

Abstract
Recent experimental evidence suggests that the low dopamine conditions in Parkinson's disease (PD) cause motor impairment through aberrant motor learning. Those data, along with computational models, suggest that this aberrant learning results from maladaptive corticostriatal plasticity and learned motor inhibition. Dopaminergic modulation of both corticostriatal long-term depression (LTD) and long-term potentiation (LTP) is proposed to be critical for these processes; however, the regulatory mechanisms underlying bidirectional corticostriatal plasticity are not fully understood. Previously, we demonstrated a key role for cAMP signaling in corticostriatal LTD. In this study, mouse brain slices were used to perform a parametric experiment that tested the impact of varying both intracellular cAMP levels and the strength of excitatory inputs on corticostriatal plasticity. Using slice electrophysiology in the dorsolateral striatum, we demonstrate that both LTP and LTD can be sequentially induced in the same D2-expressing neuron and that LTP was strongest with high intracellular cAMP and LFS, whereas LTD required low intracellular cAMP and high-frequency stimulation. Our results provide a molecular and cellular basis for regulating bidirectional corticostriatal synaptic plasticity and may help to identify novel therapeutic targets for blocking or reversing the aberrant synaptic plasticity that likely contributes to motor deficits in PD.
Keywords: cAMP, dorsolateral striatum, LTD, LTP, motor learning, synaptic plasticity
Introduction
Bidirectional synaptic plasticity is an important mechanism for learning and adapting behavior in an experience-dependent manner (Reynolds and Wickens, 2002; Jörntell and Hansel, 2006; Lovinger, 2010). At many central synapses, the strength of glutamatergic input and intracellular Ca2+ levels determine the direction and magnitude of synaptic plasticity (Malenka, 1994; Wong et al., 1999; Malenka and Bear, 2004; Gall et al., 2005). At corticostriatal synapses, the neuromodulator dopamine plays a central role in plasticity (Calabresi et al., 2007). Our earlier studies suggested that the striatal enriched adenylyl cyclase type V (AC5), which is activated by G proteins but not by Ca2+ (Guillou et al., 1999), is critical for dopamine-mediated plasticity (Kheirbek et al., 2009). However, it is not clear how dopamine-induced cAMP signaling interacts with the strength of glutamatergic input in determining the direction and magnitude of synaptic plasticity.
Phasic dopamine signaling is likely coordinated with experience-dependent patterns of cortical activity to induce persistent changes in corticostriatal synaptic strength (Graybiel et al., 1994; Costa et al., 2004). Although the contribution of striatal synaptic plasticity to motor learning has been postulated (Costa et al., 2004; Pisani et al., 2005; Calabresi et al., 2007), more recent work highlights the potential contribution of experience-dependent corticostriatal plasticity to motor impairments in Parkinson's disease (PD; Kreitzer and Malenka, 2007; Shen et al., 2008; Beeler et al., 2010; Beeler et al., 2012). Dopamine acutely alters medium spiny neuron (MSN) excitability in the striatonigral and striatopallidal pathways (Nicola et al., 2000; Surmeier et al., 2007) and these acute effects are the basis for classic models of PD (Penney and Young, 1986; Albin et al., 1989; DeLong, 1990; Mink, 1996). However, dopamine regulation of striatal synaptic plasticity could affect future motor performance in a persistent manner. Alteration in corticostriatal plasticity has only recently been considered to contribute to the motor symptoms of PD. Accumulating evidence suggests that dopamine denervation can induce an inversion in the direction of plasticity at corticostriatal synapses, particularly in the striatopallidal pathway, such that conditions that normally induce long-term depression (LTD) will instead yield long-term potentiation (LTP; Kreitzer and Malenka, 2007; Shen et al., 2008; Fino et al., 2010). Dopamine denervation or blockade results in D2-mediated aberrant learning, which is experience and task specific (Wiecki et al., 2009; Beeler et al., 2010; Beeler et al., 2012).
These observations suggest that dopamine denervation may induce inappropriate potentiation in the inhibitory striatopallidal pathway that gradually deteriorates motor performance via learned motor inhibition (Wiecki and Frank, 2010; Beeler, 2011; Zhuang et al., 2013). In this model, bidirectional corticostriatal plasticity is modulated by both dopamine and excitatory input strength. Because dopamine signaling via cAMP contributes to synaptic plasticity (Shen et al., 2008; Kheirbek et al., 2009; Lerner and Kreitzer, 2012), we systematically tested the effects of modifying intracellular cAMP levels and afferent stimulation strength to assess the permissive conditions for LTP and LTD of the excitatory inputs to striatopallidal neurons. Our data suggest that dopamine-regulated changes in intracellular cAMP is a critical control point for enhancing or suppressing excitatory inputs contingent upon the level of afferent activity.
Materials and Methods
Mice.
All animal experiments were approved by the Institutional Animal Care and Use Committee at The University of Chicago. Male and female adult (4–8 weeks old) hemizygous Drd2-enchanced green fluorescent protein (EGFP) bacterial artificial chromosome (BAC) transgenic mice (obtained from the Mutant Mouse Regional Resource Center) backcrossed onto a C57BL/6 background were used in all experiments. Drd2-EGFP homozygotes were identified from test crosses and then crossed with C57BL/6 mice to produce hemizygotes. The transgene, EGFP, in the Drd2 EGFP BAC transgenic mice is expressed under the regulatory control of DA D2 receptors (Gong et al., 2003), which allows for the visual identification of D2-expressing MSNs.
Slice preparation.
