

Abstract
The discrete arrangement of voltage-gated K+ (Kv) channels in axons may impart functional advantages in action potential (AP) signaling yet, in compact cell types, the organization of Kv channels is poorly understood. We find that in cerebellar stellate cell interneurons of mice, the composition and influence of Kv channels populating the axon is diverse and depends on location allowing axonal compartments to differentially control APs in a local manner. Kv1 channels determine AP repolarization at the spike initiation site but not at more distal sites, limiting the expression of use-dependent spike broadening to the most proximal axon region, likely a key attribute informing spiking phenotype. Local control of AP repolarization at presynaptic boutons depends on Kv3 channels keeping APs brief, thus limiting Ca2+ influx and synaptic strength. These observations suggest that AP repolarization is tuned by the local influence of distinct Kv channel types, and this organization enhances the functional segregation of axonal compartments.
Introduction
Axons are organized into subdomains with each compartment distinct in its role in excitation. Within these compartments axons must initiate action potentials (APs), propagate the resulting spikes to release sites, and direct Ca2+ entry to mediate neurotransmission. Kv channels are important determinants of axonal membrane excitability contributing to AP shape, initiation, and spike patterning. As axonal compartments are differentiated based on function, the complements of Kv channels that support AP signaling in each compartment may be distinct and organized in a manner specific to that function (Dodson and Forsythe, 2004). Despite the diversity of fast-activating Kv channels, observations from L5 pyramidal cells indicate that conductances from Kv1-containing channels predominantly determine AP repolarization throughout the axon arbor with their availability influencing diverse functions including repetitive spiking and neurotransmission (Kole et al., 2007; Shu et al., 2007; Foust et al., 2011). Whether axonal AP repolarization is similarly controlled by the monotypic influence of a single Kv family in other neuron types has not been extensively examined.
Local inhibitory interneurons often incorporate a different constellation of fast-activating Kv channels in their axons. In the cerebellum, direct recordings from the specialized presynaptic pinceau terminals of molecular layer interneurons (MLIs) show currents mediated by both Kv3- and Kv1-containing channels (Southan and Robertson, 2000) consistent with their local expression in these structures (Wang et al., 1994; Veh et al., 1995; Laube et al., 1996; Bobik et al., 2004). However, dendrotoxin (DTX)-sensitive Kv1 channels do not contribute to AP-evoked Ca2+ entry at MLI presynaptic sites (Tan and Llano, 1999) indicating a role in excitability other than spike repolarization. Kv1 channels are also prominently expressed in the axon initial segment (AIS) of these cells (Lorincz and Nusser, 2008), a specialized axonal domain without previous description of Kv3 channel expression. It may be that axons of MLIs rely on a mosaic of Kv channel types to direct spike repolarization with their influence dependent not only on their distinct biophysical properties but also on their subcellular distribution, thereby differentiating the spike-response characteristics in the compact axon arbors of this cell type.
We directly examined for the Kv channel subtypes that determine AP repolarization in different axonal regions of cerebellar stellate cell (SC) MLIs using two-photon (2P) voltage-sensitive dye (VSD) imaging and photolysis of a caged Kv channel blocker (Zayat et al., 2003) to measure spike waveform and precisely alter channel availability in a regional manner. Our results indicate that AP repolarization is locally determined by either Kv1 or Kv3 at the AIS and release sites, respectively, and that this arrangement uncouples activity-dependent control of spiking output at the AIS from neurotransmission at presynaptic sites.
Materials and Methods
Slice preparation and pharmacology.
Parasagittal slices from cerebellar vermi were prepared from C57BL/6J mice (postnatal day [P]15–21 or P28–P35) of either sex in accordance with Max Planck Florida Institute for Neuroscience Animal Care and Use Committee-approved protocols. Following isoflurane anesthesia, mice were decapitated and the cerebellum was isolated by dissection. Brain slices (200 μm) were sectioned using a vibroslicer in an ice-cold solution containing the following (in mm): 87 NaCl, 25 NaHO3, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 10 glucose, and 7 sucrose. Slices were transferred to an incubation chamber containing the following (in mm): 119 NaCl, 26.2 NaHO3, 2.5 KCl, 1 NaH2PO4, 3 CaCl2, and 11 glucose and maintained at 34°C for 30 min and then at room temperature (RT; 23−25°C) until use. For whole-cell recording, slices were placed in a submersion chamber and continuously superfused with the same solution at RT or at 32°C where noted. All solutions were oxygenated with carbogen gas (95% O2, 5% CO2) to equilibrium.
GABAA, NMDA, and AMPA receptor-mediated synaptic responses were blocked with the following (in μm): 100 picrotoxin, 10 r-CPP, and 10 NBQX, respectively (Tocris Bioscience). Picrotoxin was omitted during paired recording of synaptically connected SCs while r-CPP and NBQX were omitted in experiments involving stimulation of parallel fibers. K+ channels were inhibited with 1 μm BDS-I, 100–200 nm DTX-I, 100 nm iberiotoxin (IBTX; Alomone Labs and Peptides International), and 500 μm TEA tetraethylammonium (TEA; Sigma).
Electrophysiology.
SCs located in the outer two-thirds of the molecular layer were targeted for patching using gradient-contrast infrared video microscopy. A Multiclamp 700B amplifier (Molecular Devices) was used for electrophysiological recording. Analog signals were low-pass filtered at 2–10 kHz and digitized at 20–50 kHz at 16-bit resolution (Digidata 1440A; Molecular Devices) using pClamp software (Molecular Devices). Pipette capacitance was neutralized and electrode series resistance compensated using bridge balance in current-clamp mode. For imaging experiments, borosilicate patch pipettes (4–6 MΩ) contained a solution of the following (in mm): 128 potassium gluconate, 2 KCl, 9 HEPES, 4 MgCl2, 4 NaATP, and 0.5 NaGTP. Cells were filled through the patch pipette with the VSD di-2-AN(F)EPPTEA (L. Loew, University of Connecticut Health Center) in whole-cell mode (30 μm) for 0.25–2.0 h before data acquisition to allow for axonal diffusion. For combined voltage and Ca2+ imaging experiments, Oregon Green BAPTA-1 or 6F (Life Technologies) was also included in the internal solution (100–200 μm). For VSD imaging experiments performed in voltage-clamp mode, SCs were filled instead with an internal solution containing the following (in mm): 145 CsCl, 10 HEPES, 4 NaATP, 0.3 NaGTP, and 5 EGTA; 1 μm TTX (Tocris Bioscience) blocked Na+ channels by bath application. To avoid high background florescence during VSD imaging, patch pipettes were front loaded with a small amount of dye-free internal solution, and then back filled with internal solution that included the VSD. Axonal boutons were optically targeted for cell-attached electrophysiological recording by first filling SCs during whole-cell somatic recording with an internal solution containing a morphological indicator (Alexa 594; 60 μm) and, after dye equilibration (10–30 min), visually identifying a bouton amenable for loose-seal patching using 2P imaging. VSDs were omitted from these experiments to preclude the possibility of VSD-induced alteration of AP waveform. The axonal patch pipette contained external recording solution and was coated with BSA-Alexa 594 (Life Technologies). This allowed for simultaneous visualization of both the pipette and the targeted bouton, thereby enabling online visual guidance and establishment of loose-seal voltage-clamp recordings (Ishikawa et al., 2010). For the cell-attached configuration, the holding potential was set such that there was no bias current, therefore the resting membrane potential at the bouton should not be influenced by recording pipette (Perkins, 2006).
For whole-cell recordings, single APs were elicited using short current injections (5 ms, 200–400 pA) at the soma from cells held at −83 mV (corrected for liquid junction potential, LJP), except where noted. The membrane potential at axonal sites is expected to be similar to that at the soma due to DC coupling. Prolonged square-pulse current injections (52–270 pA; 1 s; repeated at 0.077 Hz) induced sustained firing. During this stimulus procedure, the maximal firing rate was identified by incrementally increasing the injected current until the rate of sustained firing saturated (Imax). Loss of spiking often occurred when the cell was excited beyond Imax and was likely due to Na+ channel inactivation; these data were not included in the analysis.
For synaptically connected paired recording, postsynaptic SCs were filled with an internal solution containing the following (in mm): 135 KCl, 10 HEPES, 4 NaATP, 0.3 NaGTP, and 5 EGTA such that GABAA receptor-mediated IPSCs were resolved as inward currents; cell pairs were rejected from analysis if Rs changed by >20% in the postsynaptic cell during the course of an experiment. Presynaptic SCs were maintained in the cell-attached recording configuration (>500 MΩ seal resistance) with a solution containing the following (in mm): 145 potassium methanesulfonate, 5 KCl, 5 HEPES, and 1 EGTA. Cells were stimulated to spike by brief current injections (5–25 ms); a constant bias current prevented spontaneous spiking.
We used an extracellular glass pipette, filled with saline and Alexa 594 (50 μm), to stimulate the presynaptic parallel fibers (PFs) of granule cells. The stimulating pipette was visually positioned adjacent to a dendrite of a postsynaptic SC, also filled with Alexa 594 (50 μm by patch pipette), using 2P microscopy. In this way, we could avoid blocking Kv channels on PFs by exciting presynaptic inputs that were spatially restricted from the SC AIS, the region targeted for ruthenium bipyridine-4AP (Rubi-4AP)-induced photolysis. Extracellular stimulus intensity, controlled by a constant voltage isolator (DS2A; Digitimer), varied from 9 to 24 V (10–100 μs per pulse) and was adjusted such that APs were initiated in the postsynaptic SC during the first EPSP. All electrophysiological analysis was performed using AxoGraph (AxoGraph Scientific).
