

Abstract
The sleep disorder narcolepsy results from loss of hypothalamic orexin/hypocretin neurons. Although narcolepsy onset is usually postpubertal, current mouse models involve loss of either orexin peptides or orexin neurons from birth. To create a model of orexin/hypocretin deficiency with closer fidelity to human narcolepsy, diphtheria toxin A (DTA) was expressed in orexin neurons under control of the Tet-off system. Upon doxycycline removal from the diet of postpubertal orexin-tTA;TetO DTA mice, orexin neurodegeneration was rapid, with 80% cell loss within 7 d, and resulted in disrupted sleep architecture. Cataplexy, the pathognomic symptom of narcolepsy, occurred by 14 d when ∼5% of the orexin neurons remained. Cataplexy frequency increased for at least 11 weeks after doxycycline. Temporary doxycycline removal followed by reintroduction after several days enabled partial lesion of orexin neurons. DTA-induced orexin neurodegeneration caused a body weight increase without a change in food consumption, mimicking metabolic aspects of human narcolepsy. Because the orexin/hypocretin system has been implicated in the control of metabolism and addiction as well as sleep/wake regulation, orexin-tTA; TetO DTA mice are a novel model in which to study these functions, for pharmacological studies of cataplexy, and to study network reorganization as orexin input is lost.
Keywords: diphtheria toxin A fragment, hypocretin, model mice, narcolepsy, orexin, transgenic
Introduction
Narcolepsy is a chronic sleep disorder characterized by excessive daytime sleepiness, fragmentation of sleep/wakefulness, hypnagogic hallucinations, sleep paralysis, and cataplexy. In humans, the onset of narcolepsy is typically in adolescence or early adulthood. Because definitive diagnosis may take as long as a decade, it is difficult to study the development of symptomatology after initial onset of the disorder.
The etiology of narcolepsy is a loss of the hypothalamic neurons that express the orexin/hypocretin neuropeptides (Sakurai et al., 1998; de Lecea et al., 1998). Although the cause of orexin neuron loss remains to be determined, genetic linkage of narcolepsy with human leukocyte antigen (HLA)-DQB1*0602 (Mignot et al., 1997), immune system gene polymorphisms (Mahlios et al., 2013), and orexin-reactive CD4+ T cells in narcolepsy patients (De la Herrán-Arita et al., 2013) suggest an autoimmune mechanism in which the orexin neurons are targeted.
Although narcoleptic dogs, which bear a mutation in the orexin 2 receptor (OX2R) gene (Lin et al., 1999), have been extensively used for pharmacological (Nishino and Mignot, 1997) as well genetic studies, the discovery of the orexin/hypocretin neuropeptides opened the door to creation of novel animal models of this disease. Prepro-orexin knock-out mice were reported to show fragmentation of sleep/wakefulness and behavioral arrests that are similar to cataplexy (Chemelli et al., 1999). Although orexin 1 receptor (OX1R) null mice showed a mild abnormality in sleep/wake behavior, OX2R null mice showed clear narcolepsy-like symptomatology (Willie et al., 2003) and OX1R and OX2R double null mutant mice showed even more severe symptoms (Kisanuki et al., 2001; Kalogiannis et al., 2011). The loss of orexin cells characteristic of human narcolepsy has been modeled in orexin/ataxin-3 mice in which the neurotoxic polyglutamine repeat of the Ataxin-3 protein is coupled to the prepro-orexin promoter (Hara et al., 2001).
These animal models have helped elucidate the role of orexin neurons in sleep/wakefulness regulation, metabolism, and addiction. However, current animal models lack the orexin peptides, receptors, or neurons from birth and thus do not replicate the typical postpubertal onset of this disorder in humans. Furthermore, current mouse models have limited utility in the development of novel pharmacological treatments for narcolepsy because cataplexy events are relatively infrequent. Accordingly, behavioral (España et al., 2007), dietary (Oishi et al., 2013), and pharmacological (Black et al., 2013) approaches have been implemented in attempts to elevate cataplexy levels. To create a model with closer fidelity to human narcolepsy, we used the Tet-off system in which expression of diphtheria toxin A (DTA) in orexin neurons was controlled by the presence of doxycycline (DOX). We find robust cataplexy as well as disrupted sleep architecture and weight gain in this model. Because the orexin/hypocretin system has been implicated in the control of metabolism and addiction as well as sleep/wake regulation, orexin-tTA; TetO DTA mice are a novel model in which to study these functions, for pharmacological studies of sleep/wake fragmentation or cataplexy, and to understand the process of network reorganization as orexin input is lost.
Materials and Methods
Animal usage.
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committees of the Research Institute of Environmental Medicine Nagoya University and at SRI International and were in accordance with National Institutes of Health guidelines. All efforts were made to minimize animal suffering or discomfort and to reduce the number of animals used.
Animals.
Orexin-tTA mice (Tabuchi et al., 2013), which express tTA exclusively in orexin neurons under the control of human prepro-orexin promoter (Sakurai et al., 1999), were bred with TetO DTA fragment mice (B6.Cg-Tg(tetO-DTA)1Gfi/J, The Jackson Laboratory) to generate orexin-tTA; TetO DTA mice. The transgenic construct to generate orexin-tTA transgenic mice was made by substituting the nLacZ gene (SalI-BamHI fragment) of the orexin/nLacZ transgenic construct (Sakurai et al., 1999) with 0.7 kb of the mammalianized tetracycline-controlled transcriptional activator (tTA) fragment (Inamura et al., 2012). The transgene was excised and microinjected into pronuclei of fertilized mouse eggs (C57BL/6 mice) to generate transgenic founders. Founders were bred with C57BL/6J mice (Clea-Japan) to produce stable orexin-tTA transgenic lines. A total of 9 orexin-tTA transgene-positive founders were obtained. In situ hybridization analysis of the N1 generation revealed that lines 29, C5, and G5 showed the highest tTA mRNA expression. TetO DTA mice express DTA under the control of a tetracycline operator. In these double transgenic mice, DTA expression occurs in the orexin neurons in the absence of DOX. Both orexin-tTA mice and TetO DTA mice were on the C57BL/6J genetic background.
DOX-containing chow (DOX chow) was made by adding 10% DOX powder (Kyoritsu Seiyaku) to normal chow (Labo MR Stock, Nosan) at a final concentration of 100 mg/kg. Labo MR stock was provided during DOX(−) period. Mating pairs of orexin-tTA mice and TetO DTA mice were fed with DOX-containing chow (DOX(+) condition) from the day of mating. During the prenatal and early postnatal periods, DOX was supplied via maternal circulation or lactation, respectively. After weaning, orexin-tTA; TetO DTA mice were fed with DOX (+) chow until the day of the experiment.
Experimental protocol 1: Induction of orexin cell loss in orexin-tTA; TetO DTA mice.
Male and female orexin-tTA; TetO DTA mice were maintained from weaning to 10 weeks of age on DOX(+) chow. At 10 weeks, DOX(+) chow was removed and replaced with Labo MR stock food (DOX(−) condition) for up to 13 weeks. At 0 d (n = 5), 1.5 d (n = 3), 3.5 d (n = 3), 1 week (n = 4), 2 weeks (n = 3), 4 weeks (n = 22), or 13 weeks (n = 4) after DOX removal, mice were perfused and brains were sliced, immunostained for orexin, and cell counts obtained as described below. Orexin-tTA (n = 7) and TetO DTA (n = 4) mice, maintained from weaning on DOX(+) chow, were also killed at 10 weeks of age as a comparison.
Experimental protocol 2: Characterization of sleep/wake parameters during orexin cell loss in orexin-tTA; TetO DTA mice.
For sleep/wake studies, male mice raised on DOX(+) chow were implanted with EEG and EMG electrodes for polysomnographic recording at 10 weeks of age as described below. At 12 weeks, DOX(+) chow was replaced with DOX(−) chow. EEG and EMG recordings occurred approximately weekly from age 12 to 25 weeks (0–13 weeks after DOX removal). EEG and EMG parameters were compared with orexin/ataxin-3 mice (Hara et al., 2001) and to orexin-tTA; TetO DTA mice raised from birth on DOX(−) chow (the D(−) group).
Experimental protocol 3: Induction of partial orexin cell loss in orexin-tTA; TetO DTA mice.
To reduce the number of orexin neurons without elimination of the entire cell population, DOX(+) chow was removed at 10 weeks of age and replaced with Labo MR stock food (DOX(−) condition) for 1.5 or 3.5 d, after which DOX(+) chow was reintroduced (1.5 d + restoration of DOX [RD]; n = 3 and 3.5 d + RD; n = 3, respectively). Mice were then killed and perfused for immunohistochemical analyses at 14 weeks of age. In another variation of the protocol, DOX chow was removed at 12 weeks of age and reintroduced at 14 weeks (DOX(−)2 weeks + RD; n = 6); mice were then killed at 25 weeks of age.
Experimental protocol 4: Partial orexin cell loss and cataplexy assessment.
To maintain the number of orexin neurons at 5% of the control number, DOX(+) chow was removed at 12 weeks of age and replaced with Labo MR stock food (DOX(−) condition) for 2 weeks, after which DOX(+) chow was reintroduced for 13 weeks until mice were 25 weeks old. Cataplexy bouts after DOX(−) were monitored.
Experimental protocol 5: Food-elicited cataplexy test.
In an attempt to increase cataplexy above basal levels, chocolate 1.8 ± 0.5 g (milk chocolate, Meiji) was made available along with DOX(−) chow at light offset (20:00) to mice at 4 weeks and 15 weeks after removal of DOX from the diet. Cataplexy levels were compared with those observed on the day before chocolate availability.
Experimental protocol 6: Body weight, food and water consumption, and activity measures.