Mice were anesthetized with isoflurane and decapitated into cold oxygenated (95% O2 and CO2) sucrose artificial CSF (ACSF). Coronal slices (250 μm) containing striatal tissue were taken from hemizygous Drd2-EGFP BAC transgenic mice using a vibratome. Then slices were transferred and incubated for 30–60 min in a perfusion holding chamber at 32–34°C in oxygenated ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1 MgCl2(6H2O), 2.5 CaCl2, 20 glucose, 1 NaH2PO4, 25 NaHCO3, and 1 ascorbic acid (Kheirbek et al., 2009).
Electrophysiology.
During recording, slices were perfused (3–5 ml/min) with ACSF without ascorbic acid and incubated at 37°C. The cells were first visualized using infrared-differential interference contrast microscopy with a fixed stage upright microscope. Then, D2 MSNs in the dorsolateral striatum were identified using epifluorescence microscopy (X-cite series 120Q; EXPO Photonic Solutions) and whole-cell voltage-clamp recordings were performed (Vm = −70 mV). Recording pipettes (4–7 MΩ) were filled with a cesium-gluconate internal solution containing the following (in mm): 120 Cs gluconate, 10 TEA-Cl, 10 HEPES, 10 glucose, 5 NaCl, 5 Qx-314, 4 ATP-K, 1.1 EGTA, and 0.3 GTP-Na. All experiments were conducted in the presence of 50 μm picrotoxin, a GABAA receptor antagonist. Data were acquired with a Multiclamp 700A amplifier; signals were digitized at 10 kHz with a Digidata 1440A and viewed with pCLAMP 9.2 software (Molecular Devices). The series resistance (<25 MΩ) was recorded and monitored throughout the recording. The input resistance of the cell was monitored online (<30% variation) with an 11 mV, 100 ms depolarizing pulse 1000 ms after the last paired stimuli. For stimulation of corticostriatal terminals, a bipolar tungsten electrode with a 500 μm tip separation was placed in the corpus callosum or striatum dorsolaterally to the recording location. Paired test pulses separated by 25 ms were delivered via the Master-8 stimulator (A.M.P.I.) at 0.05 Hz. EPSC amplitudes were determined by the response to the first stimulation (P1). The paired-pulse ratio (PPR) was calculated as P2/P1. Stimulus intensity ranged from 0.01 to 1 mA with a duration of 200 μs. After 5–10 min of stable baseline recording, one of two conditioning stimuli was applied. The HFS conditioning stimulus, consisting of 4 1 s 100 Hz trains, was administered while the postsynaptic cell was depolarized to 0 mV. LFS conditioning stimuli consisted of stimulation once at −70 mV and then once during a depolarizing step to +40 mV, and this was repeated 5 times at 0.5 Hz. For the AMPA/NMDA ratio, the average AMPA peak amplitude and time to peak were determined from five evoked EPSCs at −70 mV; the average NMDA peak amplitude was determined by sampling a 10 ms window 40 ms after the peak of the AMPA EPSC, from five evoked EPSCs at +40 mV. Pharmacological agents were bath applied, except the Sp-Adenosine 3′,5′-cyclic monophosphorothioate (Sp-cAMP) and Rp-Adenosine 3′,5′-cyclic monophosphorothioate (Rp-cAMP), which were applied via the recording electrode. Only one recording was performed per brain slice. Chemicals were purchased from Sigma-Aldrich except for the following: Sp-cAMP (Tocris Bioscience), d-AP5 (Tocris Bioscience and Abcam), NKY80 (EMD4 Biosciences), and U73122 (Tocris Bioscience).
The data for the electrophysiological experiments are reported as mean ± SEM and are expressed as the normalized value of the baseline (2–5 min) before the conditioning stimulus, unless otherwise stated. Changes in synaptic strength were determined by comparing the EPSC amplitude 2–5 min before and the last 2–5 min after stimulation by a paired t test. To determine group differences in EPSC amplitude (LTD, LTP, or no change), a Student's unpaired t test was used.
Results
Low intracellular cAMP is permissive for LTD in striatopallidal MSNs
To facilitate identification of striatopallidal MSNs, we used a transgenic mouse line expressing EGFP under the control of the DRD2 receptor regulatory sequence (Gong et al., 2003). Hemizygous mice expressing this transgene backcrossed onto C57BL/6 background show normal motor learning (Nelson et al., 2012). Similar animals were used in our studies to test the effects of D2 receptor activation on the reliability of LTD induction after HFS of the glutamatergic inputs to these neurons in the dorsolateral striatum, the sensorimotor region of the striatum that is important for stimulus-response learning (Costa et al., 2004; Yin et al., 2009). In control slices, HFS paired with postsynaptic depolarization induced LTD in 67% of D2-expressing MSNs (Fig. 1A). An increase in PPR was observed only in those cells exhibiting LTD (Fig. 1B), which is consistent with endocannabinoid-mediated alteration of presynaptic release probability (Choi and Lovinger, 1997; Gerdeman et al., 2002; Kheirbek et al., 2009). Administration of the D2 receptor antagonist sulpiride completely abolished LTD (Fig. 1C,D), which is consistent with previous reports (Wang et al., 2006; Kreitzer and Malenka, 2007; Shen et al., 2008; Bagetta et al., 2011). When the D2 receptor agonist quinpirole was applied, HFS induced LTD in 100% of the cells tested (Fig. 1E,F), with concomitant increases in PPR. Therefore, D2 receptor activation increased the reliability of HFS-induced LTD. These data are in agreement with the well established role of D2 receptors in LTD induction (Shen et al., 2008; Kheirbek et al., 2009; Lerner and Kreitzer, 2012). D2 receptor coupling to Giα suggests that a decrease in cAMP is important for LTD; however, D2-mediated PLC signaling can also contribute to LTD induction (Fino et al., 2005; Yin and Lovinger, 2006; Lerner and Kreitzer, 2012). To test this pathway, we included the PLC inhibitor U73122 in the patch pipette while the D2 receptor agonist quinpirole was bath applied. Under this condition, HFS still induced reliable LTD (Fig. 1G), with a trend toward an increase in PPR in the cells that showed LTD (Fig. 1H). Together, these findings suggest that reduced intracellular cAMP supports LTD induction under these recording conditions.