Two-photon imaging.
Imaging experiments were performed with a 2P laser scanning microscope (Prairie Instruments) using an Olympus upright microscope, objective (60×, 1.0 NA), and oil-immersion condenser (1.4 NA). A Ti:sapphire laser (Coherent) provided excitation. Emitted fluorescence was simultaneously collected by GaAsP photomultiplier modules (Hamamatsu) in both epifluorescence and transfluorescence pathways and chromatically separated using a t560lpxr dichroic and et640/120-2p and et510/80-2p bandpass filters (Chroma). In experiments using 473 nm laser light for uncaging, the microscope primary dichroic (720LP) was replaced with a mirror that passed both blue (∼450–480 nm) and NIR (>675 nm) bands (z470/561/NIR Trans; Chroma).
VSD fluorescence measurements were obtained with 1040 nm excitation (1020 nm for simultaneous VSD and calcium indicator dye imaging) at 10 kHz (Acker et al., 2011). Optical AP traces were processed off-line with a Butterworth filter (Fpass, 900 Hz; Fstop, 925 Hz) to reduce variability due to high-frequency shot noise. Responses were aligned together with the somatically recorded APs to avoid temporal jitter, and averaged (typically 15–50 traces) for each axonal site and/or test condition. Partitioning of the VSD within internal membranes leaves baseline fluorescence highly variable within axons. As internal membranes have no direct sensitivity to external membrane voltage, these chromophores do not contribute to voltage-induced fluorescence changes, therefore small changes in imaging location during an experiment result in appreciable differences in the apparent magnitude of voltage-induced fluorescence changes relative to baseline. For this reason, we did not quantify changes in AP amplitude in different conditions and, in figures, averaged responses were normalized to facilitate comparison of waveform duration. AP duration was determined using the full-width at half-maximal amplitude (FWHM) of the waveform. Fluorescence changes for Ca2+ imaging (average 15–25 trials per condition) were quantified as (ΔF/F), where the peak of the AP-evoked Ca2+ transient was determined from a linear fit of the fluorescence decay following single APs.
RuBi-4AP uncaging.
RuBi-4AP (Zayat et al., 2003; Abcam) was continuously recirculated in the bath using a peristaltic pump. A 473 nm laser light (0.8 mW at the objective) was used to uncage Rubi-4AP (150 μm or 300 μm). Closely spaced uncaging points were directed to each side of a targeted cellular region (<1 μm from the membrane; 50 total points). Photomultiplier tubes were mechanically shuttered during simultaneous VSD imaging and uncaging experiments. The sequence of uncaging points was randomized along the targeted region of axon. RuBi-4AP was tested for purity by monitoring for changes in AP shape in the absence of laser illumination following bath perfusion.
Immunohistochemistry.
In several cells, Lucifer yellow (LY; ∼0.2% w/v) was included in the internal solution and following equilibration (>20 min) were subsequently fixed for 1 h in 4% PFA (0.1 m PB with 15% v/v picric acid) at RT. For Ankyrin-G (AnkG) immunohistochemistry, slices were subjected to a 15 min pepsin digest (0.2 mg/ml in 0.1 m PB at RT; Lorincz and Nusser, 2008), then blocked for 1 h (10% v/v NGS in 0.1 m PB). Sequential incubation in mouse anti-AnkG (1:800; Antibodies Inc.) for 1 h at 4°C and rabbit anti-LY (1:800; Life Technologies) for 72 h at 4°C was used to immunolabel AnkG and intensify LY staining, respectively. Following repeated washing in 0.1 m PB, slices were incubated in secondary antibodies, Alexa 633-conjugated anti-mouse for AnkG and Alexa 488-conjugated anti-rabbit for LY (Life Technologies), for 1 h at RT. All antibodies were diluted in 0.1 m PB with 2% v/v NGS and 0.1% v/v Triton X-100. Slices were washed and mounted on glass slides using SlowFade (Life Technologies). Image stacks were taken with a confocal laser scanning microscope (Zeiss LSM780) using sequential multichannel acquisition. For AIS length measurements, the distal ends of AIS segments were defined using a threshold where the average pixel intensity attenuated below 10% of maximum.
Cav channel model.
A kinetic model of Cav channel gating was created from a previously published report (Li et al., 2007). The model used a six-state Markovian chain with individual voltage-dependent rate constants derived for P/Q-, N-, and R-type Cav channels. Channel stoichiometry (ratios 44:17:5, respectively) was set based on quantitative estimates from hippocampal mossy fiber boutons (Li et al., 2007). Ca2+ influx was estimated from the integral of the combined open probability (Po) for each of these channels and approximates a summed current if a similar conductance is assumed for each channel.
Statistical analysis.
Reported values are mean ± SEM. Excel (Microsoft) and GraphPad Prism (GraphPad Software) were used for statistical analysis. Normality was determined with the D'Agostino–Pearson omnibus test. Two-sided t tests (unpaired or paired where appropriate) and Mann–Whitney tests were used for parametric and nonparametric datasets, respectively. Group data were compared with one-way or two-way ANOVA and significance determined with Tukey's or Sidak's multiple-comparison tests, respectively. Differences were deemed significant with α values of p < 0.05.
Results
Distinct Kv channel subtypes regionally determine AP repolarization in axons
To measure AP waveforms in axons of cerebellar interneurons, we filled SCs during whole-cell recordings with the VSD di-2-AN(F)EPPTEA (30 μm; Acker et al., 2011) and, following equilibration (0.25–2 h), imaged with 2P microscopy. APs were evoked by somatic current injection in cells held in current-clamp mode. Optical recordings of APs were resolved in axons by statically parking the 2P beam at a diffraction-limited point of interest and illuminating that spot continuously during acquisition. Responses were filtered off-line and averaged over multiple trials (typically 15–50; Fig. 1A). Somatic APs recorded using electrophysiology with a dye-free internal solution were no different from spikes recorded from the same cell after repatching with a solution containing the VSD (Fig. 1B1; 1.06 ± 0.03 and 1.01 ± 0.02 of control for duration and amplitude, respectively; n = 4, p = 0.12, and p = 0.66, paired t test; Rs in repatched cells 1.02 ± 0.05 of initial patch, p = 0.91, paired t test). Furthermore, somatic APs were unaltered by long-duration intracellular dialysis with the VSD (Fig. 1B2,B3). These data indicate that the VSD did not perturb neural membrane properties. To test the temporal resolution of this VSD recording method, we voltage-clamped SCs using an AP-like command waveform (880 μs half-width) and measured the optically resolved voltage responses at the soma. VSD responses closely resembled the command waveform (Fig. 1C,D). We further probed temporal sensitivity in several ways. First, the duration of the command waveform was increased slightly compared with control. This change was finely tracked in the optical response (Fig. 1E–G). Similarly, VSD imaging tracked command waveforms resembling fast APs (e.g., 500 μs; Fig. 1H). Also, in current-clamp mode, optically resolved APs in axons were faster at elevated temperature (32°C) compared to room temperature (within-cell comparison from the same recoding site; 0.89 ± 0.03 of control [24°C]; n = 6, p = 0.02, paired t test) indicating that VSD imaging can resolve physiologically induced AP waveform changes. Last, we made optically targeted subcellular electrophysiological recordings of APs from axonal boutons using a loose-seal, cell-attached configuration to determine whether VSD imaging accurately reports spike waveform in these structures (Fig. 1I; distance from axon hillock; range 61–96 μm). Suprathreshold current injection through a second electrode, a whole-cell recording pipette at the soma, elicited APs in the attached axon as registered in voltage clamp by rapid inward and outward currents that were time locked to the somatic spike (Fig. 1J). Axonal AP duration was quantified by peak-to-peak measurement of these currents (Sabatini and Regehr, 1997; Yang and Wang, 2006), a method that closely approximates somatic AP half-width as determined by control recordings at the soma using the same recording configuration (Fig. 1K,L). In a separate set of experiments, VSD imaging was used to measure spike duration (FWHM) in SC boutons resulting in half-width values that closely matched those from electrophysiological recording (Fig. 1L).Together, these results indicate that VSD imaging accurately reports fast voltage changes across neural membranes, including axons, without detectable influence on AP signaling.
Figure 1.