Male orexin-tTA; TetO DTA mice, orexin-tTA mice, and TetO DTA mice were maintained from weaning to 10 weeks of age on DOX(+) chow. At 10 weeks, DOX(+) chow was removed and replaced with Labo MR stock food (DOX(−) condition) for up to 4 weeks. Daily measurements of body weight and food and water consumption were initiated 3 d before the DOX(−) condition was imposed; measurements occurred at 8:00 A.M. each day. Spontaneous activity was continuously monitored by a weight sensor (Actracer-2000, Arco System) positioned under the cage. Blood samples (200 μl) were taken from the tail vein before DOX(−) at 10 weeks and after 4 weeks DOX(−). Blood glucose levels were measured by a glucose sensor (G sensor, Glucocard DIA meter, Arkley) on 5 μl serum samples. Serum levels of insulin (ELISA kit, Morinaga Institute of Biological Science, Yokohama, Japan), leptin (ELISA kit, Morinaga Institute of Biological Science), and free fatty acids (Free fatty acid C kit, Wako) were determined; insulin and leptin concentrations were determined as the mean value of triplet measurements.
Immunohistochemical studies.
Male and female mice were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused sequentially with 15 ml of chilled saline and 15 ml of chilled 10% formalin solution (Wako). Brains were isolated and immersed in formalin solution for 24 h at 4°C, followed by a 30% sucrose solution at 4°C for at least 2 d. Brains were quickly frozen in embedding solution (Sakura Finetek). Coronal sections containing the lateral hypothalamic area, dorsal raphe (DR), and locus ceruleus (LC) were cut on a cryostat (CM3050 S, Leica) at a thickness of 40 μm. Sections were incubated for 40 min in phosphate buffer containing 0.3% H2O2 to inactivate endogenous peroxidase. After washing three times in PBS containing 0.25% Triton X-100 and 1% BSA fraction V (PBS-BX), sections were incubated in PBS-BX for 24 h at 4°C with goat anti-orexin IgG antibody (Santa Cruz Biotechnology), guinea pig anti-5-hydroxytryptamine transporter IgG antibody (Frontier Institute), rabbit anti-tyrosine hydroxylase IgG antibody (Millipore), rabbit anti-melanin-concentrating hormone (MCH) IgG antibody (Sigma), or rabbit anti-IbaI IgG antibody (Wako). After several washes in PBS-BX, sections were incubated overnight with biotinylated horse anti-goat IgG antibody (Vector Laboratories), biotinylated goat anti-guinea pig IgG antibody (Vector Laboratories), or biotinylated goat anti-rabbit IgG antibody (Vector Laboratories) in PBS-BX for 1 h at room temperature. Sections were then washed with PBS-BX and reacted with avidin-biotin peroxidase complex (Vector Laboratories) diluted in PBS-BX for 1 h at room temperature. Bound peroxidase was visualized by DAB (Merck) with 0.0015% H2O2, resulting in a golden-brown reaction product. For double staining, bound peroxidase was visualized by 0.01 m imidazole acetate buffer containing 0.05% 3,3-diaminobenzidine tetrahydrochloride (Dako), 0.0015% H2O2 and 2.5% nickel ammonium sulfate (Wako), resulting in a black reaction product. For orexin and NeuN double staining, sections were incubated with mouse anti-NeuN IgG antibody (Millipore) and goat anti-orexin IgG antibody (Santa Cruz Biotechnology) for 24 h at 4°C. These sections were incubated with CF488-labeled donkey anti-mouse IgG (Biotium) and CF 647-labeled donkey anti-goat IgG (Biotium) for 1 h at room temperature. Antisera concentrations are listed in Table 1. Sections were mounted and examined with a microscope (BZ-9000, Keyence). To confirm the specificity of antisera, incubations without primary antibody were conducted as a negative control in each experiment, and the expected absence of signal was confirmed (data not shown). Approximately 10 slices from a 1 in 4 series were selected for counting of orexin and MCH neurons. NeuN-ir cells and orexin-ir cells were counted in a region (counting box width 363 μm, height 273 μm) within the lateral hypothalamic area. The size and area of orexin neurons, axon fibers, and IbaI-ir cell were automatically calculated using image analysis software (Hybrid cell count, Keyence).
Table 1.
Reagents used to localize cell types of interest
| Target | Primary antiserum | Secondary antibody |
|---|---|---|
| Orexin neurons | Goat anti-orexin A (C-19) (1:2000, Santa Cruz Biotechnology) | Biotinylated horse anti-goat (1:1000, Vector Laboratories) |
| Serotonergic neurons | Guinea pig anti-HTT (1:1000, Frontier Institute) | Biotinylated goat anti-guinea pig (1:1000, Vector Laboratories) |
| Noradrenergic neurons | Rabbit anti-TH (1:2000, Millipore) | Biotinylated goat anti-rabbit (1:1000, Vector Laboratories) |
| MCH neurons | Rabbit anti-MCH (1:4000, Sigma) | Biotinylated goat anti-rabbit (1:1000, Vector Laboratories) |
| Microglia | Rabbit anti-IbaI (1:500, Wako) | Biotinylated goat anti-rabbit (1:1000, Vector Laboratories) |
| Orexin neurons | Goat anti-orexin A (C-19) (1:1000, Santa Cruz Biotechnology) | CF 647-labeled donkey anti-goat (1:500, Biotium) |
| Neurons | Mouse anti-NeuN (1:500, Millipore) | CF 488-labeled donkey anti-mouse (1:1000, Biotium) |
EEG/EMG surgery, recording, and analyses.
Male mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and implanted with EEG and EMG electrodes for polysomnographic recording at 10 weeks of age. Mice were then housed separately for at least 7 d recovery before recording. Continuous EEG and EMG recordings were conducted through a slip ring (Air Precision, Le Pressis Robinson) designed so that the movement of the mouse was unrestricted. EEG and EMG signals were amplified (AB-610J, Nihon Koden), filtered (EEG 1.5–30 Hz; EMG 15–300 Hz), digitized at a sampling rate of 128 Hz, and recorded using SleepSign version 3 (Kissei Comtec). The animal's behavior was monitored through a CCD video camera and recorded on a computer synchronized with EEG and EMG recording using the SleepSign video option system (Kissei Comtec). During the dark period, activity was monitored by infrared sensor. EEG/EMG records were automatically scored in 4 s epochs and classified as wakefulness, SWS, or REM sleep by SleepSign software according to standard criteria (Tobler et al., 1997; Yamanaka et al., 2002). Vigilance state classifications assigned by SleepSign were examined visually and corrected. The same individual, blinded to genotype and experimental condition, scored all EEG/EMG recordings.
Spectral analysis of the EEG was performed by fast Fourier transform (sampled at 128 Hz). This analysis yielded power spectra profiles over a 0–40 Hz window with a 1 Hz resolution for the δ (1–5 Hz), θ (6–10 Hz), α (10–13 Hz), and β (13–25 Hz) bandwidths. An average EEG spectral profile was calculated from EEG power densities in each frequency bin and expressed as average power values for the δ, θ, α, and β bandwidths for each state.
Cataplexy was scored according to the consensus definition of cataplexy (Scammell et al., 2009) based on four criteria: an abrupt episode of EMG atonia lasting ≥10 s, immobility during the episode, predominance of θ activity during the episode, and >40 s of wakefulness preceding the episode. In some very rare cases, mice briefly came out of cataplexy only to have another attack within 10–30 s; such a sequence was scored as two bouts of cataplexy.
Statistical analyses.
Data were analyzed by one-way ANOVA followed by Fisher's protected least significant difference test and t test using OriginPro 8.2 software (OriginLab). p <0.05 was considered statistically significant.
Results
Control of orexin neurons using the Tet-off system
To produce orexin neuron-specific degeneration in adulthood, DTA was expressed in orexin neurons using the Tet-off system (Fig. 1A). Orexin-tTA mice (Tabuchi et al., 2013; Tsunematsu et al., 2013), in which tTA is exclusively expressed in orexin neurons under control of human prepro-orexin promoter, were bred with TetO DTA mice to generate orexin-tTA; TetO DTA mice. In these double transgenic mice, tTA induces DTA expression by binding to the TetO sequence. Expression of DTA, which induces cell death by inhibiting protein synthesis (Lee et al., 1998), was controlled by the presence or absence of DOX in the diet because tTA loses its ability to bind to the TetO sequence in the presence of DOX. Thus, tTA-induced DTA expression was initiated by replacing DOX(+) chow with DOX(−) chow.
Figure 1.