Figure 1.

Corticostriatal LTD in striatopallidal MSNs in dorsolateral striatum. A, EPSC amplitudes were normalized to baseline, averaged, and plotted versus the time of the recording. HFS induces LTD (n = 12 cells/slices, t(11) = 3.35, p = 0.006), Inset, Representative traces before (solid line) and after (dashed line) HFS. Scale bar, 100 pA, 10 ms. B, There is an increase in the PPR from cells that responded to HFS with a significant depression of EPSC amplitude after HFS (n = 8 cells/slices, t(7) = −2.55, p = 0.038). A subset of cells did not show LTD, and the PPR from these cells were not different after HFS (n = 4 cells/slices, t(3) = −0.86, p = 0.452). C, D, HFS induces no LTD in the presence of the D2 receptor antagonist sulpiride (10 μm; n = 6 cells/slices, t(5) = −0.09, p = 0.93; C) and the PPR was not different in sulpiride-pretreated neurons (t(5) = −1.11, p = 0.32; D). E, F, HFS results in consistent LTD in the presence of the D2 receptor agonist quinpirole (10 μm; n = 6 cells/slices, t(5) = 9.09, p = 0.0002; E), HFS-induced LTD in quinpirole-treated neurons is accompanied by an increase in PPR (n = 6 cells/slices, t(5) = −2.44, p = 0.058; F). G, HFS induces LTD when the cell is dialyzed with the PLC inhibitor U73122 (10 μm) in the presence of extracellular quinpirole (10 μm; n = 6 cells/slices, t(5) = −3.25, p = 0.023). H, This LTD is accompanied by a trend toward an increase in PPR in the subset of cells that show LTD (n = 5 cells/slices, t(4) = −2.45, p = 0.07). The LTD magnitude (t(11) = 1.87, p = 0.09) and prevalence (5/6 with quinpirole and the PLC inhibitor vs 6/6 with quinpirole alone, p = 0.3 by χ2) were not different from control recordings without the PLC inhibitor.
To test directly the role of intracellular cAMP concentration in LTD induction, we used electrode solutions containing different concentrations of Sp-cAMP, a nonhydrolyzable cAMP analog that activates protein kinase A (PKA). HFS induced LTD only with the lowest Sp-cAMP concentration tested, 2 μm (Fig. 2A). Both 20 and 500 μm Sp-cAMP blocked LTD induction (Fig. 2B,C). Inhibition of PKA activity with Rp-cAMP in the recording electrode also inhibited LTD induction (100 μm; Fig. 2D). These data support the idea that there is a permissive window of intracellular cAMP that facilitates LTD induction in striatopallidal MSNs.
Figure 2.
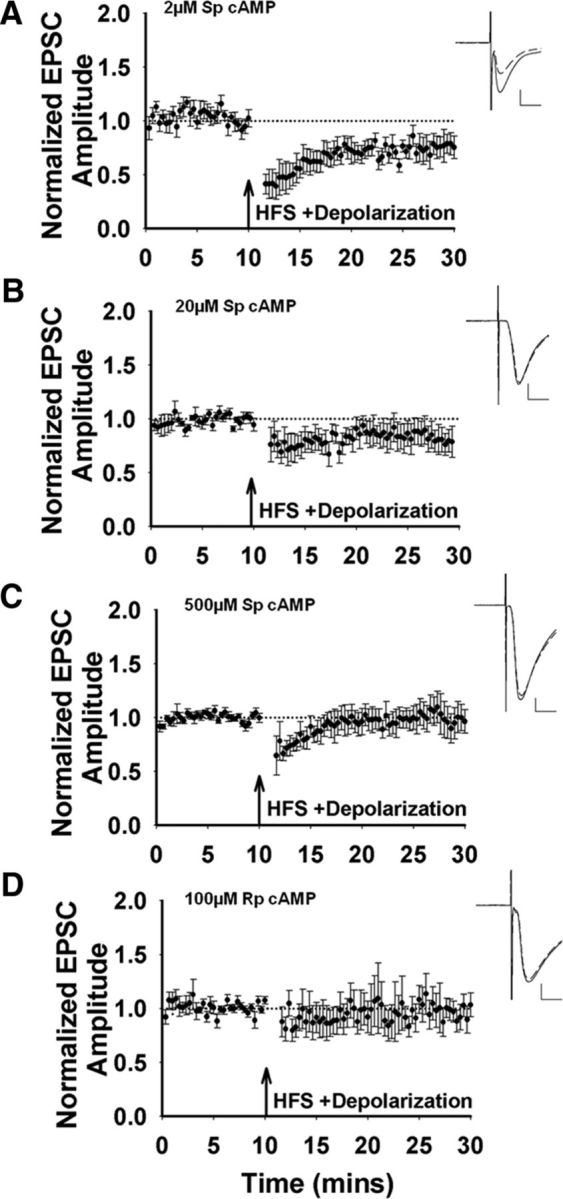
cAMP modulation of corticostriatal LTD. A, HFS induces LTD when the cell is dialyzed with 2 μm Sp-cAMP (n = 6 cells/slices, t(5) = 2.77, p = 0.04). Inset, Representative traces before (solid line) and after HFS (dashed line). Scale bar, 100 pA, 10 ms. B, HFS induces no change in synaptic strength with 20 μm Sp-cAMP in the recording pipette (n = 5 cells/slices, t(4) = 0.89, p = 0.43). C, LTD is also inhibited by 500 μm Sp-cAMP in the recording pipette (n = 5 cells/slices, t(4) = 0.37, p = 0.73). D, On average, there is no change in synaptic transmission when 100 μm Rp-cAMP is included within the patch pipette (n = 8 cells/slices, t(7) = 0.63, p = 0.55).
