

Abstract
Radical S-adenosyl-L-methionine (SAM) enzymes are a large and diverse superfamily with functions ranging from enzyme activation through a single H atom abstraction to complex organic and metal cofactor synthesis involving a series of steps. Though these enzymes carry out a variety of functions, they share common structural and mechanistic characteristics. All of them contain a site-differentiated [4Fe-4S] cluster, ligated by a CX3CX2C or similar motif, which binds SAM at the unique iron. The [4Fe-4S]1+ state of the cluster reductively cleaves SAM to produce a 5’-deoxyadenosyl radical, which serves to initiate the diverse reactions catalyzed by these enzymes. Recent highlights in the understanding of radical SAM enzymes will be presented, with a particular emphasis on enzymes catalyzing methylation and methythiolation reactions.
Structure and function of radical SAM enzymes
Radical S-adenosylmethionine (SAM or AdoMet) enzymes are part of a rapidly growing and very large superfamily first identified in 2001.1 This superfamily contains thousands of enzymes, whose functionality is extremely diverse; known functions include assembly of complex cofactors, enzyme activation, methylation and methylthiolation, DNA repair, sulfur insertion, and tRNA modification as well as numerous others.2, 3 Radical SAM enzymes are characterized by a CX3CX2C motif or variations thereof, in which the Cys thiolates coordinate three irons of a [4Fe-4S]2+/1+ cluster. The fourth iron of the [4Fe-4S] cluster is coordinated by the amino and carboxy groups of SAM, and is generally referred to as the unique iron due to its distinct ligand environment.4, 5 The [4Fe-4S]1+ state of the cluster reductively cleaves SAM to create a 5’-deoxyadenosyl (dAdo) radical and methionine (Met).6 The dAdo radical abstracts a hydrogen atom from the substrate to form a substrate-based radical that functions to carry out specific reactions dependent on each individual enzyme.
Structurally characterized members of the superfamily reveal a conserved core domain consisting of a full (α/β)8 or partial (α/β)6 TIM barrel fold (Figure 1).7, 8 The variance of the TIM barrel completeness and openness correlates with the substrate size, with the less complete, more open barrels present in enzymes acting upon larger substrates. Within this barrel the [4Fe-4S] cluster is located on a loop containing the CX3CX2C motif at the N-terminal end of the core domain. The opening of the barrel that provides access to the active site can be closed off by other protein components or the substrate, allowing for a more controlled reaction of the radical(s) produced during turnover.
Figure 1.

Representative structures of Radical SAM enzymes. Left: Structure of the pyruvate formate-lyase activating enzyme (PFL-AE), which acts on a 170 kDa protein substrate and has a partial (α/β)6 TIM barrel fold (PDB ID 3CB8). Right: Structure of biotin synthase (BioB), which acts on a small molecule substrate (dethiobiotin) and has a complete (α/β)8 TIM barrel fold (PDB ID 1R30)
Apart from this core domain, radical SAM enzymes can also contain additional elements that aid in their function. In some instances the enzyme contains a second binding motif for an additional Fe-S cluster. A second cluster is usually found in radical SAM enzymes involved in sulfur insertion (Figure 1, right).9-13 Evidence suggests that this second cluster is in fact the source of the sulfur atom incorporated into the substrate. Secondary clusters could also be used to transfer electrons. Radical SAM enzymes that modify RNA also have a supplementary domain at either the N- or C-terminus.14, 15 This domain is thought to be necessary for substrate recognition and intereaction.14, 15
Regioselectivity of S-C bond cleavage
Although there are three S-C bonds in SAM, one each to the methyl, the methionine γ-carbon, and the 5’-carbon, until recently it appeared that radical SAM enzymes catalyzed the reductive cleavage of only the S-C(5’) bond of SAM, and subsequently exploited the 5’-deoxyadenosyl radical for direct H atom abstraction (Table 1). Evidence for the involvement of the 5’-dAdo radical includes, in a number of enzymes, the stoichiometric production of 5’-deoxyadenosine and methionine as products of turnover16-20, label transfer from substrate into 5’-dAdo19-27, and the characterization by EPR of a stabilized allylic 5’-dAdo radical intermediate formed when the SAM analog 3’,4’-anhydro-SAM (anSAM) was used in place of SAM in the reaction of lysine 2,3-aminomutase28, 29. Evidence has been mounting, however, for the involvement of alternate S-C cleavage events upon the enzymatic reductive cleavage of SAM in certain enzymes.
Table 1.
S-adenosylmethionine (SAM), left, has three S-C bonds. The radical produced upon homolytic cleavage of each of these bonds is indicated (with A representing adenosine), as are representative enzymes where known.
| S-Adenosylmethionine | Bord Cleaved | Radical Produced | Enzyme Examples |
|---|---|---|---|
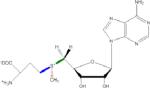
|
|
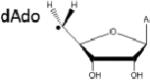
|
Radical SAM enzymes: Mtases MTTases |