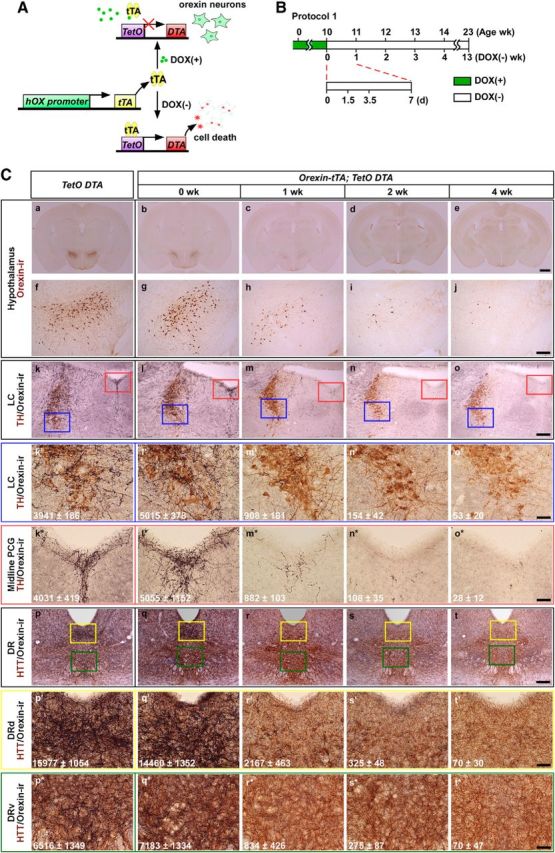
Induction of orexin neuron-specific cell death using the tet-off system. A, In the presence of DOX, DTA expression is suppressed in orexin-tTA; TetO DTA mice and orexin neurons are normal. In the absence of DOX, DTA is expressed and orexin cell death results. B, Schematic illustrating Experimental protocol 1. Green bar represents mice were fed chow containing DOX until 10 weeks of age (DOX(+) condition); open bar represents mice were fed chow without DOX (DOX(−) condition), which induces DTA expression. C, Immunostaining of orexin neurons and projections to terminal fields. Orexin neurons stained in brown (a–j) and terminals in black (k–t). Top, Orexin-ir neurons in the hypothalamus (a–j) under different conditions. Bottom, Orexin projection fields in the LC (k–o,k′–o′,k*–o*) and DR (p–t, p′–t′, p*–t*) in the corresponding conditions. Noradrenergic neurons in the LC were stained with anti-tyrosine hydroxylase (TH) in brown (k–o, k′–o′, k*–o*). Higher magnification of LC (blue rectangle in k–o) is shown in k′–o′. k–o, Red rectangle represents the midline pontine central gray (PCG); a higher magnification of this area is shown in k*–o*. Serotonergic neurons in the DR were stained with anti-5-hydroxytryptamine transporter (HTT) in brown (p–t, p′–t′, p*–t*). Yellow and green rectangles represent the dorsal part of the DR (DRd) and the ventral DR (DRv), respectively. Higher magnification of these areas is shown in p′–t′ and p*–t*, respectively. k′–t′, k*–t*, Numbers indicate the area of orexin-ir fibers in the region (counting box width 363 μm × height 273 μm) measured by image analysis software. Area (μm2) is mean ± SEM (n = 3–5). 0 week, 1 week, 2 weeks, and 4 weeks indicate the number of weeks that orexin-tTA; TetO DTA mice were in the DOX(−) condition before perfusion, as indicated in B. Littermate monogenic TetO DTA mice were used as controls. Scale bars: Ca–Ce, 1 mm; Cf–Ct, 200 μm; Ck′–Ct′, Ck*–Ct*, 50 μm.
To determine whether orexin cell death could be controlled by DOX in orexin-tTA; TetO DTA mice, in Experimental protocol 1, the number of orexin neurons was counted after DOX removal from the diet, as illustrated in Figure 1B. At 10 weeks of age on DOX(+) chow, the number of orexin neurons in orexin-tTA; TetO DTA mice was 891 ± 54 (n = 5), which was comparable with monogenic littermate TetO DTA (897 ± 32; n = 4) or orexin-tTA (889 ± 54; n = 7) control mice. Throughout the experiments, the number of neurons reported is a sum of the neurons counted in every fourth brain section (brains were sliced into 40 μm sections). Neither the shape nor the size of orexin neurons was altered at this age. The area of orexin neurons in control (TetO DTA), 0 week, 1 week, 2 weeks, and 4 weeks was 185 ± 21 μm2, 155 ± 14 μm2, 170 ± 16 μm2, 163 ± 25 μm2, and 196 ± 26 μm2, respectively (n = 13–73, p > 0.3, ANOVA, no significantly difference). These results indicated that Tet-off system worked in the presence of DOX without leakage of DTA expression in orexin neurons until at least 10 weeks of age. After replacing DOX(+) chow with DOX(−) chow, the number of orexin neurons dramatically decreased in orexin-tTA; TetO DTA mice (Figs. 1Cb–e,Cg–j and 2A). The number of orexin neurons after 1.5 d (36 h), 3.5 d, 1 week, 2 weeks, 4 weeks, and 13 weeks DOX(−) decreased to 572 ± 68 (64.2%, n = 5), 323 ± 3 (36.2%, n = 6), 127 ± 26 (14.3%, n = 4), 47 ± 7 (5.2%, n = 3), 25 ± 2 (2.8%, n = 22), and 7 ± 1 (0.8%, n = 4), respectively. Post hoc tests revealed significant reductions in all groups relative to 0 week. Figure 2B presents the rostrocaudal pattern of orexin neuron ablation after DOX removal. Orexin neurons are distributed −1.0 mm to bregma −2.0 mm from bregma, with the peak population occurring at bregma −1.3 mm. The number of orexin neurons rostral-caudally decreased uniformly after DOX removal. However, the number of orexin neurons in the lateral hypothalamus decreased faster than the medial population (Fig. 2C).
Figure 2.

Transition of the number of orexin-ir cell bodies. A, Bar graph showing the number of orexin-ir cell bodies in the hypothalamus at various time points after removal of DOX from the diet. Number on the top of bar indicates number of mice in each experiment. B, Number of orexin-ir cell bodies in coronal sections at multiple anterior–posterior levels relative to bregma. C, Number of orexin-ir neurons in the lateral part and the medial part of the lateral hypothalamus after DOX removal. Inset, The fornix was used to delineate the lateral and medial orexin populations. *p < 0.05 versus TetO DTA. Values are mean ± SEM.
Orexin neurons densely innervate monoaminergic nuclei, such as the noradrenergic LC and serotonergic DR nucleus, which play an important role in the regulation of sleep/wakefulness. In conjunction with loss of orexin cell bodies in the hypothalamus, orexin neuron nerve endings in terminal fields in the LC and DR decreased as well. By 4 weeks after removal of DOX, orexin nerve endings almost completely disappeared from these projection sites (LC: Fig. 1Cl–o,l′–o′; midline pontine central gray: Fig. 1Cl*–o*; DR: Fig. 1Cq–t,q′–t′,q*–t*). Although orexin nerve endings gradually decreased after removal of DOX, the number of noradrenergic neurons and serotonergic neurons was not altered in either of these nuclei.
To confirm whether DTA-induced cell death induction was restricted to the orexin neurons, MCH neurons were stained and counted (Fig. 3A). MCH neurons are distributed in the lateral hypothalamic area but do not colocalize with orexin (Brischoux et al., 2001). The number of MCH neurons was unaffected by DOX removal for 4 weeks in orexin-tTA; TetO DTA mice (Fig. 3B). Even though MCH neurons are located near the orexin neurons, no signs of unhealthiness, such as shrinkage of MCH cell bodies or unclear edges of MCH neurons, were observed. To determine whether any other neurons located near orexin cells were affected, sections of the tuberal hypothalamus were stained with an anti-NeuN antibody. NeuN-ir cells and orexin-ir cells were counted (counting box width 363 μm × height 273 μm) in the area where orexin neurons are densely distributed. The number of nonorexin neurons (NeuN-ir cells − orexin-ir cells) in TetO DTA mice and orexin-tTA; TetO DTA mice at DOX(−) 6 weeks was 67 ± 8 (NeuN-ir cells: 79 ± 8; orexin-ir cells: 12 ± 0.1, n = 3) and 65 ± 7 (NeuN-ir cells: 65 ± 9; orexin-ir cells: 0.2 ± 0.1) (n = 4, p = 0.848, Student's t test), respectively. These results confirmed that the Tet-off system functioned correctly in the transgenic mice and that orexin cell bodies and nerve terminals were specifically ablated after removing DOX from the chow.
Figure 3.

MCH neurons were intact after ablation of orexin neurons in orexin-tTA; TetO DTA mice. A, MCH neurons (brown) were stained with anti-MCH antibody. Bottom panels, Higher magnification of the region indicated by the square in the top panel. Scale bars: top, 1 mm; bottom, 200 μm. B, Number of MCH-ir cell bodies in the hypothalamus of orexin-tTA; TetO DTA mice after 4 weeks DOX(−) versus 0 week DOX(−) or controls. Values are mean ± SEM (n = 3 or 4).
To determine the effect of orexin neuron cell death on other cells in the surrounding hypothalamus, immunostaining with IbaI, a marker for microglial cells, which act as scavengers in the brain (Graeber et al., 1998; Ohsawa et al., 2000; Hanisch and Kettenmann, 2007), was conducted. Double staining with anti-orexin and anti-IbaI antisera was performed at 0, 1, 2, and 4 weeks after removal of DOX (Fig. 4). IbaI-microglial cell number increased in the lateral hypothalamic area by 2 weeks in the DOX(−) condition, suggesting that microglial cells were activated to remove dead cell bodies by phagocytosis. Microglial cell activation was restricted to the local area where the orexin neurons were distributed; microglial cells in other hypothalamic regions, such as the ventral medial hypothalamus, were unaffected by DOX removal. Activated microglial cells returned to normal shape and number by 4 weeks after DOX removal.
Figure 4.

Immunohistochemistry indicating activation of microglia cells in hypothalamus after DOX removal from diet (0, 1, 2, and 4 weeks after DOX(−)). Orexin neurons (brown) were stained with anti-orexin antibody. Microglia cells (black) were stained with anti-IbaI antibody. Orange and blue rectangles in the top panels are shown in middle (orange, lateral hypothalamic area [LHA]) and bottom (blue, ventromedial hypothalamus [VMH]). The numbers in each panel indicate the area of IbaI-ir cell in the region measured by image analysis software. Area (μm2) is mean ± SEM (n = 3). Scale bars: top row, 500 μm; middle row, 50 μm; bottom row, 50 μm.