LFS potentiates excitatory inputs to striatopallidal MSNs
To assess the cAMP modulation of corticostriatal potentiation, we tested an LFS protocol combined with MSN depolarization. This protocol consisted of 5-step depolarizations to +40 mV for 550 ms at a frequency of 0.5 Hz, combined with stimulation of inputs once within and once between each of these depolarizations (Fig. 3A). This LFS protocol increased EPSC amplitude (Fig. 3). We then tested the contribution of intracellular cAMP to LFS induced potentiation. With 2 μm Sp-cAMP in the recording electrode (Fig. 3B), the only condition that produced LTD after HFS, potentiation was weak (Fig. 3B). Increasing the intracellular Sp-cAMP to 20 and 500 μm resulted in potentiation (Fig. 3C,D), the concentrations that corresponded to no HFS-induced LTD. The EPSC amplitude at 500 μm Sp-cAMP was less compared with 20 μm, suggesting an optimal range of cAMP levels for potentiation, as observed for LTD. When the pipette solution included the PKA inhibitor Rp-cAMP, the synapses were strongly potentiated in response to LFS (Fig. 3E). These data suggest a window of low intracellular cAMP concentration that restricts potentiation, presumably favoring LTD, and levels above or below this range will promote potentiation by low-frequency inputs.
Figure 3.
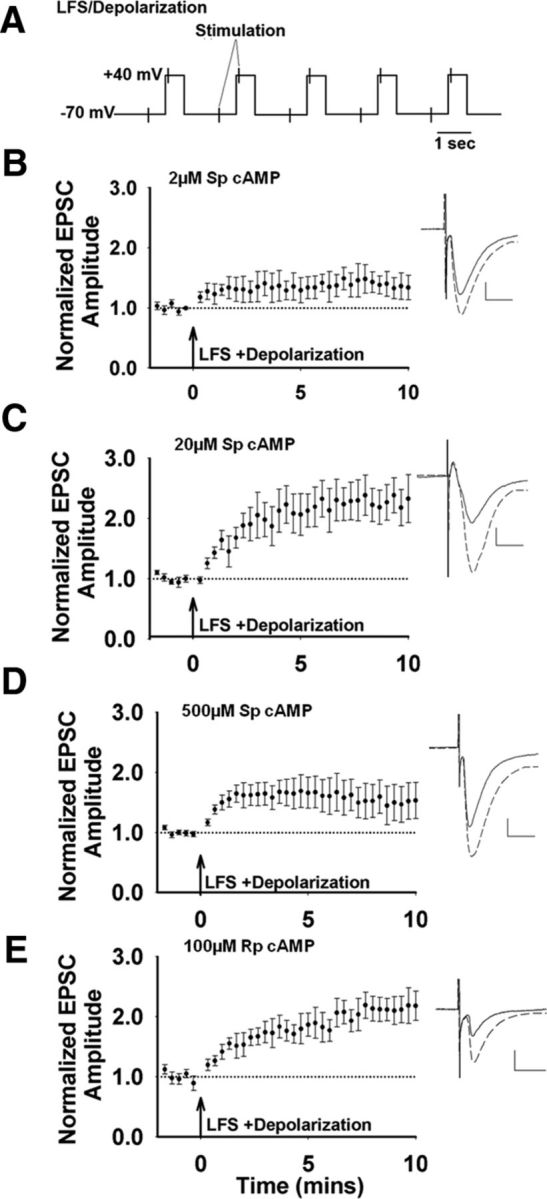
cAMP modulation of corticostriatal potentiation. A, Schematic of LFS/depolarization protocol. B, 2 μm Sp-cAMP in the recording pipette paired with LFS results in a weak potentiation (n = 5 cells/slices, t(4) = −1.89, p = 0.13). Inset, Representative traces before (solid line) and after LFS (dashed line); Scale bar 100 pA, 10 ms. C, With 20 μm Sp-cAMP in the recording pipette, LFS induces a robust increase in the normalized peak amplitude (n = 7 cells/slices, t(6) = −5.85, p = 0.001). D, With 500 μm Sp-cAMP in the pipette, LFS induces a small potentiation (n = 5 cells/slices, t(4) = −1.77, p = 0.15). E, With the PKA inhibitor, Rp-cAMP (100 μm) in the recording pipette, LFS induces a potentiation of EPSC amplitudes (n = 6 cells/slices, t(5) = −4.06, p = 0.01).
LFS-induced potentiation is NMDA-receptor-dependent LTP
We next tested whether LFS-induced potentiation is long lasting and can be considered LTP. With 20 μm Sp-cAMP in the recording electrode, LFS induced a long-lasting potentiation, which is consistent with LTP (Fig. 4A). When Sp-cAMP is not included in the recording solution, LFS did not induce LTP (Fig. 4B). When we included 20 μm Sp-cAMP in the pipette without presynaptic or postsynaptic stimulation, there was no change in EPSC amplitude (Fig. 4C). Postsynaptic depolarization mimics the upstate of MSNs, which activates l-type calcium channels and enables NMDA receptor function (Carter and Sabatini, 2004). When the neurons were depolarized without additional afferent stimulation other than the test stimuli, we saw a slight trend toward potentiation that was not statistically different from baseline (Fig. 4D). Application of AP-5, an NMDA receptor antagonist, eliminated LFS-induced LTP (Fig. 4E), indicating that this potentiation is NMDA receptor dependent, which is consistent with previous observations of striatal LTP (Calabresi et al., 1992b; Dang et al., 2006; Shen et al., 2008; Fino et al., 2010). The data demonstrate an NMDA-receptor-dependent corticostriatal LTP that is dependent upon LFS combined with postsynaptic depolarization and permissive intracellular cAMP concentrations.
