecTag runs. This change causes Cys residues to account for 160 Da rather than 103 Da, reflecting their modification by iodoacetamide.
For the Sprung data set (15) on the Core 2 Quad CPU test machine, this process took about two hours per file when a subset database produced by a fully specific search was used. The more comprehensive subset database produced by a semi-specific search brought the required time up to 8 h per file.
Other forms in the IDPicker window can be minimized by clicking the pin on the top-right section of the panel title bar. Modifications may be highlighted based on how many times they were observed in the given data set: green for more than 10, blue for more than 50, and red if the modification was seen over 100 times. To show only common modifications click “Unimod Filter” in the top right and check all category boxes, leaving “Show Hidden” unchecked. If a modification number is bold it contains a list of possible Unimod explanations in the tooltip.
Initially only the Unimod annotations labeled as “unhidden” will appear in the pop-up box. To show all identified modifications in the list click the checkbox in the upper-left corner. Rounded modification masses must exactly match the Unimod entries to be recognized. Increasing the “Round to Nearest” number will increase the likelihood a modification is recognized by Unimod, but decrease the overall specificity.
For best results run job again with “Database Search” and selected modifications as dynamic mods, using the subset FASTA. Both the database search and tag search results can be loaded into IDPicker 3 at the same time to create a combined report.
References
- 1.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Yates JR, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 3.Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun Mass Spectrom. 2003;17:2310–2316. doi: 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- 4.Mann M, Wilm M. Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994;66:4390–4399. doi: 10.1021/ac00096a002. [DOI] [PubMed] [Google Scholar]
- 5.Tabb DL, Saraf A, Yates JR. GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal Chem. 2003;75:6415–6421. doi: 10.1021/ac0347462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner S, Shu H, Frank A, et al. InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal Chem. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 7.Tsur D, Tanner S, Zandi E, Bafna V, Pevzner PA. Identification of post-translational modifications by blind search of mass spectra. Nat Biotechnol. 2005;23:1562–1567. doi: 10.1038/nbt1168. [DOI] [PubMed] [Google Scholar]
- 8.Dasari S, Chambers MC, Codreanu SG, et al. Sequence tagging reveals unexpected modifications in toxicoproteomics. Chem Res Toxicol. 2011;24:204–216. doi: 10.1021/tx100275t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J Proteome Res. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabb DL, Ma Z-Q, Martin DB, Ham A-JL, Chambers MC. DirecTag: accurate sequence tags from peptide MS/MS through statistical scoring. J Proteome Res. 2008;7:3838–3846. doi: 10.1021/pr800154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dasari S, Chambers MC, Slebos RJ, Zimmerman LJ, Ham A-JL, Tabb DL. TagRecon: high-throughput mutation identification through sequence tagging. J Proteome Res. 2010;9:1716–1726. doi: 10.1021/pr900850m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007;6:3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Z-Q, Dasari S, Chambers MC, et al. IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J Proteome Res. 2009;8:3872–3881. doi: 10.1021/pr900360j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holman JD, Ma Z-Q, Tabb DL. Identifying proteomic LC-MS/MS data sets with Bumbershoot and IDPicker. Current protocols in bioinformatics/editorial board, Andreas D. Baxevanis. 2012;Chapter 13(Unit 13.17) doi: 10.1002/0471250953.bi1317s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sprung RW, Jr, Brock JWC, Tanksley JP, et al. Equivalence of protein inventories obtained from formalin-fixed paraffin-embedded and frozen tissue in multidimensional liquid chromatography-tandem mass spectrometry shotgun proteomic analysis. Mol Cell Proteomics. 2009;8:1988–1998. doi: 10.1074/mcp.M800518-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]