Alteration of sleep/wakefulness patterns in conjunction with ablation of orexin neurons
Orexin-tTA; TetO DTA mice allowed us to study the relationship between the number of orexin neurons and sleep/wakefulness regulation before and after DOX removal, as illustrated in Experimental protocol 2 (Fig. 5A). EEG and EMG electrodes were implanted at 10 weeks of age, and mice were habituated for at least 1 week after surgery. At 12 weeks of age, DOX(−) chow was supplied for 13 weeks. Control recordings were conducted the day before replacing DOX(+) chow with DOX(−) chow. Orexin/ataxin-3 mice, in which orexin neurons degenerate because of expression of the Ataxin-3 protein under control of prepro-orexin promoter (Hara et al., 2001), were used as a comparison. To determine the effect of orexin neuron degeneration during early postnatal development, orexin-tTA; TetO DTA mice fed DOX(−) chow from birth were also studied (D(−) condition; Fig. 5A). Although the D(−) condition is similar to orexin/ataxin-3 mice, degeneration may be faster and more severe in the D(−) group. When killed at 12 weeks of age, the number of remaining orexin neurons of the D(−) group and orexin/ataxin-3 mice was 2 ± 1 (n = 8) and 39 ± 6 (n = 5), respectively (Fig. 5B).
Figure 5.

Analysis of sleep and wakefulness in orexin-tTA; TetO DTA mice. A, Schematic illustrating Experimental protocol 2. EEG and EMG electrodes were implanted at 10 weeks of age. Control recordings for orexin-tTA; TetO DTA mice occurred between 11 and 12 weeks of age. At 12 weeks of age, DOX was removed from the chow for the subsequent 13 weeks. In contrast, orexin-tTA; TetO DTA D(−) mice were raised from birth without DOX in the chow. In this strain (referred to as D(−) mice), EEG and EMG were recorded between 11 and 12 weeks as a comparison. Green bar represents DOX(+) condition; open bar represents DOX(−) condition. B, Number of orexin-ir cell bodies counted at the end of experiments. C, Example hypnograms from orexin-tTA; TetO DTA mice (control and 1, 2, 4, and 13 weeks in DOX(−) condition) and orexin/ataxin-3 (AT) mice. Clock time and light-dark period are indicated at the bottom of the hypnogram (light on 8:00–20:00 h; light off 20:00–8:00 h). D, Time spent in wakefulness, SWS, REM sleep, and cataplexy in the dark (top) and light (bottom) periods. In orexin-tTA; TetO DTA mice, 24 h recordings occurred at 1, 2, 3, 4, 7, 8, 9, 10, 11, and 13 weeks after DOX removal from the diet. *p < 0.05 versus control. Values are mean ± SEM (n = 5–10). D(−), orexin-tTA; TetO DTA mice raised from birth in DOX(−) condition. Ctr, Control; AT, orexin/ataxin-3 mice; Wake or W, wakefulness; SWS or S, slow-wave sleep; REM or R, REM sleep; C, cataplexy.
Sleep/wakefulness states were determined as wakefulness, slow-wave sleep (SWS), and REM sleep from EEG and EMG recordings. Figure 5C shows representative hypnograms from orexin-tTA; TetO DTA mice during ablation of orexin neurons (control, 1, 2, 4, and 13 weeks) and orexin/ataxin-3 mice. At 1 week DOX(−), fragmentation of wakefulness was particularly observed in the early dark period that progressed in conjunction with ablation of orexin neurons. Cataplexy was observed as early as 2 weeks after DOX removal. At 13 weeks DOX(−), sleep/wake fragmentation and cataplexy bout frequency were greatly increased. In contrast, orexin/ataxin-3 mice exhibited a mild phenotype compared with orexin-tTA; TetO DTA mice.
The time spent in each state was compared with the control (0 week) period (Fig. 5D). During the dark period, the total time spent in wakefulness was significantly reduced from 3 to 10 weeks after DOX removal. Interestingly, however, the amount of wakefulness recovered by 11 weeks DOX(−). On the other hand, the total time spent in SWS did not significantly change, except during the dark period at 4 weeks after DOX removal. The total time spent in REM sleep significantly decreased during the light period after 2 weeks, and there was a trend toward reduced REM sleep during the dark period as well. The time spent in cataplexy significantly increased after 3 weeks DOX(−) during both the dark and the light periods, which may be related to the reduced time in wakefulness and REM sleep after 3 weeks DOX(−). The transition frequency between sleep/wake states significantly increased during the dark period between 1 and 13 weeks compared with control (Fig. 6A) but not in the light period. Transition frequency during the dark period was further analyzed in detail (Fig. 6B). Transition frequency to each state was counted during ablation of orexin neurons. At DOX(−) 1 week, transition frequency from wakefulness to SWS was significantly increased. Transition frequency between wakefulness and SWS was dramatically increased until DOX(−) 4 weeks. The increase in cataplexy frequency was followed by a decrease in transitions to REM sleep. Orexin-ataxin3 mice showed severe sleep/wake fragmentation with fewer transitions to cataplexy compared with orexin-tTA; TetO DTA mice. Although the distribution of EEG power within each state was unaffected by orexin neuron loss (Fig. 7A), spectral power in most bandwidths significantly increased after DOX removal (Fig. 7B).
Figure 6.

The frequency of stage transitions. A, The frequency of transitions between all states across the 24 h in the control condition and 1, 2, 3, 4, 7, 8, 9, 10, 11, and 13 weeks after DOX removal in orexin-tTA; TetO DTA mice, in D(−) mice, and in AT mice during the dark (left) and light (right) periods. B, The frequency of transitions between each stage during the dark period in the control condition and 1, 2, 4, and 13 weeks after DOX removal in orexin-tTA; TetO DTA mice, and in AT mice. The numbers indicate the frequency of transitions between each stage. *p < 0.05 versus control. Values are mean ± SEM (n = 5–10). D(−), orexin-tTA; TetO DTA mice raised from birth in DOX(−) condition. Ctr, Control; AT, orexin/ataxin-3 mice; W, wakefulness; S, slow-wave sleep; R, REM sleep; C, cataplexy.
Figure 7.
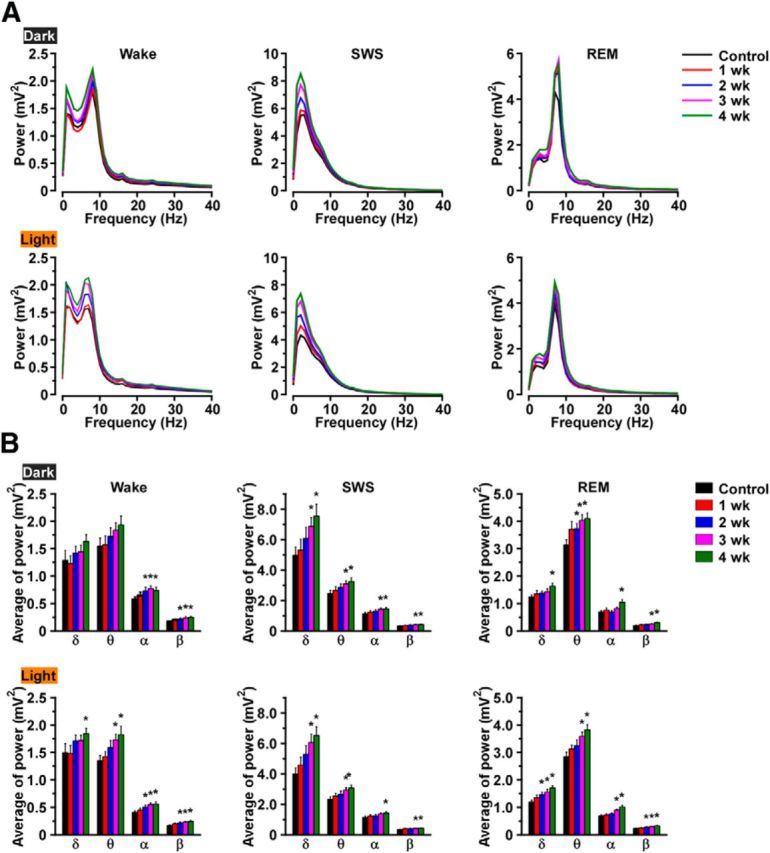
EEG power spectra during wakefulness, SWS, and REM sleep in orexin-tTA; TetO DTA mice during the first 4 weeks after DOX removal. A, Average EEG power spectra during wakefulness (left), SWS (middle), and REM sleep (right) in the dark (top) and light (bottom) periods (n = 8). B, Average power in theδ, θ, α, and β bands in each stage of sleep and wakefulness (n = 8). *p < 0.05 versus control. Values are mean ± SEM. Control, EEG recordings conducted before DOX(−). EEG recordings were conducted for 4 weeks after removal of DOX.
Cataplexy in orexin-tTA; TetO DTA mice versus orexin/ataxin-3 mice.