Figure 4.

LFS and corticostriatal LTP. All neurons were dialyzed with 20 μm Sp-cAMP to promote potentiation, except in B. Arrow indicates the time of the conditioning stimulus. Inset, Representative traces before (solid line) and after conditioning stimulus (dashed line). In the case where no conditioning stimulus was given, the solid line represents the first 10 min and the dashed line represents the last 10 min of recording. Scale bar, 100 pA, 10 ms. A, LFS paired with depolarization induced robust LTP (n = 6 cells/slices, t(5) = −2.84, p = 0.04). B, In the absence of 20 μm Sp-cAMP, LFS/depolarization does not increase EPSC amplitude (n = 5 cells/slices, t(4) = −1.18, p = 0.30). C, In the absence of any stimulation, 20 μm Sp-cAMP in the internal solution did not induce a change in EPSC amplitude (n = 5 cells/slices, t(4) = −0.61, p = 0.58). D, Depolarization without LFS induced a weak trend toward LTP (n = 5 cells/slices, t(4) = −2.63, p = 0.06). E, Bath application of the NMDA antagonist d-AP5 (50 μm) inhibited LTP induction (n = 5 cells/slices, t(4) = 0.96, p = 0.39).
Sequential induction of potentiation and depression in striatopallidal MSNs
Corticostriatal plasticity in D2-expressing MSNs is bidirectional (Shen et al., 2008) and the results presented indicate that cAMP signaling can regulate both potentiation and depression. However, it is not known whether LTD and LTP can be induced in the same cell. Therefore, we tested whether these two forms of synaptic plasticity can be induced in the same D2-expressing MSNs. Cells were dialyzed with either 2 or 20 μm Sp-cAMP to support either LTD or LTP induction, respectively. After gaining whole-cell access, the neurons were initially subjected to LFS (Fig. 3A), EPSCs were monitored for 10 min, and then HFS was applied.
Figure 5, A and B, shows representative examples of EPSC amplitudes recorded from 2 neurons with either 2 or 20 μm Sp-cAMP in the recording electrode. EPSC amplitudes were normalized to the baseline immediately preceding each stimulation protocol and plotted separately (Fig. 5C,D). As illustrated, 2 μm Sp-cAMP in the recording electrode resulted in a weak, LFS-induced potentiation, followed by a strong, HFS-induced depression of excitatory inputs (Fig. 5C). In contrast, 20 μm Sp-cAMP supported strong potentiation and did not support HFS-induced depression (Fig. 5D). To assess changes in postsynaptic AMPA receptor expression, we measured the AMPA/NMDA ratio. Under both 2 and 20 μm Sp-cAMP conditions, LFS-induced potentiation of EPSC amplitudes was associated with an increase in the AMPA/NMDA ratio, which is consistent with increased postsynaptic expression of AMPA receptors and LTP induction (Fig. 5C,D, panel 3). This change persisted after administration of HFS, suggesting that HFS-induced synaptic depression does not alter AMPA receptor expression. Under conditions that support only LTD (2 μm Sp-cAMP and HFS), when the AMPA/NMDA ratio was tested immediately before the HFS and then 20 min later, there was a trend toward an increase in the ratio that was not statistically significant (data not shown; n = 7 cells, t(6) = −1.82, p = 0.12). The lack of a decrease in this ratio supports the idea that HFS-induced LTD is predominantly a presynaptic phenomenon that does not alter postsynaptic AMPA receptor expression.
Figure 5.

cAMP modulation of HFS- and LFS-induced corticostriatal plasticity in the same D2-expressing MSNs. Shown is a single-cell example of EPSC amplitudes recorded after LFS and HFS with 2 μm (A) or 20 μm (B) Sp-cAMP administered via the recording pipette. Inset, Representative traces before (solid line) and after (dashed line) LFS/HFS. Scale bar, 100 pA, 10 ms. Note that LFS-induced potentiation data (C1, D1) are the same results presented in Figure 3, B and C. C1, LFS induces a weak potentiation with 2 μm Sp-cAMP in the recording pipette. C2, HFS stimulation, administered 10 min after the LFS, induces robust LTD (n = 5 cells/slices, t(4) = 6.66, p = 0.003). C3, Average AMPA/NMDA ratio 21 min from the start of the recording (t(4) = −3.56, p = 0.02). Inset, Representative traces of AMPA and NMDA EPSC. Scale bar 50 pA, 10 ms. Vertical dotted lines indicate the sampling window after the decay of the AMPA receptor-mediated EPSC, when the NMDA EPSC amplitude was assessed. D1, With 20 μm Sp-cAMP in the recording pipette, LFS induces a robust potentiation. D2, Subsequent HFS has no effect of EPSC amplitude (n = 7 cells/slices, t(6) = −0.03, p = 0.97). D3, This potentiation of EPSC amplitude is accompanied by an increase in the AMPA/NMDA ratio (21 min after the beginning of the recording; t(6) = −2.60 p = 0.04).