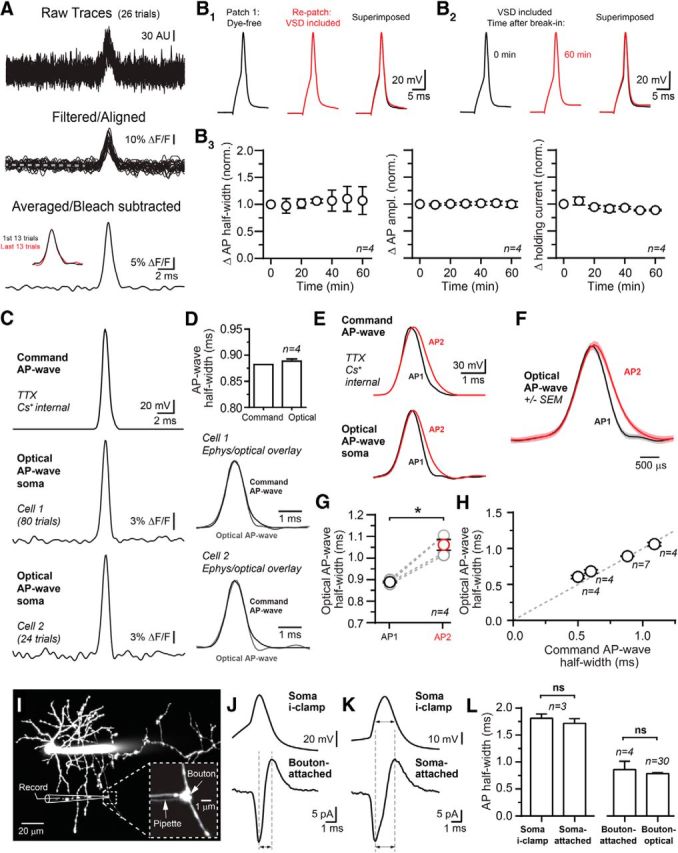
Two-photon VSD imaging in SC interneurons. A, Raw traces show AP-evoked voltage transients recorded in an axon of an SC during point measurement of fluorescence. Following acquisition, traces were digitally filtered off-line and aligned to remove any jitter using the peak of APs recorded at the soma with electrophysiology. Trials were averaged and photobleaching subtracted using a trend line extrapolated from a slope fit during the baseline period indicated by the dashed line. Inset shows that responses were stable throughout the recording. B1–B3, On the upper left, APs recorded at the soma using electrophysiology prior to and after repatching with an internal solution containing VSD. In a second cell shown on the right, somatic APs recorded immediately after whole-cell break-in and following prolonged dialysis with an internal solution containing VSD. Below are summary graphs showing that somatic AP duration and amplitude as well as holding current were unchanged during VSD dialysis. Rs remained ±15% of initial value, measured by bridge balance, throughout the experiment. C, Two examples of AP-like voltage transients recorded using VSD imaging at the somata of voltage-clamped SCs. The voltage-clamp command waveform, simulating an AP, is shown at the top. D, The half-widths of the AP-like voltage transients were nearly identical to the command waveforms. Below, voltage transients are superimposed with the command waveforms. E, AP-like voltage transients recorded from the soma of a voltage-clamped SC. The half-width of the second command waveform (AP2; red) was prolonged by 1.23 compared with the first waveform (AP1; black). F, AP-like voltage transients from the example on the left including response SEM. G, Summary plots showing that small changes in command waveform duration are accurately reported using VSD recording; *p < 0.05 with paired t test. H, Linearity of the VSD optical response, measured at the soma, for AP-like voltage commands of varying width. All points were significantly different from one another; *p < 0.05 by one-way ANOVA. Unity is indicated by the dashed line. I, A 2P fluorescence image of a cerebellar SC filled with the volume indicator dye Alexa 594 (60 μm) via the whole-cell somatic pipette. In the magnified view, a fluorescently coated second pipette (BSA-Alexa 594) is used for loose-seal, cell-attached recording from an axonal bouton. J, Simultaneous current-clamp and cell-attached recordings of aligned and average APs from the soma and bouton of the SC shown on the left. Dashed lines indicate the peak-to-peak measurements used to assess AP duration. K, Simultaneous current-clamp and cell-attached recordings from SC soma. Arrows indicate the half-duration for APs measured with each recording configuration. L, Summary data of AP half-widths from simultaneous current-clamp and cell-attached recordings. At boutons, similar AP half-width values were obtained with electrical recording and VSD imaging.
Although APs were observed throughout the axonal arbor, our analysis focused on spikes recorded in two specialized axonal locations, boutons–the morphological swellings encountered along an axon that demarcate presynaptic sites of release–and the AIS, a specialized region important for spike initiation (Clark et al., 2009; Bender and Trussell, 2012; Fig. 2A). To confirm that our AIS-targeted recordings were in general correspondence to the actual position of AIS, we immunolabeled SCs, filled with LY (n = 5) during whole-cell recording, with the AIS marker protein AnkG (Fig. 2B). Labeling revealed short AIS segments (13.7 ± 2.1 μm) that began shortly after the axon hillock (4.6 ± 1.6 μm). Correspondingly, by comparing the timing between APs recorded at the soma with the patch electrode and the optically resolved axonal spike (Fig. 2C), it was apparent that spikes occurred in the axon first with the shortest latencies in the most proximal segment, ∼11 μm from the hillock (Fig. 2D). Thus, the AP initiation zone in SCs clearly corresponds to a region in register with the anatomically identified AIS demarcated by AnkG immunolabeling, indicating that our VSD recordings in the most proximal axon region (measurement range 5–25 μm from the axon hillock) overlapped the AIS.
Figure 2.

AP repolarization is determined by distinct Kv channel subtypes in axonal subregions. A, A 2P fluorescence image of an SC filled with the green volume indicator dye Alexa 488 (20 μm). An axonal bouton and the AIS, targeted for voltage imaging by inclusion of the red VSD di-2-AN(F)EPPTEA (30 μm), are shown in the magnified views. B, Immunohistochemical labeling for AnkG (red) in the molecular layer of the cerebellum. An axon protruding from an SC, filled with LY (green) during whole-cell recording, is labeled for AnkG (indicated by white arrows). C, Comparison of AP latency for spikes simultaneously recorded in the soma with electrophysiology (black) and at the AIS (15 μm) with 2P VSD imaging (red). APs were elicited by somatic current injection. These traces are the average of multiple trials and have been normalized to the peak of the AP to facilitate comparison. D, Group data (n = 61 cells) showing the onset latency of the axonal AP relative to the somatic spike (0 μm). On the y-axis are latency differences from simultaneous electrophysiological and 2P VSD measurements of somatic APs. The red bar indicates the average position of the AIS, determined using AnkG immunolabeling. E, For the SC shown on the left, APs recorded at the soma with electrophysiology and at the AIS and a bouton using 2P VSD imaging. Each trace is the average of multiple trials. F, Superimposed APs, recorded at the AIS in control (black) and following bath application of either DTX (100 nm) or BDS-I (1 μm). The peak amplitudes of the averaged APs are normalized to facilitate comparison. G, APs recorded from axonal boutons in control (black) and following application of either DTX or BDS-I. H, AP repolarization at the AIS was prolonged by DTX but not by BDS-I. Data are mean ± SEM; *p < 0.05 by one-way ANOVA. I, Pharmacological profile of AP repolarization at presynaptic boutons. Data are mean ± SEM; *p < 0.05 by one-way ANOVA.
Comparison of spike waveforms (Fig. 2E) showed that APs were shorter in duration in boutons than in the AIS (785 ± 18 μs; n = 30 and 969 ± 48 μs; n = 19; for boutons and AIS, respectively; p < 0.01; Mann–Whitney test), a result also observed in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C (711 ± 21 μs, n = 5 and 1223 ± 93 μs, n = 5; p < 0.01; boutons and AIS, respectively) and in older mice (P28–P35; 741 ± 19 μs, n = 11 and 1094 ± 88 μs, n = 9; p < 0.01; boutons and AIS, respectively). These findings indicate that AP duration in axons of SCs varies depending on location likely reflecting local differences in the passive and active electrical features of these regions.
Kv channels are well known to set the rate AP repolarization and therefore are a major determinant of AP duration. Kv1 channels are located in the AIS of many neuron types and these channels influence the shape of the resulting axonal spike (Kole et al., 2007; Foust et al., 2011). To determine whether Kv1-mediated conductances contribute to spike repolarization in the AIS of SCs, we used the Kv1-specific blocker DTX-I (100 nm), which blocks Kv1 channels containing Kv1.1, 1.2, and 1.6 subunits (Coetzee et al., 1999). Bath-applied DTX substantially broadened APs in the AIS (Fig. 2F,H) indicating that a Kv1-mediated conductance directs spike repolarization in this region, a result consistent with the expression pattern of Kv1.1 and Kv1.2 subunits in the proximal axons of cerebellar interneurons (Lorincz and Nusser, 2008). This effect was not due to a somatic influence since electrically recorded APs were unchanged in width following DTX application (1.06 ± 0.03 of control, n = 18, p = 0.49; paired t test). Kv3 channels are abundant in many interneuron types including parvalbumin-expressing neocortical and hippocampal interneurons and drive fast AP repolarization (Erisir et al., 1999; Lien and Jonas, 2003). To directly test for the additional contribution of Kv3 conductances in mediating AP repolarization in the AIS, we used the Kv3-specific modulator BDS-I (Yeung et al., 2005). However, BDS-I (1 μm) did not affect AP repolarization in the AIS (Fig. 2F,H; also 0.93 ± 0.04 of control in P28–P35 mice, n = 5, p = 0.12; paired t test) indicating that Kv1, but not Kv3, channels likely play an important role in defining the spike-response properties governed by the AIS in SCs.