In humans, narcolepsy is diagnosed with and without cataplexy; approximately half of narcoleptic patients report cataplexy. Although symptoms in narcoleptic patients without cataplexy are milder than in narcolepsy with cataplexy (Sasai et al., 2009), the cause of this difference is incompletely understood. Consequently, cataplexy-like behavioral arrests in orexin-tTA; TetO DTA mice during ablation of orexin neurons were analyzed in detail. First, we determined the proportion of orexin neurons that were lost before cataplexy being observed. Cataplexy was initially observed at 2 weeks DOX(−) in both the dark (3.0 ± 0.9 bouts) and light (0.6 ± 0.3 bouts) periods (Fig. 8A), by which time ∼95% of the orexin neurons were lost (Fig. 2A). Cataplexy bout frequency dramatically increased from 2 to 11 weeks DOX(−) in both the dark and light periods (Fig. 8A). During this period, the proportion of orexin neurons decreased from 5% to 1% (Fig. 2A). At 11 weeks DOX(−), the number of cataplexy bouts in the dark period and the light period was 56.2 ± 7.7 bouts (n = 5, p < 0.001 vs 2 weeks, ANOVA) and 15.0 ± 2.2 bouts (n = 5, p < 0.001 vs 2 weeks, ANOVA), respectively. On the other hand, the number of cataplexy bouts in the D(−) group and orexin/ataxin-3 mice was much lower than orexin-tTA; TetO DTA mice at 13 weeks DOX(−), even though the number of remaining orexin neurons was much less than that of 13 weeks DOX(−) (Fig. 5B). During the dark period, the number of cataplexy bouts in the D(−) group and in orexin/ataxin-3 mice was 17.6 ± 2.6 bouts (n = 8, p < 0.001 vs 13 weeks, ANOVA) and 6.7 ± 2.6 bouts (n = 5, p < 0.001 vs 13 weeks, ANOVA), respectively. During the light period, the number of cataplexy bouts in the D(−) group and orexin/ataxin-3 mice was 6.1 ± 1.4 bouts (n = 8, p = 0.005 vs 13 weeks, ANOVA) and 0.5 ± 0.4 bouts (n = 5, p < 0.001 vs 13 weeks, ANOVA), respectively.
Figure 8.

A, Cataplexy bout frequency in the dark (left) and light (right) periods as orexin neurodegeneration progresses in orexin-tTA; TetO DTA mice (n = 5–10). D(−) and AT mice plotted as a comparison. B, Durations of individual cataplexy bouts plotted as circles in the dark (left) and light (right) periods (n = 5–10). C, EEG power spectra during cataplexy in orexin-tTA; TetO DTA mice during the dark (left) and light (right) periods (n = 8). D, The average power of the δ, θ, α, and β bands during cataplexy in the dark (left) and light (right) periods (n = 8). Values are mean ± SEM.
The mean cataplexy bout duration did not change in either the dark or the light periods as cataplexy frequency increased (Fig. 8B). During the dark period, the mean cataplexy duration at 2, 9, and 13 weeks was 64.0 ± 10.7 s (30 bouts), 78.0 ± 4.8 s (188 bouts, p = 0.27 vs 2 weeks, ANOVA), and 58.8 ± 3.4 s (265 bouts, p = 0.68 vs 2 weeks, ANOVA), respectively. During the light period, the mean cataplexy duration at 2, 9, and 13 weeks was 114.0 ± 21.3 s (6 bouts), 85.1 ± 9.3 s (57 bouts, p = 0.32 vs 2 weeks, ANOVA), and 73.5 ± 7.0 s (69 bouts, p = 0.16 vs 2 weeks, ANOVA), respectively. The mean duration of D(−) group and orexin/ataxin-3 mice was significantly longer compared with 13 weeks in either the dark or the light periods. In the dark period, the mean duration of D(−) group and orexin/ataxin-3 mice was 100.1 ± 3.5 s (463 bouts, p < 0.001 vs 13 weeks, ANOVA) and 101.1 ± 6.7 s (100 bouts, p < 0.001 vs 13 weeks, ANOVA), respectively. In the light period, mean cataplexy duration of D(−) group and orexin/ataxin-3 mice was 99.8 ± 5.9 s (167 bouts, p = 0.007 vs 13 weeks, ANOVA) and 101.1 ± 40.6 s (7 bouts, p = 0.30 vs 13 weeks, ANOVA), respectively.
To determine whether the characteristics of cataplexy changed as the orexin neurons degenerated, the EEG spectrum was compared between the initial occurrence of cataplexy and subsequent weeks. Figure 8C, D shows that the EEG power spectrum and the average power of each bandwidth did not change during cataplexy between the dark and light periods nor over the course of weeks 2–4 DOX(−) when the number of orexin neurons declined from ∼5% to ∼3% of control values (Fig. 2A).
Figure 9 presents the diurnal distribution of cataplexy bouts as the orexin neurons degenerated. Cataplexy bout frequency was greater during the dark (active) period and highest during the first half of the dark period. Bout frequency tended to increase during the last 2 h of light period. Similar trends were observed in D(−) and orexin/ataxin-3 mice, although cataplexy rarely occurred during the light period in orexin/ataxin-3 mice.
Figure 9.

Hourly distribution of cataplexy bouts across the 24 h period in orexin-tTA; TetO DTA mice at 4 time points after DOX removal: A, 2 weeks; B, 4 weeks; C, 8 weeks; D, 13 weeks. E, D(−). F, AT mice plotted as a comparison. Horizontal axis represents clock time. Gray and orange bars represent dark and light periods, respectively. Values are mean ± SEM (n = 5–10).
These results indicate that cataplexy appeared when ∼95% of orexin neurons were lost. Ablation of orexin neurons in adulthood induced much more severe cataplexy episodes than ablation from birth, suggesting compensation during postnatal development. Although cataplexy bout frequency dramatically increased with age, the electrophysiological properties of cataplexy were unaltered. Last, cataplexy bout occurrence was not uniform throughout the day but rather was related to circadian time.
Partial ablation of orexin neurons
The Tet-off system enables reversible control of gene expression in the presence or absence of DOX. In theory, this feature would enable partial ablation of orexin neurons by restoration of DOX (RD) after mice were subjected to the DOX(−) condition for a period of time. Experimental protocol 3 (Fig. 10A) determined whether this occurred in orexin-tTA; TetO DTA mice. After being raised from birth in the presence of DOX(+) chow, mice were fed with DOX(−) chow for 1.5 or 3.5 d at 10 weeks of age and then either killed (1.5 d − RD and 3.5 d − RD groups; n = 3 each) or fed DOX(+) chow for 4 weeks until age 14 weeks (1.5 d + RD and 3.5 d + RD groups; n = 3 each) and then killed. Figure 10B shows that the number and shape of orexin neurons in 3.5 d + RD were comparable with those of 3.5 d RD. Figure 10C, D shows that the number of orexin neurons remaining 4 weeks after DOX restoration was comparable with the DOX(−) group killed before DOX(+) restoration at 10 weeks of age. The number of orexin neurons in the 1.5 d + RD and 3.5 d + RD groups was 597 ± 52 (p = 0.599 vs 1.5 d, ANOVA) and 328 ± 31 (p = 0.817 vs 3.5 d, ANOVA), respectively, indicating that ablation of orexin neurons was arrested by DOX restoration and that the magnitude of ablation was dependent on the duration of DOX removal. Analysis of the remaining orexin neurons revealed that the distribution of these cells in the DOX restoration groups coincided with that of the corresponding DOX(−) groups (Fig. 10D), indicating the absence of an orexin neuron subpopulation that was differentially sensitive to DTA. These results confirmed that the number of remaining orexin neurons could be controlled by varying the DOX(−) period and subsequent restoration of DOX.
Figure 10.

Partial lesion of orexin neurons induced by manipulation of DOX chow availability. A, Schematic illustrating DOX chow availability in different experimental groups in Experimental protocol 3. Green bar represents DOX(+); open bar represents DOX(−). B, Immunostaining of orexin neurons. Inset, Higher magnification of single neurons indicated by the red arrowhead. C, Bar graph showing the number of orexin-ir cell bodies in the hypothalamus in different conditions indicated in A (n = 3–7). D, Distribution of orexin-ir cell bodies in coronal sections at multiple anterior–posterior levels relative to bregma (n = 3–7). Scale bars: B, 200 μm; inset, 20 μm.
Partial ablation of orexin neurons and cataplexy bout frequency
As indicated above and in Figure 8, cataplexy bout frequency at 11 weeks was ∼30-fold greater than the values observed after 2 weeks of DOX(−). This increase might be caused by further reduction of the number of orexin neurons from 5% to 1% (Fig. 2A). Alternatively, the 95% decrease in the number of orexin neurons induced by 2 weeks of DOX(−) might be sufficient to not only trigger cataplexy but also to cause the increase in cataplexy bout frequency over time. To distinguish between these possibilities, the number of orexin neurons was maintained ∼5% using the experimental protocol illustrated in Figure 11A, and cataplexy bout frequency was monitored. Mice were fed DOX(+) chow from birth to 12 weeks of age, DOX(−) chow for 2 weeks (which induced 95% loss of orexin neurons; see Fig. 2A), and then DOX(+) chow was restored for 11 weeks (2 weeks + RD group; n = 6) to arrest further degeneration. Cataplexy bout frequency was monitored for 11 weeks after DOX restoration and compared with the cataplexy frequency of mice in which DOX was permanently removed from the diet at 12 weeks of age (DOX(−) group). For both groups, the cataplexy bout frequency at 2 weeks DOX(−) was used as a reference value. Figure 11B shows that cataplexy bout frequency did not increase if the number of orexin neurons was maintained at 5% in the 2 weeks + RD group in contrast to the cataplexy increase that occurred in the DOX(−) group. The cataplexy bout increment in the 2 weeks + RD group at 11 weeks after DOX restoration was −0.33 ± 4.35 (n = 6).
Figure 11.

A, Schematic illustrating design of DOX(−) 2 weeks + RD trial in Experimental protocol 4. DOX was removed from diet at 12 weeks of age for 2 weeks followed by DOX replacement for 10 weeks. Green bar represents DOX(+); open bar represents DOX(−) conditions. B, Increment in the number of cataplexy bouts during the dark (left) and light (right) periods (n = 6) relative to the number of bouts at 2 weeks of DOX(−). Horizontal axis represents time after DOX(−). C, The number of orexin-ir cell bodies at the end of the DOX(−) 2 weeks + RD trial, with DOX(−) for 2 weeks and 13 weeks plotted as a comparison. D, Number of orexin-ir cells distributed in the lateral and the medial portions of the hypothalamus using the fornix to delineate the border. *p < 0.05 versus 2 weeks + RD. Values are mean ± SEM.