These data emphasize the independent mechanisms underlying these two forms of plasticity. Under these conditions, LTD induction is unaffected by the prior potentiation, because the magnitude of depression is similar with and without preceding LFS (cf. Fig. 2A,C). In contrast, under 20 μm Sp-cAMP conditions, LFS induced a robust potentiation of synaptic transmission and a large increase in the AMPA/NMDA ratio (Fig. 5D), whereas subsequent HFS did not induce LTD (Fig. 5D). The data demonstrate that both potentiation and depression can occur within the same neuron and that they do not reverse each other mechanistically. In addition, both cAMP signaling and the activity of excitatory inputs will favor one form of plasticity over the other—that is, conditions that favor potentiation suppress LTD induction and vice versa.
Discussion
The striatopallidal or D2 pathway is thought to be principally involved in the motor deficits associated with the hypodopamine conditions in PD (Penney and Young, 1986; Albin et al., 1989; DeLong, 1990; Mink, 1996). Recently published studies suggest that aberrant corticostriatal potentiation in the striatopallidal pathway in the absence of dopamine contributes significantly to motor impairments in PD, whereas dopamine replacement restores both LTD in this pathway and motor performance (Beeler et al., 2010; Wiecki and Frank, 2010; Beeler et al., 2012). Both of these processes are experience dependent. Previous studies suggest that bidirectional corticostriatal plasticity in the striatopallidal pathway could be modulated by both dopamine signaling and excitatory input strength. To test this, we manipulated cAMP levels via the recording electrode and revealed that HFS-induced LTD occurs most prominently within a low concentration range (Table 1). This form of plasticity was lost when the cAMP levels were higher. This is in agreement with our earlier studies (Kheirbek et al., 2009), as well as published studies by others (Lerner and Kreitzer, 2012). These findings suggest that the high prevalence and magnitude of LTD seen in the presence of the D2 receptor agonist quinpirole is mediated by suppression of cAMP to permissive levels in the presence of strong excitatory input. This is further supported by the observation that quinpirole still facilitates HFS-induced LTD in the presence of a PLC inhibitor, as reported previously (Lerner and Kreitzer, 2012).
Table 1.
Summary of average changes in synaptic strength
| Treatment | % Baseline | n | p |
|---|---|---|---|
| HFS | LTD | ||
| Control | 76.7 ± 6.3 | 12 | 0.006* |
| Quinpirole (10 μm) | 68.5 ± 4.0 | 6 | 0.0002* |
| U73122 (10 μm), quinpirole (10 μm) | 73.7 ± 5.2 | 6 | 0.023* |
| Sulpiride (10 μm) | 98.1 ± 3.4 | 6 | 0.93 |
| Sp-cAMP (2 μm) | 79.8 ± 7.4 | 6 | 0.04* |
| Sp-cAMP (20 μm) | 83.2 ± 11.3 | 5 | 0.43 |
| Sp-cAMP (500 μm) | 97.0 ± 7.7 | 5 | 0.73 |
| Rp-cAMP (100 μm) | 98.3 ± 12.3 | 8 | 0.55 |
| LFS | Potentiation | ||
| Sp-cAMP (2 μm) | 136.1 ± 18.4 | 5 | 0.13 |
| Sp-cAMP (20 μm) | 227.6 ± 29.3 | 7 | 0.001* |
| Sp-cAMP (500 μm) | 149.3 ± 30.2 | 5 | 0.15 |
| Rp-cAMP (100 μm) | 220.2 ± 17.1 | 6 | 0.01* |
| LTP | |||
| No Sp-cAMP | 104.2 ± 10.4 | 5 | 0.30 |
| Sp-cAMP (20 μm) | 176.7 ± 43.9 | 6 | 0.04* |
| Sp-cAMP (20 μm), no stimulation | 89.4 ± 10.0 | 5 | 0.58 |
| Sp-cAMP (20 μm), depolarization only | 124.8 ± 17.6 | 5 | 0.06 |
| Sp-cAMP (20 μm) + d-APV | 87.5 ± 11.2 | 5 | 0.39 |
The treatment conditions and procedures for assessing changes in synaptic strength are described in the Materials and Methods.
*p < 0.05.
In contrast to the widely observed LTD associated with HFS in striatopallidal MSNs, applying LFS paired with postsynaptic depolarization induces LTP. This LTP is NMDA receptor dependent, which is consistent with previous studies (Calabresi et al., 1992b; Dang et al., 2006; Flajolet et al., 2008; Shen et al., 2008; Fino et al., 2010), and is the strongest at intracellular cAMP concentrations outside the range that supports LTD (Table 1). Conversely, the concentration that produced the weakest LFS-induced LTP also produced the strongest HFS-induced LTD. This reduced potentiation was not due to a competing depression because blocking LTP with an NMDA receptor antagonist did not unmask any underlying LTD. These data demonstrate that, under permissive cAMP conditions and LFS, striatopallidal LTP can be induced in slice preparations under whole-cell recording conditions without the use of Mg2+ free solutions (Calabresi et al., 1992b) or neurotrophic factors (Flajolet et al., 2008; Jia et al., 2010), suggesting that intracellular cAMP and the frequency of afferent stimulation are critical determinants for corticostriatal plasticity in the striatopallidal pathway.
GABA transmission contributes to striatal neural networks, which in turn can affect the direction of excitatory synaptic plasticity (Fino et al., 2005). We did not assess the impact of GABA transmission on cAMP modulation of synaptic plasticity because GABAA receptors were inhibited in all of our recordings. We expect that, along with the suppression of excitability, GABA modulation of the valence and magnitude of plasticity is also related to its effects on cAMP levels, which is a topic for future investigations.