Remarkably, bath-applied DTX had no effect on AP repolarization at axonal boutons (Fig. 2G,I), regardless of their location (measurement range 41–109 μm from the axon hillock; also 0.99 ± 0.10 of control in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C; n = 4, p = 0.74; paired t test). Similar results were obtained in older mice (P28–P35; DTX 1.20 ± 0.03 of control, n = 5; p < 0.01; paired t test and 1.04 ± 0.04 of control, n = 6, p = 0.37; paired t test, AIS and bouton, respectively). Therefore, a K+ channel conductance, which is pharmacologically distinct from that in the AIS, must direct repolarization at boutons. Kv3 channels are particularly abundant in the axonal pinceau of cerebellar basket cell interneurons, a closely related cell-type to SCs, and mediate fast-activating currents in these structures (Veh et al., 1995; Laube et al., 1996; Southan and Robertson, 2000; Bobik et al., 2004). We found that a conductance with a pharmacological profile consistent with Kv3 channels directs AP repolarization at SC boutons. First, bath application of the Kv3 modulator BDS-I (1 μΜ) significantly broadened APs recorded at SC boutons (Fig. 2G,I). Additionally, TEA (500 μm), a high-sensitivity blocker of Kv3 channels (IC50 = ∼200 μm; Coetzee et al., 1999), similarly increased the duration of APs at boutons (Fig. 2I). In a separate set of experiments, we included the large-conductance Ca2+-activated K+ (BK)-specific blocker IBTX (100 nm) in the bath to occlude the TEA-dependent block of fast-activating BK channels (Coetzee et al., 1999). In this condition, TEA (500 μm) continued to induce spike broadening in boutons (Fig. 2I; also 1.30 ± 0.09 of control [in IBTX] in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C; n = 4, p = 0.03; paired t test), a result also observed in older mice (P28–P35; 1.20 ± 0.04 of control, n = 5; p = 0.01; paired t test). Thus, SCs employ two different types of Kv-mediated conductances to repolarize APs along their axons, Kv1-containing channels at the AIS and Kv3-containing channels at boutons.
Local control of AP repolarization in boutons
Following Kv3 channel block, AP duration is still comparatively brief in boutons (986 ± 32 μs, n = 10) suggesting that other K+ channels may be recruited in this condition. At the calyx of Held, Kv1 channels do not normally inform AP repolarization in basal conditions but can contribute to repolarization following Kv3 inhibition (Ishikawa et al., 2003). To test if Kv3 channel activity at SC boutons limits Kv1 channel recruitment in a similar manner, we applied DTX in the presence of TEA (500 μm). Block of Kv3 channels unmasked a DTX-induced increase in AP duration in boutons (Fig. 3A,B). In L5 pyramidal cells, DTX-induced spike broadening in the AIS is thought to directly increase the spike width in boutons, albeit in a distance-dependent manner, apparently due to propagation dependent on a long axonal length constant (Kole et al., 2007; but see Foust et al., 2011). This raises the possibility that, when Kv3 channels are blocked in SCs, AP broadening in the bouton reflects the specific loss of Kv1 channels in the AIS rather than a local effect at the bouton. However, we found that DTX-induced broadening was not related to distance from the axon hillock (Fig. 3C).
Figure 3.

Local determination of AP repolarization in axons. A, APs recorded at a bouton in a background of 500 μm TEA (black) and following application of 100 nm DTX. Traces are the average of many trials and are shown normalized to the peak of the AP to facilitate comparison. B, Summary of DTX-induced AP widening in TEA at boutons. Data are mean ± SEM; *p < 0.05 by unpaired t test. C, In a background of TEA, lack of dependence of DTX-induced AP widening at a bouton with distance from the axon hillock. Fit of the linear regression is indicated by the dashed line. D, Diagram depicting the experimental configuration. AP recording sites included both the AIS and an axonal bouton located in the same field of view to eliminate need for objective refocusing. Local photolysis of RuBi-4AP (bath applied at 150 μm) was limited to the AIS. Uncaging pulse trains (highlighted in red) were repeated five times at 0.33 Hz before each imaging trial. An imaging trial (highlighted in blue) consisted of five APs stimulated at 0.33 Hz. Measurements of AP waveform were made iteratively between these two sites in control and following RuBi-4AP uncaging. E, APs recorded at the AIS in control (black) and immediately following local photolysis of RuBi-4AP at the AIS. In the same cell, APs were also recorded at a bouton following RuBi-4AP photolysis at the AIS. F, Effect of AIS-directed RuBi-4AP photolysis on AP duration for spikes recorded at the AIS. In addition, control experiments show that laser pulses alone have no effect on AP repolarization and that RuBi-4AP has no basal effect on AP duration without laser-induced uncaging. Data are mean ± SEM; *p < 0.05 by one-way ANOVA. G, Effect of AIS-directed RuBi-4AP photolysis on AP repolarization at the AIS and boutons in basal conditions and in a background of TEA (500 μm). Data are mean ± SEM; *p < 0.05 by paired t test. The distance of the bouton recording position relative to the hillock is indicated below in parenthesis. H, Recording configuration showing that a short region of an axon branch was targeted for local Rubi-4AP photolysis. VSD responses were recorded in alternating trials from boutons located in either the targeted region or on a distal axon branch. I, APs recorded in control (black) and following Rubi-4AP photolysis (red) at bouton locations. J, AP widening, induced at target boutons by Rubi-4AP photolysis, was highly reduced in boutons located on distal axon branches. Data are mean ± SEM; *p < 0.05 by paired t test.
To more directly assess this possibility, we used local photolysis of the caged Kv channel blocker RuBi-4AP (150 μm; Zayat et al., 2003) to inhibit Kv channels in a spatially restricted manner at the AIS (<10 μm; Fig. 6) and examine the extent to which this perturbation affects AP duration in boutons (Fig. 3D). Laser pulses (473 nm; 0.8 mW; 0.5 ms pulses at 1 kHz) delivered to closely spaced uncaging points (n = 25) on each side of the proximal axon (11.7 ± 1.6 μm in length; starting 5.4 ± 1.3 μm from the axon hillock) increased AP width in the AIS (Fig. 3E,F) indicating partial loss of Kv channel-mediated repolarization in this region. This result was occluded by DTX (200 nm; Fig. 3F) indicating that RuBi-4AP photolysis blocks Kv1 channels at the AIS. At boutons, AP width was unaffected by local inhibition of Kv1 channels at the AIS (Fig. 3E,G). This shows that, in basal conditions, control of spike repolarization in boutons is uncoupled from the AIS. We observed a similar result when Kv3 channels were blocked by 500 μm TEA (Fig. 3G). Together, these data indicate that while Kv1 channels are locally available at boutons, conductances from these channels do not normally participate in AP repolarization in these compartments due to the availability of Kv3 channels. Yet, even in the absence of Kv3 conductances, control of AP repolarization in boutons remains uncoupled from the AIS as active spike propagation, supported by fast-activating conductances, is known to permit local shaping of AP duration even over short distances (Stuart and Häusser, 1994). The apparent short length constant of AP broadening in this condition likely reflects the influence of AP shaping by remaining active conductances, including those of Kv1-containing channels in boutons.
Figure 6.
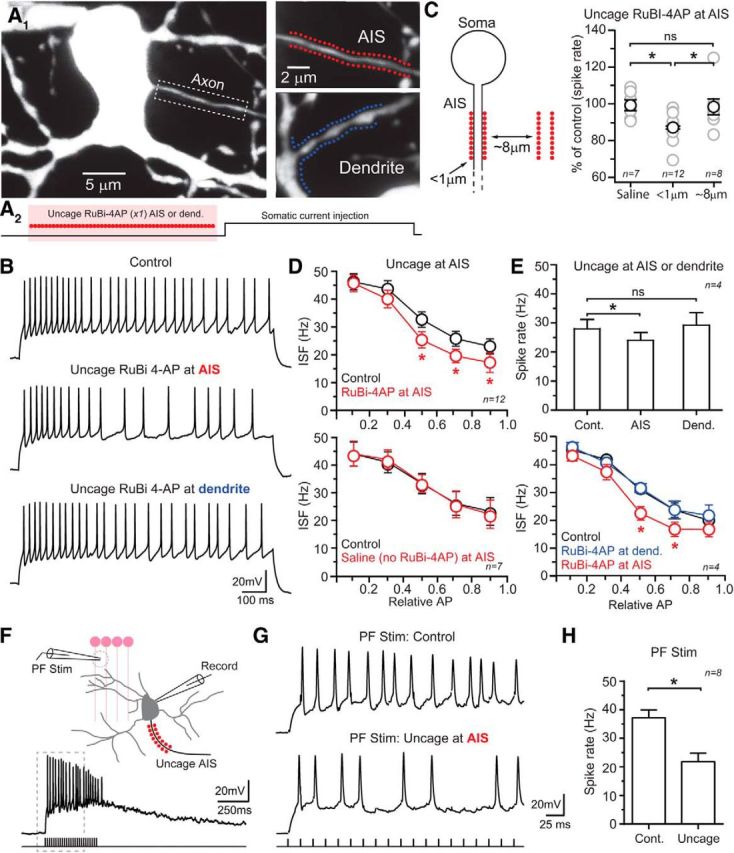
Kv1 channels in the AIS contribute to spiking phenotype. A1, Images of an SC with uncaging locations demarcated by points in the magnified views for either the AIS (red) or a dendrite (blue). A2, Diagram depicting the experimental configuration. Local photolysis of RuBi-4AP (300 μm) directly preceded somatic current injection (25 ms). Uncaging pulses, highlighted in red, are indicated by filled circles. B, Sustained firing, induced by somatic current injection, in control and immediately following RuBi-4AP photolysis at either the AIS or along a dendritic segment as illustrated in the images shown above. C, The average steady-state spiking frequency is reduced when photolysis of RuBi-4AP is directed immediately adjacent to the AIS. Moving the location of uncaging a short-distance lateral to the orientation of the axon demonstrates that area affected by photolyzed RuBi-4AP occurs in a spatially restricted manner. Data are mean ± SEM; *p < 0.05 by one-way ANOVA. D, Summary plots showing ISF for spike trains measured in control and immediately after photolysis of RuBi-4AP at the AIS. Data are mean ± SEM; *p < 0.05 by two-way ANOVA. In the bottom plot, laser pulses alone have no effect on ISF. For these plots, APs were normalized for total number and then binned based on their relative position in the spike train. E, Spiking measurements obtained following RuBi-4AP uncaging at two locations in the same cell including the AIS and at a dendritic (Dend.) site. Steady-state firing was induced by somatic current injection. Data are mean ± SEM; *p < 0.05 by one-way ANOVA in top plot, two-way ANOVA in the bottom plot. F, Diagram depicting the experimental recording configuration used for extracellular PF stimulation with a resulting postsynaptic SC response, recorded in control, shown below. G, Spike firing induced by repetitive PF stimulation (Stim.) in control and following AIS-targeted Rubi-4AP photolysis. An expanded view is shown with stimulus artifacts blanked for clarity. H, Summary graph showing AIS-directed Rubi-4AP photolysis reduces spiking. Data are mean ± SEM; *p < 0.05 by paired t test. Cont., control.