At the end of the experiment, the number of remaining orexin neurons in the 2 weeks + RD group was counted (Fig. 11C) and found to be 44 ± 3 (n = 6). As reported in Figure 2A, at 2 weeks DOX(−), the number of remaining orexin neurons was 47 ± 7 (n = 3; p = 0.598, unpaired t test). Furthermore, from medial to lateral, analyses showed a similar bias in the ablation of orexin neurons both in 2 weeks − RD and 2 weeks + RD (Fig. 11D). Because cataplexy continued to increase in the DOX(−) group but not in the 2 weeks + RD group (Fig. 11B), these results indicate that the dramatic increase in cataplexy bout frequency in the DOX(−) group results from the decrease in the number of orexin neurons from 5% to 1%.
Food-elicited cataplexy test
The food-elicited cataplexy test was established to quantify cataplexy intensity in canine narcolepsy models (Babcock et al., 1976). It has previously been reported that palatable food significantly increases cataplexy bout frequency in narcoleptic mice (Clark et al., 2009; Oishi et al., 2013). To determine whether orexin-tTA; TetO DTA mice showed a response similar to that which occurs in other mouse narcolepsy models, orexin-tTA; TetO DTA mice were fed 1.8 ± 0.5 g chocolate in addition to DOX(−) chow at the beginning of the dark period at 4 weeks and 15 weeks of DOX(−). Mice fed with chocolate at 4 weeks DOX(−) (4 weeks + chocolate) showed a 3.6-fold increase in cataplexy bouts in the dark period compared with 4 weeks (Fig. 12A). In the 15 weeks + chocolate group, cataplexy bouts increased 1.6-fold in the dark period compared with 15 weeks (Fig. 12A). However, no significant differences were observed during the light period in either group. The distribution of cataplexy bouts across the day is shown in Figure 12B. Cataplexy bouts gradually increased after mice had chocolate. The effect of chocolate lasted for 12–13 h. These results confirmed that palatable food increased cataplexy in orexin-tTA; TetO DTA mice as occurs in other mouse narcolepsy models.
Figure 12.
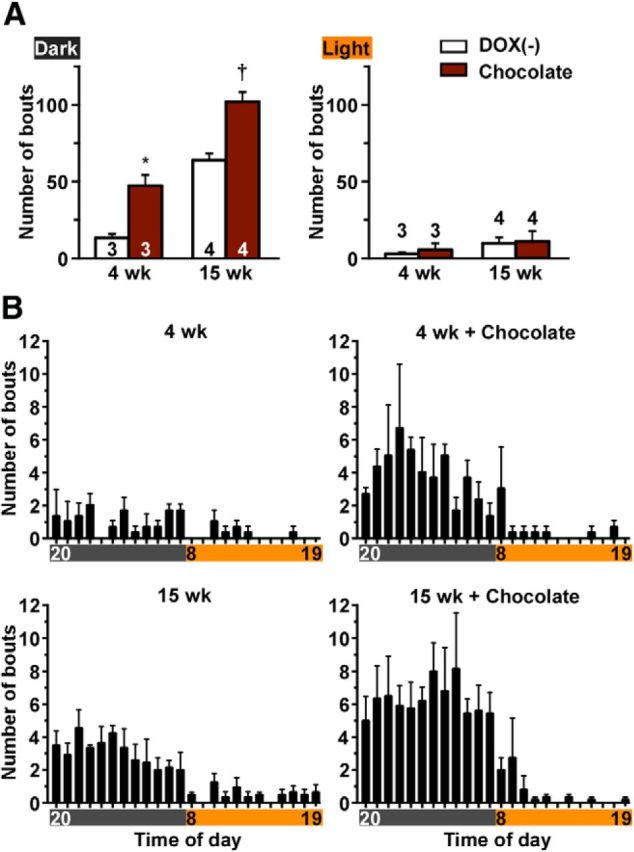
Palatable food induces cataplexy in orexin-tTA; TetO DTA mice. In Experimental protocol 5, chocolate (1.8 g) was made available at the beginning of the dark period. Orexin-tTA; TetO DTA mice DOX (−) for 4 weeks and 15 weeks were subjects for this experiment. A, Number of cataplexy bouts induced by chocolate in the dark (left) and the light (right) periods. B, Hourly distribution of cataplexy bouts across the 24 h period in orexin-tTA; TetO DTA mice in the presence or absence of chocolate. *p < 0.05 versus DOX(−) 4 weeks. †p < 0.05 versus DOX(−) 15 weeks. Values are mean ± SEM.
Metabolic measurements in orexin-tTA; TetO DTA mice
Because the activity of orexin neurons is modulated by metabolic factors, such as glucose, leptin, and ghrelin (Yamanaka et al., 2003), orexin neurons are thought to be involved in energy and fluid homeostasis as well as sleep/wakefulness regulation. In addition, orexin/ataxin-3 mice become obese despite eating less food than wild-type mice (Hara et al., 2005). Narcoleptic patients also show metabolic abnormalities, including a higher body mass index than normal subjects (Honda et al., 1986; Schuld et al., 2000). Consequently, orexin-tTA; TetO DTA mice were subjected to measurement of body weight, food and water consumption, and spontaneous activity during ablation of orexin neurons. Monogenic orexin-tTA or TetO DTA littermate mice were used as controls. The body weight of orexin-tTA; TetO DTA mice began to increase shortly after DOX removal and was significantly higher by 2 weeks after DOX removal (Fig. 13A). Body weight of orexin-tTA; TetO DTA mice at 4 weeks after DOX removal was 30.1 ± 1.1 g (n = 9) versus 26.2 ± 1.0 g (n = 9) for age-matched control mice (p = 0.01, unpaired t test). The incremental increase of body weight became significant by 5 d after DOX removal (Fig. 13B) despite the fact that food consumption was unaltered (Fig. 13C). On the other hand, water intake significantly decreased in orexin-tTA; TetO DTA mice by 4 d after DOX removal (Fig. 13D). These results suggest that the increase in body weight was not caused by overeating or overdrinking.
Figure 13.

Abnormalities of energy and fluid homeostasis and spontaneous activity in orexin-tTA; TetO DTA mice. A, Body weight of orexin-tTA; TetO DTA mice for 4 weeks after DOX removal from the diet; both orexin-tTA and TetO DTA littermates were used as controls. *p < 0.05 versus control (n = 9). B, Increment of body weight in orexin-tTA; TetO DTA versus control mice. Body weight on day −3 is set as 100% (n = 9). *p < 0.05 versus control. C, Cumulative food consumption in orexin-tTA; TetO DTA versus control mice for 4 weeks after DOX removal (n = 9). D, Cumulative water consumption in orexin-tTA; TetO DTA and control mice for 4 weeks after DOX removal (n = 9). E, Mean activity of control mice during light and dark periods and the 24 h average for 4 weeks after DOX removal (n = 9). F, Mean activity of orexin-tTA; TetO DTA mice during light and dark periods and the 24 h average for 4 weeks after DOX removal (n = 9). *p < 0.05 versus DOX(+). Values are mean ± SEM.
To further study the mechanism of weight gain in orexin-tTA; TetO DTA mice, blood-borne metabolic markers were measured in the same individuals before and after DOX removal (Table 2). Insulin and leptin concentrations increased significantly in orexin-tTA; TetO DTA mice at 4 weeks after DOX removal compared with age-matched control mice. However, there were no significant changes in the levels of glucose or free fatty acids. Spontaneous activity was also analyzed to reveal the cause of the body weight increase. In control mice, there was no difference in the level of spontaneous activity in either the light period or the dark period after DOX removal (Fig. 13E). In contrast, orexin-tTA; TetO DTA mice became hypoactive after DOX removal with a decrease in spontaneous activity evident in the light period by 1 week after DOX removal (Fig. 13F). Spontaneous activity in the dark period significantly decreased by 3 weeks after DOX removal.
Table 2.
Serum levels of metabolic parameters in two mouse strains before and after DOX removal from the diet
| Insulin (ng/ml) | Leptin (ng/ml) | FFA (mEq/L) | Glucose (mg/dl) | |
|---|---|---|---|---|
| Orexin-tTA DOX(+) (n = 7) | 1.1 ± 0.3 | 3.0 ± 0.5 | 1.0 ± 0.1 | 128.6 ± 6.0 |
| Orexin-tTA DOX(−) (n = 7) | 0.8 ± 0.2 | 1.8 ± 0.3 | 0.8 ± 0.1 | 126.0 ± 8.9 |
| Orexin-tTA; TetO DTA DOX(+) (n = 6) | 1.5 ± 0.4 | 3.9 ± 0.6 | 1.3 ± 0.1 | 117.8 ± 3.9 |
| Orexin-tTA; TetO DTA DOX(−) (n = 6) | 1.9 ± 0.4* | 4.2 ± 1.0* | 1.0 ± 0.1 | 110.5 ± 6.6 |
*p < 0.05, Orexin-tTA DOX(−) versus Orexin-tTA; TetO DTA DOX(−).
Discussion
In the present study, we generated a novel mouse model of narcolepsy, orexin-tTA; TetO DTA mice, and analyzed the relationship between the number of orexin neurons and symptoms of narcolepsy. The timing of orexin neuron degeneration was controlled using the Tet-off system. By replacing DOX(+) chow with DOX(−) chow, orexin neurons were ablated within a couple of weeks. The specificity of orexin neuron ablation was confirmed because the coextensive MCH neurons as well as nonorexin-ir/Neu-N-ir cells were intact; consequently, DTA expressed in the orexin neurons apparently did not affect neurons in the surrounding hypothalamus. This conclusion is consistent with the fact that DTA does not induce cell death when applied extracellularly in the absence of diphtheria toxin B fragment, which helps DTA to enter the cytoplasm (Murphy, 2011).