Our data show that D2-expressing MSNs can exhibit an LFS-induced potentiation followed by an HFS-induced LTD in the same neuron. Moreover, we observed that the LTP-associated increase in AMPA/NMDA ratios remained elevated after LTD induction. This dual direction of plasticity was most obvious at 2 μm Sp-cAMP and was not observed under cAMP concentrations that favored strong LTP induction. Consistent with the widespread view that corticostriatal LTP and LTD are mediated by postsynaptic and presynaptic mechanisms, respectively (Choi and Lovinger, 1997; Gerdeman et al., 2002; Ronesi et al., 2004), these data support the idea that these two forms of plasticity are not mutually exclusive and coexist within the same cell, as was speculated recently (Lovinger, 2010). Although these two types of plasticity functionally oppose one another, our data suggest that potentiation and depression do not necessarily reverse the physiological changes induced by each. Whereas other processes such as depotentiation (Picconi et al., 2003) and dedepression may reverse LTP and LTD, the conditions for those processes will be an important subject for future studies.
Our studies demonstrate both cAMP signaling and the intensity of excitatory inputs will favor one form of plasticity over the other—that is, conditions that favor potentiation suppress LTD induction and vice versa. Because cAMP is a cardinal subcellular effector for dopamine signaling, these observations are consistent with the idea that dopamine signaling through the cAMP pathway regulates the direction of striatal plasticity (Reynolds and Wickens, 2002; Calabresi et al., 2007). High concentrations of dopamine that reduce cAMP via D2 activation facilitate LTD in the striatopallidal pathway (Calabresi et al., 1995; Ochi et al., 1995). In contrast, low dopamine levels reduce D2 activation, increasing intracellular cAMP and favoring LTP, as suggested here. The “three factor rule” of striatal plasticity states that synaptic strength is regulated by presynaptic and postsynaptic activity in combination with dopamine (Calabresi et al., 1992a,1992b; Lovinger et al., 1993; Walsh, 1993; Reynolds and Wickens, 2002). Our findings elaborate upon this, suggesting that intracellular cAMP is a key mediator of the dopamine regulation of plasticity in response to excitatory input strength.
In many brain areas, strong activation of excitatory synapses leads to potentiation and weaker stimulation induces depression (Malenka and Bear, 2004; Jörntell and Hansel, 2006). In striatum, this relationship is less clear. Within the striatopallidal pathway, it has been amply demonstrated that HFS yields synaptic depression (Calabresi et al., 1992a; Lovinger et al., 1993; Pawlak and Kerr, 2008). Our finding that LFS favors potentiation suggests that the relationship between the direction of synaptic plasticity and the strength of afferent activity is inverted within the striatopallidal pathway and is also regulated by cAMP. At other synapses in the brain, mGluRs located outside the synapse have been shown to contribute to input-specific induction of synaptic plasticity (Lu et al., 1997; Anwyl, 1999; Bellone et al., 2008; Le Vasseur et al., 2008). Striatal MSNs express mGluR5 receptors (Testa et al., 1994; Pin and Duvoisin, 1995; Kerner et al., 1997) and it has been reported that activation of mGluR5 receptors contributes significantly to HFS-induced LTD in striatal MSNs (Sung et al., 2001; Kreitzer and Malenka, 2005). Consistent with this idea, the prevalence of LTP increases when mGluR activity is inhibited (Dos Santos Villar and Walsh, 1999). It is likely that HFS induces sufficient glutamate release to activate extrasynaptic mGluR5 receptors and that LFS will not. Further studies are needed to identify the possible contribution of mGluR5 activity to LTP induction at these synapses.
The aberrant learning hypothesis suggests that dopamine denervation induces inappropriate inhibitory learning that contributes to progressive deterioration of motor function in PD (Beeler et al., 2010; Wiecki and Frank, 2010; Beeler, 2011; Beeler et al., 2012; Zhuang et al., 2013). Although the underlying cellular mechanisms remain to be elucidated, the present study suggests that alterations in intracellular cAMP signaling secondary to dopamine denervation or blockade may induce a shift in bidirectional regulation of synaptic plasticity at striatopallidal synapses. By maintaining intracellular cAMP outside concentrations permissive for LTD, decreased dopamine tone inappropriately favors LTP and inhibitory learning. The mechanisms underlying this aberrant learning and plasticity may present promising targets for the development of more effective treatments for PD.
Footnotes
This work was supported by the National Institutes of Health (Grant DA019695 to S.M.A., Grant DA25875 to J.A.B., Grant DA07255 to D.S.M., and Grant NS083383 to X.Z.).