We also observed uncoupled control of AP repolarization between boutons located on separate branches of the axon arbor. Rubi-4AP uncaging targeting a small region of an axon branch (6.5 ± 2.1 μm in length; 50 points, 0.8 mW per 0.5 ms pulse) increased spike duration in a test bouton within the targeted region (Fig. 3H–J), an increase similar to that induced by bath application of 4AP (1.22 ± 0.05 of control; n = 6, p < 0.01; paired t test) at a concentration that is generally selective for Kv3 channels (30 μm; Coetzee et al., 1999; Alle et al., 2011). However, in alternating trials, APs measured at a second bouton located on an axon branch distal to the first (average distance between boutons 31.9 ± 7.4 μm) were largely unchanged despite Rubi-4AP-induced AP widening at the more proximal location (Fig. 3I,J; also observed in older mice [P28–P35] test bouton 1.21 ± 0.05 of control; distal bouton 1.03 ± 0.05; n = 4, p = 0.03; paired t test; average distance between boutons 19.9 ± 3.5 μm). Together, these results indicate that locally available K+ conductances within an axon branch impart regional control of spike repolarization allowing for uncoupled control of spike repolarization from the AIS as well as other axon branches.
Local expression of use-dependent AP broadening at the AIS
The uncoupled influence of Kv1 channels on AP repolarization at the AIS of SCs suggests that Kv1-mediated conductances may play a prominent role in determining the response properties of AP signaling at the AIS independent of other axonal regions. In hippocampal granule cells, repetitive spiking can induce cumulative broadening of APs at mossy fiber boutons due to use-dependent inactivation of Kv1 channels (Geiger and Jonas, 2000). Therefore, in SCs, the AIS may be particularly susceptible to activity-dependent prolongation of spike waveform. We examined AP shape in axons of SCs during repetitive spiking induced by brief somatic current injections (Fig. 4A) at a near-physiological firing frequency (40 Hz; Ruigrok et al., 2011). During the course of high-frequency firing, AP duration in the AIS became progressively longer indicating a reduction in the rate of repolarization. AP broadening was apparent by the 20th AP (Fig. 4B,D; also observed in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C; 20th AP 1.35 ± 0.03 of first AP; n = 4, p = 0.02; paired t test; in older mice [P28–P35]; 20th AP 1.36 ± 0.02 of first AP; n = 3, p = 0.03; paired t test; and last, at a resting potential [−54 ± 2 mV] just below the threshold for spontaneous spiking [−50 ± 1 mV]; 20th AP 1.44 ± 0.06 of first AP; n = 4; p < 0.01; paired t test) highlighting the rapid onset of use-dependent spike prolongation at this frequency. Broadening continued with additional APs at an average rate of 1.04 ± 0.25% per spike by the 80th AP (Fig. 4E), a rate similar to that observed at the mossy fiber bouton when stimulated at a comparable frequency (Geiger and Jonas, 2000). Activity-induced AP broadening was occluded in the presence of DTX (200 nm; Fig. 4B,D), suggesting that use-dependent inactivation of Kv1channels strongly contributes to the cumulative AP-broadening observed at the AIS.
Figure 4.

Activity-dependent broadening of APs in the AIS. A, A high-frequency train of APs (40 Hz) elicited by a series of brief current injections at the soma. B, The first (black) and the twentieth (red) APs in a spike train are shown superimposed for recordings made at the AIS in basal conditions and in DTX (200 nm). APs in each condition, averaged over many trials, were obtained from two different cells. C, Superimposed APs recorded at a bouton in basal conditions or with TEA (500 μm) in the bath. Averaged APs in each condition are from two different cells. D, Summary showing the effect of repeated firing on AP duration for spikes recorded at the AIS or boutons. Data are mean ± SEM; *p < 0.05 by one-way ANOVA. E, Plot of the increase in AP duration during repeated spiking (40 Hz) in the AIS and boutons. Significant differences in AP duration with spike number, compared with the first AP at either the AIS or boutons; *p < 0.05 by one-way ANOVA.
In contrast to the AIS, APs at boutons were resistant to broadening during high-frequency firing (Fig. 4C–E; also observed in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C; 20th AP 0.96 ± 0.06 of first AP; n = 4, p = 0.48; paired t test; and in older mice [P28–P35]; 20th AP 0.95 ± 0.05 of first AP; n = 4; p = 0.38 paired t test). This suggests that local control of spike repolarization by Kv3-mediated conductances ensures relatively brief APs at release sites, even after repeated use. To determine whether AP duration in boutons becomes susceptible to activity-induced broadening in the absence of Kv3 conductances, we repeated this experiment in TEA (500 μm). In this condition, high-frequency firing (40 Hz) broadened APs at boutons (Fig. 4C,D), a result consistent with use-dependent inactivation of Kv1 conductances that mediate spike repolarization when boutons are deprived of Kv3. With Kv3 channels blocked (500 μm TEA), the addition of DTX (200 nm) occluded spike broadening induced by repetitive firing in boutons (DTX 20th AP 1.02 ± 0.08 of first AP; p = 0.78 paired t test) similar to the result observed at the AIS. Together, these results indicate a unique feature of SC axon function. The location-dependent influence of two distinct classes of Kv channels on AP repolarization, Kv1 in the AIS and Kv3 at boutons, segregates the susceptibility of use-dependent spike plasticity to the AIS.
The AIS as a key regulatory site of spike firing
As the AP initiation site in SCs, the AIS is a privileged cellular location where the organization and influence of Kv1 channels may confer a high degree of specificity in determining key attributes of excitability such as the spiking pattern during sustained stimulation. To study steady-state firing properties, we used square-pulse current injections (1 s) to elicit the maximally sustained firing frequency. To find this frequency, currents were increased stepwise (5–10 pA/step) from a near-threshold level until reaching saturation (Imax), just before spiking failure. Spiking failure is a likely consequence of Na+ channel inactivation following high-intensity stimulation (Carter and Regehr, 2000). When stimulated at the maximum frequency, SCs fired continuously (50.1 ± 6.4 Hz, n = 16) with a modest amount of spike-frequency accommodation [Fig. 5A,C,D; instantaneous spike firing (ISF) 64.8 ± 5.6 and 41.2 ± 6.1 Hz, for the initial and last 20% of spikes in the train, respectively; n = 16, p < 0.01 by t test] indicating a use-dependent change in AP firing properties.
Figure 5.

Kv1 channels inform spike rate and pattern. A, Maximum sustained AP firing induced by prolonged somatic current injection in control, TEA (500 μm) and, following wash, DTX (100 nm). B, The average steady-state spiking frequency, recorded during maximum sustained AP firing, in TEA, or DTX. Data are mean ± SEM; *p < 0.05 by unpaired t test. C, ISF plotted in AP sequence for the trials illustrated on the left. A decrease in ISF is indicative of spike-frequency accommodation. D, Summary plots showing ISF for SCs recorded in control and following TEA or DTX. APs, normalized for total spike number, were binned according to their relative position in the spike train. Data are mean ± SEM with significance differences between controls and matched pharmacological conditions, *p < 0.05 by two-way ANOVA. Cont., control.