Although it has been reported that regulation of gene expression of Tet system can be problematic, we conclude that leakage of DTA in the presence of DOX was not observed in our application of the Tet-off system because the number of orexin neurons in the presence of DOX was comparable to that of control mice. In the absence of DOX, DTA expression was strongly induced in orexin neurons as the number of orexin neurons was reduced to 64.2% only 1.5 d after removal of DOX. Neuronal loss was more rapid in the lateral part of the orexin population than in the medial portion. This difference might be because tTA expression level differed among orexin neurons. Alternatively, sensitivity to DTA might be different between the medial and lateral orexin neuron populations. On the other hand, DTA expression was reversibly regulated by DOX because restoration of DOX terminated DTA expression, which enabled control of the number of remaining orexin neurons. These results indicate that the Tet-off system in orexin-tTA; TetO DTA mice enabled control of both the timing of cell death as well as the number of orexin neurons. These features will allow investigators to produce models for different types of narcolepsy, such narcolepsy with and without cataplexy.
Relationship between the number of orexin neurons and narcolepsy symptoms
Using orexin-tTA; TetO DTA mice, we were able to study the progression of narcoleptic symptoms during ablation of orexin neurons by comparing behavior and physiology before, during, and after ablation of orexin neurons within the same individual animal. The first symptom observed was fragmentation of sleep/wakefulness. This symptom was especially prominent in the early dark period at 1 week after DOX(−) by which time ∼86% of orexin neurons had been ablated. The first cataplexy bout was observed 2 weeks after DOX(−) when ∼95% of orexin neurons had been ablated. Although 95% orexin neuron loss was necessary to trigger cataplexy, further orexin neuron loss exacerbated sleep/wakefulness fragmentation and dramatically increased cataplexy bout frequency. These observations indicate that a small difference in the number of remaining orexin neurons produced a large difference in symptomatology. Conversely, these results imply that a small increase in the number of orexin neurons, the efficacy of orexin neuron function, or orexin concentration in the CSF might prevent the most severe symptoms of narcolepsy. Low levels of orexin restoration might prevent fragmentation of sleep/wakefulness, in particular.
Narcolepsy is divided into two types based on the presence or absence of cataplexy. These distinct symptomatologies might be caused by a small difference in the number of orexin neurons remaining, a difference of the pattern of degeneration of orexin neurons, or differential efficacy of neurotransmission of the remaining orexin cells. In humans, excessive sleepiness (fragmentation of sleep/wakefulness) is usually the first symptom of narcolepsy, and cataplexy develops after the onset of sleepiness (Okun et al., 2002). Thus, orexin-tTA; TetO DTA mice reproduce the order of symptom appearance in narcoleptic patients. Orexin-tTA; TetO DTA mice showed a greater number of cataplexy bouts compared with orexin/ataxin-3 mice or to the D(−) group in which orexin neuron cell death was initiated early in the postnatal period. Although the number of orexin neurons remaining in D(−) group was smaller than DOX(−) 13 weeks, these mice showed milder sleep/wake fragmentation and fewer cataplexy bouts. These results strongly indicate that functional compensation occurred when orexin neurons were ablated during early postnatal development.
In this study, the number of cataplexy bouts reached a plateau after 11 weeks DOX(−). In humans, the number of sleep-onset REM periods and cataplexy frequency are reported to decrease with age (Dauvilliers et al., 2004). The decrease in cataplexy in narcoleptic patients is attributed to avoidance of emotional situations that trigger cataplexy. The saturation of cataplexy bout frequency observed in orexin-tTA; TetO DTA mice after 11 weeks DOX(−) might reflect functional adaptation of the underlying neural circuitry to loss of orexin innervation. Conversely, the increase in cataplexy frequency subsequent to orexin neuron degeneration may reflect functional reorganization of the deafferented projection sites of the orexin neurons. However, when provided with a novel and palatable food, the number of cataplexy bouts increased in orexin-tTA; TetO DTA mice even 15 weeks after DOX removal, indicating the efficacy of emotional stimuli to increase cataplexy above the plateau level. Notably, the EEG spectral features of cataplexy (Vassalli et al., 2013) are stable across all weeks of DOX(−) and between dark and light periods, and appear consistent with the EEG power density observed during cataplexy in mice that lack orexin peptides. Furthermore, EEG spectral power during wake, SWS, and REM sleep gradually increased as the orexin neurons degenerated. This observation suggests that synchronization of cortical neurons may be enhanced by orexin neuron loss.
We observed cataplexy bouts throughout a 24 h period. The frequency of cataplexy bouts in the dark period was greater than in the light period because mice are nocturnal and are primarily active during the dark and spend most of the light period asleep. However, rodents have a polyphasic sleep/wakefulness rhythm; thus, cataplexy attacks also occurred during waking in the light period. Mice tended to have many attacks during early half of dark period and at the end of light period, which seems to be correlated with the arousal rhythm.
Orexin neurons and metabolism
We observed a significant increase in body weight in orexin-tTA; TetO DTA mice subsequent to ablation of orexin neurons without any accompanying signs of overfeeding or overdrinking. Ablation of orexin neurons also induced hypoactivity. These results are in good agreement with the observation that narcoleptic patients show mild obesity without increasing caloric intake and that the activity level of narcoleptics is lower than that of control subjects (Middelkoop et al., 1995). Ablation of orexin neurons might affect blood-borne metabolic parameters as well. Insulin and leptin concentrations were higher than control mice, although free fatty acid and glucose concentrations were unaffected, suggesting that the physiological role of orexin neurons in energy homeostasis is not simple. The increased serum leptin concentration could be a result of the weight gain. The altered metabolism may be due, at least in part, to disruption of the circadian timekeeping system. The decrease in water intake is in agreement with previous reports that orexin is involved in fluid, as well as energy, homeostasis (Kunii et al., 1999; Tsunematsu et al., 2008).
Utility of orexin-tTA; TetO DTA mice
In the present study, we were able to control the number of orexin neurons and found that it was possible to create mice that varied in symptom severity from presymptomatic to severe narcolepsy simply by changing the duration of the DOX(−) period. Several drugs are in clinical use to treat various symptoms of narcolepsy (Morgenthaler et al., 2007; Aran et al., 2010; Hirai and Nishino, 2011). Because symptomatology varies among narcoleptic patients, orexin-tTA; TetO DTA mice could be a useful tool for discovery of new drugs that target symptoms associated with this disorder (Black et al., 2014). It is commonly thought that symptom progression in human narcoleptics stops at some point and become stable. However, longitudinal studies of symptom dynamics in individual patients has not been possible to date, in part, because most narcoleptics are medicated to prevent symptoms. Using orexin-tTA; TetO DTA mice, it is possible to determine the effect of aging on narcoleptic symptoms, which may reveal novel aspects of orexin deficiency with aging. These mice will also be valuable to study the process of network reorganization in terminal fields, such as the LC and DR, as orexin input is lost. The number of histaminergic neurons has recently been reported to significantly increase in human narcoleptic patients (John et al., 2013; Valko et al., 2013). Orexin neurons release glutamate (Rosin et al., 2003; Torrealba et al., 2003), dynorphin (Chou et al., 2001), and neurotensin (Furutani et al., 2013) as well as orexin. In prepro-orexin or orexin receptor gene knock-out mice, these other neurotransmitters are still released.
Although the present study primarily focused on narcolepsy, orexin neurons are involved in many physiological functions, such as addiction, reward, and the stress response. Orexin-tTA; TetO DTA mice will likely be valuable to study the regulatory mechanisms underlying these physiological functions in the future.
Footnotes
This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas “Mesoscopic Neurocircuitry” (23115103), Grant-in-Aid for Scientific Research (B) (23300142) to A.Y., the JST PRESTO program, Japan Society for Promotion of Science research fellowship for young researcher to S.T. and postdoctoral fellowship to T.T., and National Institutes of Health Grants R01 NS057464 and NS077408 to T.S.K. We thank Claire Saito and Keiko Nishimura for technical assistance.
The authors declare no competing financial interests.