The authors declare no competing financial interests.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/S0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Bagetta V, Picconi B, Marinucci S, Sgobio C, Pendolino V, Ghiglieri V, Fusco FR, Giampà C, Calabresi P. Dopamine-dependent long-term depression is expressed in striatal spiny neurons of both direct and indirect pathways: implications for Parkinson's disease. J Neurosci. 2011;31:12513–12522. doi: 10.1523/JNEUROSCI.2236-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA. Preservation of function in Parkinson's disease: what's learning got to do with it? Brain Res. 2011;1423:96–113. doi: 10.1016/j.brainres.2011.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Ding Y, Koranda J, Murakami M, Kang UJ, Zhuang X. Dopamine-dependent motor learning: insight into levodopa's long-duration response. Ann Neurol. 2010;67:639–647. doi: 10.1002/ana.21947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Frank MJ, McDaid J, Alexander E, Turkson S, Bernandez MS, McGehee DS, Zhuang X. A role for dopamine-mediated learning in the pathophysiology and treatment of Parkinson's disease. Cell Rep. 2012;2:1747–1761. doi: 10.1016/j.celrep.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C, Mameli M. Mechanisms of synaptic depression triggered by metabotropic glutamate receptors. Cell Mol Life Sci. 2008;65:2913–2923. doi: 10.1007/s00018-008-8263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992a;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci. 1992b;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Fedele E, Pisani A, Fontana G, Mercuri NB, Bernardi G, Raiteri M. Transmitter release associated with long-term synaptic depression in rat corticostriatal slices. Eur J Neurosci. 1995;7:1889–1894. doi: 10.1111/j.1460-9568.1995.tb00710.x. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–493. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Choi S, Lovinger DM. Decreased probability of neurotransmitter release underlies striatal long-term depression and postnatal development of corticostriatal synapses. Proc Natl Acad Sci U S A. 1997;94:2665–2670. doi: 10.1073/pnas.94.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa RM, Cohen D, Nicolelis MA. Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr Biol. 2004;14:1124–1134. doi: 10.1016/j.cub.2004.06.053. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-V. [DOI] [PubMed] [Google Scholar]
- Dos Santos Villar F, Walsh JP. Modulation of long-term synaptic plasticity at excitatory striatal synapses. Neuroscience. 1999;90:1031–1041. doi: 10.1016/S0306-4522(98)00504-1. [DOI] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, Wallach I, Nairn AC, Surmeier DJ, Greengard P. FGF acts as a co-transmitter through adenosine A(2A) receptor to regulate synaptic plasticity. Nat Neurosci. 2008;11:1402–1409. doi: 10.1038/nn.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall D, Prestori F, Sola E, D'Errico A, Roussel C, Forti L, Rossi P, D'Angelo E. Intracellular calcium regulation by burst discharge determines bidirectional long-term synaptic plasticity at the cerebellum input stage. J Neurosci. 2005;25:4813–4822. doi: 10.1523/JNEUROSCI.0410-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Guillou JL, Nakata H, Cooper DM. Inhibition by calcium of mammalian adenylyl cyclases. J Biol Chem. 1999;274:35539–35545. doi: 10.1074/jbc.274.50.35539. [DOI] [PubMed] [Google Scholar]
- Jia Y, Gall CM, Lynch G. Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum. J Neurosci. 2010;30:14440–14445. doi: 10.1523/JNEUROSCI.3310-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H, Hansel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 2006;52:227–238. doi: 10.1016/j.neuron.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Kerner JA, Standaert DG, Penney JB, Jr, Young AB, Landwehrmeyer GB. Expression of group one metabotropic glutamate receptor subunit mRNAs in neurochemically identified neurons in the rat neostriatum, neocortex, and hippocampus. Brain Res Mol Brain Res. 1997;48:259–269. doi: 10.1016/S0169-328X(97)00102-2. [DOI] [PubMed] [Google Scholar]
- Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X. Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci. 2009;29:12115–12124. doi: 10.1523/JNEUROSCI.3343-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Vasseur M, Ran I, Lacaille JC. Selective induction of metabotropic glutamate receptor 1- and metabotropic glutamate receptor 5-dependent chemical long-term potentiation at oriens/alveus interneuron synapses of mouse hippocampus. Neuroscience. 2008;151:28–42. doi: 10.1016/j.neuroscience.2007.09.071. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol. 1993;70:1937–1949. doi: 10.1152/jn.1993.70.5.1937. [DOI] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol. 1996;50:381–425. doi: 10.1016/S0301-0082(96)00042-1. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Hang GB, Grueter BA, Pascoli V, Lüscher C, Malenka RC, Kreitzer AC. A comparison of striatal-dependent behaviors in wild-type and hemizygous Drd1a and Drd2 BAC transgenic mice. J Neurosci. 2012;32:9119–9123. doi: 10.1523/JNEUROSCI.0224-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Ochi M, Inoue H, Koizumi S, Shibata S, Watanabe S. Long-term enhancement of dopamine release by high frequency tetanic stimulation via a N-methyl-d-aspartate-receptor-mediated pathway in rat striatum. Neuroscience. 1995;66:29–36. doi: 10.1016/0306-4522(94)00559-N. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney JB, Jr, Young AB. Striatal inhomogeneities and basal ganglia function. Mov Disord. 1986;1:3–15. doi: 10.1002/mds.870010102. [DOI] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-G. [DOI] [PubMed] [Google Scholar]
- Pisani A, Centonze D, Bernardi G, Calabresi P. Striatal synaptic plasticity: implications for motor learning and Parkinson's disease. Mov Disord. 2005;20:395–402. doi: 10.1002/mds.20394. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Wickens JR. Dopamine-dependent plasticity of corticostriatal synapses. Neural Netw. 2002;15:507–521. doi: 10.1016/S0893-6080(02)00045-X. [DOI] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM. Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol. 2001;86:2405–2412. doi: 10.1152/jn.2001.86.5.2405. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JP. Depression of excitatory synaptic input in rat striatal neurons. Brain Res. 1993;608:123–128. doi: 10.1016/0006-8993(93)90782-I. [DOI] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wiecki TV, Frank MJ. Neurocomputational models of motor and cognitive deficits in Parkinson's disease. Prog Brain Res. 2010;183:275–297. doi: 10.1016/S0079-6123(10)83014-6. [DOI] [PubMed] [Google Scholar]
- Wiecki TV, Riedinger K, von Ameln-Mayerhofer A, Schmidt WJ, Frank MJ. A neurocomputational account of catalepsy sensitization induced by D2 receptor blockade in rats: context dependency, extinction, and renewal. Psychopharmacology (Berl) 2009;204:265–277. doi: 10.1007/s00213-008-1457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–798. doi: 10.1016/S0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MRF, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat Rev Neurol. 2013;9:248–256. doi: 10.1038/nrneurol.2013.57. [DOI] [PubMed] [Google Scholar]