Kv channels are particularly important in determining spike output. For example, Kv3 channels confer a fast-spiking, nonaccommodating phenotype in a broad class of GABAergic interneurons in the brain (Erisir et al., 1999; Baranauskas et al., 2003; Lien and Jonas, 2003). However, in SCs, blocking Kv3 channels with TEA (500 μm) had no observable effect on the steady-state firing rate or on the spike-response pattern (Fig. 5A–D). In contrast, block of Kv1 channels with DTX (100 nm) reduced the steady-state firing rate resulting in fewer APs during the stimulus (Fig. 5A–C; also observed in 1.5 mm Ca2+ and 1.5 mm Mg2+ at 32°C; 0.74 ± 0.08 of control firing rate; n = 7, p = 0.03; paired t test; and in older mice [P28–P35]; 0.79 ± 0.06 of control firing rate; n = 5; p = 0.03; paired t test). In addition, the rate of spike-frequency accommodation increased dramatically compared with control before plateauing near the end of the stimulus (Fig. 5D), indicating that Kv1-mediated conductances normally moderate the onset of use-dependent spike accommodation. While the holding current was unaffected by DTX (−63.9 ± 7.7 and −56.0 ± 11.1 pA; control and DTX, respectively; n = 9; p = 0.18; paired t test), the magnitude of current necessary to reach the maximally sustained firing frequency (Imax) was reduced (121 ± 21.7 and 99.1 ± 20.4 pA; control and DTX, respectively; n = 9; p = 0.01; paired t test), a result analogous to that obtained in neocortical fast-spiking interneurons following Kv3 channel block (Erisir et al., 1999). Together, our results show that Kv1-mediated conductances strongly influence SC spiking patterns.
To determine whether Kv1 conductances specific to the AIS influence SC spiking patterns, we used local photolysis of RuBi-4AP (300 μm) to inhibit Kv1 channels at the AIS and measured the effect on sustained firing (Fig. 6A1). Laser pulses (473 nm; 0.8 mW; 8 ms pulses at 0.11 kHz) were directed to the proximal axon (25 sites on each side of the axon, visualized by inclusion of 50 μm Alexa 594; 9.7 ± 0.6 μm in length; starting 3.0 ± 1.0 from the axon hillock) just before stimulation (25 ms; Fig. 6A2). Current injections (1 s) elicited spiking slightly below the maximal firing rate (60–70% of Imax) were kept constant throughout the experiment. Photolysis of RuBi-4AP near the AIS reduced the steady-state firing rate and accelerated the onset of spike-frequency accommodation (Fig. 6B–D), a result similar to that obtained with bath-applied DTX. This effect appeared specific to the AIS. Moving the location of uncaging away from the axon (∼8 μm) did not alter the steady-state spike rate (Fig. 6C), suggesting that the area of 4AP-mediated Kv channel inhibition must be near the location of uncaging. We examined the subcellular specificity of this effect by uncaging RuBi-4AP onto proximal dendrites. However, local photolysis of RuBi-4AP onto dendrites was insufficient to alter steady-state spiking or spike accommodation (Fig. 6A1,B,E) despite within-cell comparisons showing that similar photolysis conditions at the AIS (∼2.5 points/μm; total targeted length, 19.2 ± 1.4 and 20.9 ± 0.8 μm; dendrites and AIS, respectively, n = 4; p > 0.05; paired t test) were sufficient to altering spike patterning (Fig. 6E). These observations indicate that AIS Kv conductances, most likely Kv1, play a prominent role in determining spike patterning at the AIS of SC interneurons.
To examine the role of Kv-mediated conductances in the AIS in determining spiking properties during more naturalist excitation, we stimulated the presynaptic PFs of granule cells using an extracellular pipette (60 Hz, 400 ms) thereby evoking prolonged SC excitation by summating EPSPs (Fig. 6F). Granule cells are known to maintain such high rates of prolonged firing in vivo during continuous cutaneous stimulation (Jörntell and Ekerot, 2006). In alternating trials, we used Rubi-4AP photolysis targeting the AIS of postsynaptic SCs to block Kv channels immediately before PF stimulation (Fig. 6F). Uncaging induced a reduction in the aggregate spike rate (Fig. 6G,H), similar to results obtained with steady state-current injection, reinforcing the conclusion that Kv1 channels in the AIS are a critical determinant of AP output.
Kv3 channels determine AP-evoked Ca2+ influx at release sites
At presynaptic sites of release, the AP waveform informs the efficacy of neurotransmission by directing voltage-gated Ca2+ channel (Cav) opening and, in following, the Ca2+ influx that triggers vesicle fusion and neurotransmission (Augustine et al., 1991). Therefore, at SC boutons, it is expected that Kv3 conductances mediating AP repolarization must have a prominent role in determining release efficacy. Using an experimentally derived kinetic model of Cav channel gating (Li et al., 2007), we first examined for the likely contribution of Kv3-mediated conductances in determining Cav activation using a representative AP waveform recorded from a SC bouton. Simulating the loss of Kv3-mediated repolarization by increasing AP duration (1.22 of control) enhanced Cav-mediated Ca2+ influx in a proportional manner (Fig. 7A; peak amplitude of Cav Po integral, 1.23 of control). In a complementary set of in vitro experiments, we simultaneously recorded AP waveform and the AP-evoked Ca2+ transient in the same bouton by including the spectrally separable Ca2+ indicator dye, Oregon Green BAPTA-1 (or OGB-6F), in the patch pipette together with the red VSD (Fig. 7B) and exciting both dyes at the same wavelength (1020 nm). Ca2+ influx primarily occurred after the peak of the AP suggesting that, in basal conditions, rapid Kv3-mediated repolarization limits Ca2+ influx in boutons. Indeed, slowing repolarization by blocking Kv3 channels (500 μm TEA) increased the amplitude of the AP-evoked Ca2+ transient (Fig. 7C). The increase in Ca2+ influx was proportional to the increase in AP half-width (AP half-width 1.22 ± 0.05 of control; peak Ca2+ amplitude, 1.22 ± 0.06 of control; n = 6) indicting that, like excitatory synapses (Sabatini and Regehr, 1997; Borst and Sakmann, 1999; Bischofberger et al., 2002), Ca2+ influx at presynaptic GABA release sites linearly tracks AP duration.
Figure 7.

Control of AP-evoked Ca2+ entry and neurotransmission is determined by Kv3 channels. A, An AP (black), recorded from an SC bouton, was used as the voltage command for a kinetic model of Cav channel gating. Cav channel open probability (Po) was integrated and is shown below. AP duration was prolonged (red) simulating the loss of Kv3-mediated repolarization resulting in a proportional increase in Cav channel opening. B, Simultaneous recording of spike waveform using VSD imaging and the resulting AP-evoked Ca2+ transient recorded with the green Ca2+ indicator dye, OGB-1. C, APs and the resulting AP-evoked Ca2+ transients simultaneously recorded in a bouton in control and in TEA (500 μm). D, In a paired recording from synaptically connected SCs, APs evoked by on-cell stimulation of a presynaptic SC (SC Pre.) evoke GABAA receptor-mediated IPSCs in a voltage-clamped postsynaptic SC (SC Post.). Averaged postsynaptic responses are shown in control and following bath application of TEA (500 μm). E, Average AP-evoked IPSC from a paired SC recording in control and BDS-I (1 μm). F, Spontaneous mIPSCs (0.5 μm TTX) in control and in TEA (500 μm). Cumulative probability of mIPSC amplitudes is shown in the histogram on the right. Superimposed in the inset are the averaged and aligned mIPSCs from each condition. G, A recording from synaptically connected SCs with AP-evoked IPSCs in control and DTX (100 nm). H, In a background of TEA (500 μm), AP-evoked IPSCs from a paired SC recording with responses in control and DTX (100 nm). I, Summary data from synaptically connected SC recordings showing the effect of Kv channel blockers on the amplitude of AP-evoked IPSCs. Data are mean ± SEM; *p < 0.05 by one-way ANOVA. Cont., control.
To directly test if Kv3-mediated control of the presynaptic waveform influences synaptic efficacy, we made paired electrophysiological recordings from synaptically connected SCs. APs were elicited by brief current injections in presynaptic SCs held in the cell-attached recording configuration; a constant bias current prevented these cells from spontaneously firing during recordings. Presynaptic APs elicited time-locked, GABAA receptor-mediated IPSCs in voltage-clamped postsynaptic cells (ECl ≈ 0 mV). Bath-applied TEA (500 μm) reversibly increased the amplitude of AP-evoked IPSCs (Fig. 7D,I; 1.36 ± 0.5 of control/wash average; n = 7; p = 0.02; paired t test) suggesting that Kv3 conductances limit synaptic strength. Likewise, the Kv3 modulator BDS-I (1 μm) increased IPSC amplitude to a similar extent as TEA (Fig. 7E,I). To test for a possible postsynaptic contribution of Kv3 on IPSC amplitude, we recorded spontaneous miniature IPSCs (mIPSCs; 0.5 mm TTX). However, mIPSC amplitude was slightly reduced by TEA (Fig. 7F; 0.96 ± 0.01 of control; n = 5; p = 0.03; paired t test). Thus, the increase in AP-evoked IPSC amplitude following block of Kv3 channels must be attributable to the broadening of spike waveform and increase in presynaptic Ca2+ entry. We also examined for a Kv1-mediated effect on synaptic efficacy; however, blocking Kv1 channels with DTX (100 nm) did not affect AP-evoked IPSC amplitude (Fig. 7G,I). This suggests that, despite a prominent role for Kv1 in mediating excitability at the AIS, these conductances do not normally inform synaptic efficacy likely because of the limited role of Kv1 in determining AP repolarization at boutons in basal conditions. In contrast, with Kv3 channels blocked by TEA (500 μm), DTX increased AP-evoked IPSC amplitude (Fig. 7H,I), a result in keeping with our finding that local Kv1-mediated conductances determine AP repolarization at release sites in the absence of Kv3.