References
- Aran A, Einen M, Lin L, Plazzi G, Nishino S, Mignot E. Clinical and therapeutic aspects of childhood narcolepsy-cataplexy: a retrospective study of 51 children. Sleep. 2010;33:1457–1464. doi: 10.1093/sleep/33.11.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DA, Narver EL, Dement WC, Mitler MM. Effects of imipramine, chlorimipramine, and fluoxetine on cataplexy in dogs. Pharmacol Biochem Behav. 1976;5:599–602. doi: 10.1016/0091-3057(76)90298-7. [DOI] [PubMed] [Google Scholar]
- Black SW, Morairty SR, Fisher SP, Chen TM, Warrier DR, Kilduff TS. Almorexant promotes sleep and exacerbates cataplexy in a murine model of narcolepsy. Sleep. 2013;36:325–336. doi: 10.5665/sleep.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black SW, Morairty SR, Chen TM, Leung AK, Wisor JP, Yamanaka A, Kilduff TS. GABAB agonism promotes sleep and reduces cataplexy in murine narcolepsy. J Neurosci. 2014;34:6485–6494. doi: 10.1523/JNEUROSCI.0080-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brischoux F, Fellmann D, Risold PY. Ontogenetic development of the diencephalic MCH neurons: a hypothalamic ‘MCH area’ hypothesis. Eur J Neurosci. 2001;13:1733–1744. doi: 10.1046/j.0953-816x.2001.01552.x. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/S0092-8674(00)81973-X. [DOI] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE, Mochizuki T. Feeding-elicited cataplexy in orexin knockout mice. Neuroscience. 2009;161:970–977. doi: 10.1016/j.neuroscience.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Gosselin A, Paquet J, Touchon J, Billiard M, Montplaisir J. Effect of age on MSLT results in patients with narcolepsy-cataplexy. Neurology. 2004;62:46–50. doi: 10.1212/01.WNL.0000101725.34089.1E. [DOI] [PubMed] [Google Scholar]
- De la Herrán-Arita AK, Kornum BR, Mahlios J, Jiang W, Lin L, Hou T, Macaubas C, Einen M, Plazzi G, Crowe C, Newell EW, Davis MM, Mellins ED, Mignot E. CD4+ T cell autoimmunity to hypocretin/orexin and cross-reactivity to a 2009 H1N1 influenza A epitope in narcolepsy. Sci Transl Med. 2013;5:216ra176. doi: 10.1126/scitranslmed.3007762. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España RA, McCormack SL, Mochizuki T, Scammell TE. Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep. 2007;30:1417–1425. doi: 10.1093/sleep/30.11.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furutani N, Hondo M, Kageyama H, Tsujino N, Mieda M, Yanagisawa M, Shioda S, Sakurai T. Neurotensin co-expressed in orexin-producing neurons in the lateral hypothalamus plays an important role in regulation of sleep/wakefulness states. PLoS One. 2013;8:e62391. doi: 10.1371/journal.pone.0062391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, López-Redondo F, Ikoma E, Ishikawa M, Imai Y, Nakajima K, Kreutzberg GW, Kohsaka S. The microglia/macrophage response in the neonatal rat facial nucleus following axotomy. Brain Res. 1998;813:241–253. doi: 10.1016/S0006-8993(98)00859-2. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/S0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Hara J, Yanagisawa M, Sakurai T. Difference in obesity phenotype between orexin-knockout mice and orexin neuron-deficient mice with same genetic background and environmental conditions. Neurosci Lett. 2005;380:239–242. doi: 10.1016/j.neulet.2005.01.046. [DOI] [PubMed] [Google Scholar]
- Hirai N, Nishino S. Recent advances in the treatment of narcolepsy. Curr Treat Options Neurol. 2011;13:437–457. doi: 10.1007/s11940-011-0137-6. [DOI] [PubMed] [Google Scholar]
- Honda Y, Doi Y, Ninomiya R, Ninomiya C. Increased frequency of non-insulin-dependent diabetes mellitus among narcoleptic patients. Sleep. 1986;9:254–259. doi: 10.1093/sleep/9.1.254. [DOI] [PubMed] [Google Scholar]
- Inamura N, Sugio S, Macklin WB, Tomita K, Tanaka KF, Ikenaka K. Gene induction in mature oligodendrocytes with a PLP-tTA mouse line. Genesis. 2012;50:424–428. doi: 10.1002/dvg.20808. [DOI] [PubMed] [Google Scholar]
- John J, Thannickal TC, McGregor R, Ramanathan L, Ohtsu H, Nishino S, Sakai N, Yamanaka A, Stone C, Cornford M, Siegel JM. Greatly increased numbers of histamine cells in human narcolepsy with cataplexy. Ann Neurol. 2013;74:786–793. doi: 10.1002/ana.23968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogiannis M, Hsu E, Willie JT, Chemelli RM, Kisanuki YY, Yanagisawa M, Leonard CS. Cholinergic modulation of narcoleptic attacks in double orexin receptor knockout mice. PLoS One. 2011;6:e18697. doi: 10.1371/journal.pone.0018697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki Y, Chemelli RM, Tokita S, Willie JT, Sinton CM, Yanagisawa M. Behavioral and polysomnographic characterization of orexin-1 receptor and orexin-2 receptor double knockout mice. Sleep. 2001;24:A22. [Google Scholar]
- Kunii K, Yamanaka A, Nambu T, Matsuzaki I, Goto K, Sakurai T. Orexins/hypocretins regulate drinking behaviour. Brain Res. 1999;842:256–261. doi: 10.1016/S0006-8993(99)01884-3. [DOI] [PubMed] [Google Scholar]
- Lee P, Morley G, Huang Q, Fischer A, Seiler S, Horner JW, Factor S, Vaidya D, Jalife J, Fishman GI. Conditional lineage ablation to model human diseases. Proc Natl Acad Sci U S A. 1998;95:11371–11376. doi: 10.1073/pnas.95.19.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/S0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Mahlios J, De la Herrán-Arita AK, Mignot E. The autoimmune basis of narcolepsy. Curr Opin Neurobiol. 2013;23:767–773. doi: 10.1016/j.conb.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelkoop HA, Lammers GJ, Van Hilten BJ, Ruwhof C, Pijl H, Kamphuisen HA. Circadian distribution of motor activity and immobility in narcolepsy: assessment with continuous motor activity monitoring. Psychophysiology. 1995;32:286–291. doi: 10.1111/j.1469-8986.1995.tb02957.x. [DOI] [PubMed] [Google Scholar]
- Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep. 1997;20:1012–1020. [PubMed] [Google Scholar]
- Morgenthaler TI, Kapur VK, Brown T, Swick TJ, Alessi C, Aurora RN, Boehlecke B, Chesson AL, Jr, Friedman L, Maganti R, Owens J, Pancer J, Zak R. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30:1705–1711. doi: 10.1093/sleep/30.12.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JR. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins (Basel) 2011;3:294–308. doi: 10.3390/toxins3030294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol. 1997;52:27–78. doi: 10.1016/S0301-0082(96)00070-6. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113:3073–3084. doi: 10.1242/jcs.113.17.3073. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Williams RH, Agostinelli L, Arrigoni E, Fuller PM, Mochizuki T, Saper CB, Scammell TE. Role of the medial prefrontal cortex in cataplexy. J Neurosci. 2013;33:9743–9751. doi: 10.1523/JNEUROSCI.0499-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun ML, Lin L, Pelin Z, Hong S, Mignot E. Clinical aspects of narcolepsy-cataplexy across ethnic groups. Sleep. 2002;25:27–35. doi: 10.1093/sleep/25.1.27. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG. Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol. 2003;465:593–603. doi: 10.1002/cne.10860. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Moriguchi T, Furuya K, Kajiwara N, Nakamura T, Yanagisawa M, Goto K. Structure and function of human prepro-orexin gene. J Biol Chem. 1999;274:17771–17776. doi: 10.1074/jbc.274.25.17771. [DOI] [PubMed] [Google Scholar]
- Sasai T, Inoue Y, Komada Y, Sugiura T, Matsushima E. Comparison of clinical characteristics among narcolepsy with and without cataplexy and idiopathic hypersomnia without long sleep time, focusing on HLA-DRB1(*)1501/DQB1(*)0602 finding. Sleep Med. 2009;10:961–966. doi: 10.1016/j.sleep.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C, Siegel JM. A consensus definition of cataplexy in mouse models of narcolepsy. Sleep. 2009;32:111–116. [PMC free article] [PubMed] [Google Scholar]
- Schuld A, Hebebrand J, Geller F, Pollmächer T. Increased body-mass index in patients with narcolepsy. Lancet. 2000;355:1274–1275. doi: 10.1016/S0140-6736(05)74704-8. [DOI] [PubMed] [Google Scholar]
- Tabuchi S, Tsunematsu T, Kilduff TS, Sugio S, Xu M, Tanaka KF, Takahashi S, Tominaga M, Yamanaka A. Influence of inhibitory serotonergic inputs to orexin/hypocretin neurons on the diurnal rhythm of sleep and wakefulness. Sleep. 2013;36:1391–1404. doi: 10.5665/sleep.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler I, Deboer T, Fischer M. Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci. 1997;17:1869–1879. doi: 10.1523/JNEUROSCI.17-05-01869.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrealba F, Yanagisawa M, Saper CB. Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience. 2003;119:1033–1044. doi: 10.1016/S0306-4522(03)00238-0. [DOI] [PubMed] [Google Scholar]
- Tsunematsu T, Fu LY, Yamanaka A, Ichiki K, Tanoue A, Sakurai T, van den Pol AN. Vasopressin increases locomotion through a V1a receptor in orexin/hypocretin neurons: implications for water homeostasis. J Neurosci. 2008;28:228–238. doi: 10.1523/JNEUROSCI.3490-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu T, Tanaka KF, Yamanaka A, Koizumi A. Ectopic expression of melanopsin in orexin/hypocretin neurons enables control of wakefulness of mice in vivo by blue light. Neurosci Res. 2013;75:23–28. doi: 10.1016/j.neures.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Valko PO, Gavrilov YV, Yamamoto M, Reddy H, Haybaeck J, Mignot E, Baumann CR, Scammell TE. Increase of histaminergic tuberomammillary neurons in narcolepsy. Ann Neurol. 2013;74:794–804. doi: 10.1002/ana.24019. [DOI] [PubMed] [Google Scholar]
- Vassalli A, Dellepiane JM, Emmenegger Y, Jimenez S, Vandi S, Plazzi G, Franken P, Tafti M. Electroencephalogram paroxysmal θ characterizes cataplexy in mice and children. Brain. 2013;136:1592–1608. doi: 10.1093/brain/awt069. [DOI] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in Orexin receptor-2 and Orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/S0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Tsujino N, Funahashi H, Honda K, Guan JL, Wang QP, Tominaga M, Goto K, Shioda S, Sakurai T. Orexins activate histaminergic neurons via the orexin 2 receptor. Biochem Biophys Res Commun. 2002;290:1237–1245. doi: 10.1006/bbrc.2001.6318. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/S0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]