Discussion
In this report, we show that the types of Kv channels mediating AP repolarization in SC axons vary depending on location. This implies that Kv-mediated conductances impart a highly localized influence on AP signaling likely derived from their subcellular targeting and expression. Due to this subcellular organization, axons multiply their adaptive properties by tuning excitation in one axon compartment independent of others, for example, uncoupling activity-dependent control of spiking at the AIS from neurotransmission at release sites.
Location-specific control of AP repolarization in axons by distinct Kv channel types
Using 2P VSD imaging to measure AP waveform, we observed that spike repolarization in SC axons is regionally determined by different types of Kv channels. Kv1 channels direct repolarization at the AIS and Kv3 channels direct repolarization at boutons. Whether other types of neurons also employ a diverse complement of Kv channels to regionally control spike repolarization in axons is unclear. Unlike SCs, Kv1-mediated control of AP repolarization in L5 pyramidal cells is not restricted to the AIS. Rather, this channel type determines spike shape throughout the axon arbor (Kole et al., 2007; Foust et al., 2011). In neocortical fast-spiking interneurons, Kv3 conductances not only inform release efficacy but also determine spiking rate (Erisir et al., 1999; Lau et al., 2000; Goldberg et al., 2005), a result indicative of a more global influence of Kv3 conductances fitting with the prominent expression of Kv3.1b and Kv3.2 in the both the soma and axons of this cell type (Chow et al., 1999). However, the location-specific arrangement and influence of voltage-gated ion channels, including Kv channels, is a common organizing motif in neurons critical for determining local excitability. In some cells, Kv channel subtypes partition within the AIS (Van Wart et al., 2007; Lorincz and Nusser, 2008) and, in SCs, form dense clusters on dendritic segments (Kollo et al., 2006) suggesting a precise subcellular organization on a very local scale.
Our results show that when Kv1 channels are blocked at the AIS, spike broadening is constrained to the most proximal region of axon suggesting that AP repolarization must be set by the local availability of K+ conductances and not from those at more remote locations. Furthermore, we find that AP repolarization is also locally determined within subregions beyond the AIS, including between branches within the axon arbor. The local influence of a particular Kv conductance is determined by a number of factors including channel density and the electrotonic nature of the axon including its complex geometry and passive properties, as well as the frequency content of the command voltage signal (Johnston and Wu, 1999). The spatial extent to which an AP is shaped by the local availability of active K+ conductances is currently unresolved. Nevertheless, it is clear that the Kv conductances determining spike repolarization at bouton release sites are spatially uncoupled from those at the AIS.
By using a diverse complement of Kv channel types at the AIS and boutons, axons can differentially regulate spike signaling in these two compartments. In a similar manner, dendrites frequently use ion channel gradients along their membranes to locally modify excitability multiplying their computational capacity (Nusser, 2012). In this way, use-dependent spike broadening in SCs is limited to the AIS in basal conditions because a biophysically distinct set of channels from the Kv3 family locally directs repolarization in boutons. Kv3 channels deactivate rapidly upon repolarization (Baranauskas et al., 2003; Lien and Jonas, 2003) and are likely less apt to accumulate inactivation during repetitive firing. Our observation that Kv1 conductances do not normally contribute to AP repolarization in boutons does not preclude the possibility that Kv1 channels are also expressed at these sites (Wang et al., 1994; Laube et al., 1996). With Kv3 conductances blocked, AP repolarization in boutons was set by a local Kv1 conductance and was also susceptible to use-dependent prolongation. When measured in axons, Kv3 channels are known to have faster activation kinetics, a larger single channel conductance, and a greater steady-state availability at rest compared with Kv1 channels (Alle et al., 2011). Thus, when coexpressed at the same site, AP repolarization is dominated by Kv3 with Kv1-mediated conductances controlling excitability and limiting aberrant spiking (Dodson et al., 2003; Ishikawa et al., 2003; Alle et al., 2011). Kv1 channels in boutons of SCs may function to control excitability in a similar manner.
AP waveform control at the site of initiation determines firing patterns
In myelinated axons of projection cells, APs initiate in the AIS (Kole et al., 2007; Shu et al., 2007; Foust et al., 2010). Although recent findings have established an axonal locus for spike initiation in a number of unmyelinated inhibitory interneuron types (Hu et al., 2010; Vervaeke et al., 2012; Casale and McCormick, 2011), a precise axonal location has not been identified in this broad cell class. We find that in SC interneurons, APs are generated in the proximal axon (∼11 μm from the hillock) in anatomical register with the well known AIS marker AnkG. Beyond being a spike trigger zone for AP initiation, the AIS may also play a central role in determining spiking properties (Dodson et al., 2003; Goldberg et al., 2008; Clark et al., 2009). Our observation that AIS-targeted inhibition of Kv1 channels alters the frequency and pattern of AP firing, whether induced by constant current injection or by more naturalistic PF-mediated synaptic activation, implies that K+ channel conductances in this region play a prominent role in determining spiking phenotype in SCs. Thus, our data suggest that the AP firing pattern is not only determined by the types of Kv-channels that SCs express but may also depend on their location.
An intriguing possibility is that inactivating Kv1 channels localized to the AIS play a key role in determining the spiking properties of SCs. Use-dependent inactivation of Kv1 channels during repetitive firing results in spike broadening at mossy fiber boutons in the hippocampus (Geiger et al., 2000). We also observed a similar rate of spike broadening at the AIS during repeated spiking and found that AP repolarization is determined by Kv1 channels in this region. Slower repolarization of APs in the AIS following use-dependent inactivation or pharmacological block of Kv1 may delay the recovery of Na+ channels from inactivation (Kuo and Bean, 1994) or promote opening of slowly activating K+ conductances (Giese et al., 1998) affecting spike rate. Perhaps this explains our observation that blocking Kv1 channels decreased spiking in SCs and promoted spike-frequency accommodation. Similarly, in fast-spiking interneurons, loss of Kv3 conductances decreases spiking and imposition of an inactivating Kv conductance promotes spike broadening and diminishes spike rate likely due to a complex interplay of Kv3 and recovery of Na+ channels from inactivation (Erisir et al., 1999; Lien and Jonas, 2003). But blocking Kv3 conductances in SCs did not alter spiking, a result consistent with the absence of Kv3-mediated spike repolarization in the AIS. How spike-frequency accommodation is used by interneurons in the cerebellum to encode behaviorally relevant circuit function is not clear. It may be that spike-frequency accommodation decorrelates spiking over time among SCs following PF-mediated synaptic transmission, in turn, temporally offsetting the balance of feedforward inhibition onto postsynaptic Purkinje cells altering their excitability and output.
Presynaptic Kv3 channels control neurotransmission
Our results indicate that Kv3 channels set a rapid rate of AP repolarization at boutons relative to the AIS. Although APs in SC boutons are broad compared with spikes recorded at other release sites where Kv3 channels similarly direct repolarization (Ishikawa et al., 2003; Alle et al., 2011; but see Sabatini and Regehr, 1996; Matsukawa et al., 2003), perhaps because of the small diameter (Palay and Chan-Palay, 1974) or passive properties of SC axons, Kv3 conductances still limit AP-evoked Ca2+ entry. Following Kv3 block, AP-evoked Ca2+ influx increased in a manner proportional to the increase in AP duration, similar to the relationship observed at glutamate releasing synapses (Sabatini and Regehr, 1997; Geiger and Jonas, 2000; Bischofberger et al., 2002). Increasing presynaptic Ca2+ influx by prolonging AP duration can induce both linear (Augustine, 1990) and exponential increases (Sabatini and Regehr, 1997; Borst and Sakmann, 1999) in the strength of neurotransmission depending whether the organization of Ca2+ sources and release sensors are tightly or loosely coupled (Augustine et al., 1991). We did not examine the Ca2+ coupling of release in SCs in earnest. Though it is tempting to speculate that, given the weak effect of exogenous slow-binding Ca2+ buffers on basal transmission (Christie et al., 2011), release is tightly coupled at SC synapses and therefore likely to track linearly with changes in presynaptic Ca2+ following AP waveform changes.
The kinetics of AP-evoked Ca2+ influx is critical in determining the precision of neurotransmission. By constraining Ca2+ influx and, thus, the time period when release occurs, rapid Kv3-mediated AP repolarization at SC boutons may synchronize quantal release, minimize synaptic delay, and reduce jitter (Borst and Sakmann, 1999; Fedchyshyn and Wang, 2005). In addition, the subcellular arrangement and influence of Kv3 channels at SC boutons, as opposed to Kv1, likely protects these presynaptic specializations from use-dependent spike broadening. This ensures the fidelity of the AP waveform despite ongoing activity, keeping spikes brief and limiting potentiation of transmitter release with more prolonged APs (Geiger and Jonas, 2000). Together, these properties may be important in regulating the precise timing and strength of inhibition onto postsynaptic Purkinje neurons, the sole output of the cerebellum. In Purkinje neurons, parallel fiber-mediated excitation is closely followed (∼1 ms) by feedforward inhibition from coincidently activated interneurons setting a narrow window for synaptic integration (Mittmann et al., 2005). Thus, the spiking response properties of MLIs are likely key to encoding temporal sequences necessary for coordinated movements. In the absence of Kv3, activity-induced spike broadening may widen this window disrupting the capacity to precisely represent temporal information on a millisecond timescale.
Footnotes
This work was supported by the Max Planck Society, the Max Planck Florida Institute (MPFI) for Neuroscience, and by the National Institutes of Health Grant NS083894 (J.M.C.).We thank Yul Park (MPFI) and Yishai Elyada (MPFI) for their helpful discussions and comments during the preparation of this manuscript, and Peter Jonas (IST Austria) for assistance with the kinetic model of voltage-gated calcium channels. We also thank Corey Acker (University of Connecticut Health Center) for his assistance with voltage-sensitive dye imaging.
The authors declare no competing financial interests.
References
- Acker CD, Yan P, Loew LM. Single-voxel recording of voltage transients in dendritic spines. Biophys J. 2011;101:L11–13. doi: 10.1016/j.bpj.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alle H, Kubota H, Geiger JR. Sparse but highly efficient Kv3 outpace BKCa channels in action potential repolarization at hippocampal mossy fiber boutons. J Neurosci. 2011;31:8001–8012. doi: 10.1523/JNEUROSCI.0972-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ. Regulation of transmitter release at the squid giant synapse by presynaptic delayed rectifier potassium current. J Physiol. 1990;431:343–364. doi: 10.1113/jphysiol.1990.sp018333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann NY Acad Sci. 1991;635:365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x. [DOI] [PubMed] [Google Scholar]
- Baranauskas G, Tkatch T, Nagata K, Yeh JZ, Surmeier DJ. Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat Neurosci. 2003;6:258–266. doi: 10.1038/nn1019. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. The physiology of the axon initial segment. Annu Rev Neurosci. 2012;35:249–265. doi: 10.1146/annurev-neuro-062111-150339. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, Jonas P. Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci. 2002;22:10593–10602. doi: 10.1523/JNEUROSCI.22-24-10593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobik M, Ellisman MH, Rudy B, Martone ME. Potassium channel subunit Kv3.2 and the water channel aquaporin-4 are selectively localized to cerebellar pinceau. Brain Res. 2004;1026:168–178. doi: 10.1016/j.brainres.2004.07.088. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci. 1999;354:347–355. doi: 10.1098/rstb.1999.0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale AE, McCormick DA. Active action potential propagation but not initiation in thalamic interneuron dendrites. J Neurosci. 2011;31:18289–18302. doi: 10.1523/JNEUROSCI.4417-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Erisir A, Farb C, Nadal MS, Ozaita A, Lau D, Welker E, Rudy B. K+ channel expression distinguishes subpopulations of parvalbumin- and somatostatin-containing neocortical interneurons. J Neurosci. 1999;19:9332–9345. doi: 10.1523/JNEUROSCI.19-21-09332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Chiu DN, Jahr CE. Ca2+-dependent enhancement of release by subthreshold somatic depolarization. Nat Neurosci. 2011;14:62–68. doi: 10.1038/nn.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BD, Goldberg EM, Rudy B. Electrogenic tuning of the axon initial segment. Neuroscientist. 2009;15:651–668. doi: 10.1177/1073858409341973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Forsythe ID. Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004;27:210–217. doi: 10.1016/j.tins.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Billups B, Rusznák Z, Szûcs G, Barker MC, Forsythe ID. Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol. 2003;550:27–33. doi: 10.1113/jphysiol.2003.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erisir A, Lau D, Rudy B, Leonard CS. Function of specific K+ channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J Neurophysiol. 1999;82:2476–2489. doi: 10.1152/jn.1999.82.5.2476. [DOI] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust A, Popovic M, Zecevic D, McCormick DA. Action potentials initiate in the axon initial segment and propagate through axon collaterals reliably in cerebellar Purkinje neurons. J Neurosci. 2010;30:6891–6902. doi: 10.1523/JNEUROSCI.0552-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust AJ, Yu Y, Popovic M, Zecevic D, McCormick DA. Somatic depolarization potential and Kv1 channels control spike repolarization in cortical axon collaterals and presynaptic boutons. J Neurosci. 2011;31:15490–15498. doi: 10.1523/JNEUROSCI.2752-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca2+ inflow by fast activating K+ channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/S0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Giese KP, Storm JF, Reuter D, Fedorov NB, Shao LR, Leicher T, Pongs O, Silva AJ. Reduced K+ channel inactivation, spike broadening and after-hyperpolarization in Kvb1.1-deficient mice with impaired learning. Learn Mem. 1998;5:257–273. [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- Ishikawa D, Takahashi N, Sasaki T, Usami A, Matsuki N, Ikegaya Y. Fluorescent pipettes for optically targeted patch-clamp recordings. Neural Netw. 2010;23:669–672. doi: 10.1016/j.neunet.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, Takahashi T. Distinct roles of Kv1 and Kv3 potassium channel at the calyx of Held presynaptic terminal. J Neurosci. 2003;23:10445–10453. doi: 10.1523/JNEUROSCI.23-32-10445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Wu SM-S. Foundations of cellular neurophysiology. Cambridge, MA: MIT; 1999. [Google Scholar]
- Jörntell H, Ekerot CF. Properties of somatosensory synaptic integration in cerebellar granule cells in vivo. J Neurosci. 2006;26:11786–11797. doi: 10.1523/JNEUROSCI.2939-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Kollo M, Holderith NB, Nusser Z. Novel subcellular distribution pattern of A-type K+ channels on neural surface. J Neurosci. 2006;26:2684–2691. doi: 10.1523/JNEUROSCI.5257-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Lau D, Vega-Saenz de Miera EC, Contreras D, Ozaita A, Harvey M, Chow A, Noebels JL, Paylor R, Morgan JI, Leonard CS, Rudy B. Impaired fast-spiking, suppressed cortical inhibition, and increased susceptibility to seizure in mice lacking Kv3.2 K+ channel proteins. J Neurosci. 2000;20:9071–9085. doi: 10.1523/JNEUROSCI.20-24-09071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube G, Röper J, Pitt JC, Sewing S, Kistner U, Garner CC, Pongs O, Veh RW. Ultrastructural localization of Shaker-related potassium channel subunits and synapse-associated protein 90 to septate-like junctions in rat cerebellar Pinceaux. Mol Brain Res. 1996;42:51–61. doi: 10.1016/S0169-328X(96)00120-9. [DOI] [PubMed] [Google Scholar]
- Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci. 2007;27:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Jonas P. Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. J Neurosci. 2003;23:2058–2068. doi: 10.1523/JNEUROSCI.23-06-02058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knöpfel T. Motor dysfunction and altered synaptic dysfunction at the parallel fiber-Purkinje cell synapses in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Koch U, Häusser M. Feedforward inhibition shapes the spike output of cerebellar Purkinje cells. J Physiol. 2005;563:369–378. doi: 10.1113/jphysiol.2004.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z. Differential subcellular distribution of ion channels and the diversity of neuronal function. Curr Opin Neurobiol. 2012;22:366–371. doi: 10.1016/j.conb.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. New York: Springer; 1974. [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok TJ, Hensbroek RA, Simpson JI. Spontaneous activity signatures of morphologically identified interneurons in the vestibulocerebellum. J Neurosci. 2011;31:712–724. doi: 10.1523/JNEUROSCI.1959-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Yu Y, Yang J, McCormick DA. Selective control of cortical axon spikes by an inactivating K+ current. Proc Natl Acad Sci U S A. 2007;104:11453–11458. doi: 10.1073/pnas.0702041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan AP, Robertson B. Electrophysiological characterization of voltage-gated K+ currents in cerebellar basket and Purkinje cells: Kv1 and Kv3 channel subfamilies are present in basket cell nerve terminals. J Neurosci. 2000;20:114–122. doi: 10.1523/JNEUROSCI.20-01-00114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Häusser M. Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron. 1994;13:703–712. doi: 10.1016/0896-6273(94)90037-X. [DOI] [PubMed] [Google Scholar]
- Tan YP, Llano I. Modulation of K+ channels of action potential-evoked intracellular Ca2+ concentration rises in rat cerebellar basket cell axons. J Physiol. 1999;520:65–78. doi: 10.1111/j.1469-7793.1999.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500:339–352. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- Veh RW, Lichtinghagen R, Sewing S, Wunder F, Grumbach IM, Pongs O. Immunohistochemical localization of five members of the Kv1 channel subunits: contrasting subcellular locations and neuron-specific co-localization in rat brain. Eur J Neurosci. 1995;7:2189–2205. doi: 10.1111/j.1460-9568.1995.tb00641.x. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Lorincz A, Nusser Z, Silver RA. Gap junctions compensate for sublinear dendritic integration in an inhibitory network. Science. 2012;335:1624–1628. doi: 10.1126/science.1215101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two channel proteins, to synaptic terminals, somata and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Wang LY. Amplitude and kinetics of action potential-evoked Ca2+ current and its efficacy in triggering transmitter release at the developing calyx of Held synapse. J Neurosci. 2006;26:5698–5708. doi: 10.1523/JNEUROSCI.4889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Thompson D, Wang Z, Fedida D, Robertson B. Modulation of Kv3 subfamily by the sea anemone toxin BDS: significance for CNS and biophysical studies. J Neurosci. 2005;25:8735–8745. doi: 10.1523/JNEUROSCI.2119-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayat L, Calero C, Alborés P, Baraldo L, Etchenique R. A new strategy for neurochemical photodelivery: metal-ligand heterolytic cleavage. J Am Chem Soc. 2003;125:882–883. doi: 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]