|

|
Dph2 GD-AE | |
|
|
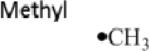
|
None Known |
In Dph2 from Pyrococcus horikoshii, which performs the first step in diphthamide biosynthesis by addition of an 3-amino-3-carboxypropyl (ACP) group to a specific histidine residue on the translational elongation factor 2 (EF2), reductive cleavage of SAM by an iron-sulfur cluster leads to cleavage of the bond between the sulfur and the methionine γ-carbon (Table 1). This cleavage results in formation of an ACP radical and methylthioadenosine (MTA). Dph2 does not contain the canonical radical SAM cysteine motif; rather, the protein is a homodimer with each monomer containing three conserved cysteines (C59, C163, and C287), each from a different domain, capable of binding a [4Fe-4S] cluster.30 The reduced [4Fe-4S]1+ state of the cluster cleaves SAM homolytically between the sulfur and methionine γ-carbon to form the ACP radical.31 The ACP radical does not mediate an H-atom abstraction, however, and instead attacks C-2 on the imidazole ring of the modified histidine residue resulting in addition to the ring.30
Evidence suggests that the B12-independent glycerol dehydratase activating enzyme (GD-AE) also catalyzes cleavage of the S-C(γ) bond rather than the S-C(5’) bond.32 GD-AE is part of a subset of radical SAM enzymes that activate glycyl radical enzymes (GREs) by H atom abstraction from a specific glycine residue of their respective substrates.33, 34 When investigating the activation of glycerol dehydratase (GD), it was discovered that instead of production of dAdo and Met, the products of SAM cleavage were MTA and 2-aminobutyrate, implicating the involvement of the ACP radical. 32
The selective cleavage of SAM between either the sulfur and 5’-carbon bond or the sulfur and methionine γ-carbon bond may be related to the alignment of the Fe-S cluster and bond to be broken.35, 36 One proposal by Kampmeier suggests that the cleavage can be thought of as a displacement reaction at the sulfonium with the unique Fe of the [4Fe-4S] cluster taking the place of the S-C bond. This would require the unique Fe as well as the S-C bond on SAM to be broken to be collinear.36 The crystal structures of several radical SAM enzymes which cleave SAM to produce dAdo and Met support this theory with the observed geometry of SAM bound in the active site.37-41 However, it remains to be seen whether the enzymes that utilize the ACP radical bind SAM in an alternate fashion as would be necessary for the Fe and S-C(γ) bond broken to be colinear. The idea that the type of radical produced is related to the alignment of SAM with the [4Fe-4S] cluster is intriguing, and further structural studies of enzymes that use SAM in different fashions will help to either confirm this hypothesis or give new understanding into how an alternate radical could be produced.
Radical SAM methylation
It has long been suspected that the radical SAM superfamily contains methylases (MTases), but it is only recently that two have begun to be characterized. The bacterial enzymes RlmN, which is endogenous and thought to be important in ribosome function, and Cfr, which is acquired and confers antibiotic resistance, are methyltransferases that act upon A2503 in 23S rRNA. RlmN methylates the C2 position42, while Cfr methylates C8 preferentially but can also act upon C2.43, 44 Both of these enzymes contain the typical CX3CX2C motif that binds a [4Fe-4S] cluster. Both enzymes also utilize two SAM molecules; one is reductively cleaved to form the dAdo radical intermediate, and the second is used as the source of the methyl group.44
The mechanism by which A2503 is methylated by RlmN has recently been explored using isotopic labels. Using S-adenosyl-L-[methyl-3H]methionine it was observed that the tritium label was incorporated into 23S rRNA, indicating that the methyl group is provided by SAM; production of dAdo, Met, and S-adenosylhomocysteine (SAH or AdoHcy) during the reaction was also detected.44 From these results a mechanism was put forth in which SAM is cleaved to form the dAdo radical.44 This radical then abstracts the C2 H from A2503 to generate a substrate radical that is then methylated by a second molecule of SAM.44 This proposal was further revised when the use of A2503 labeled with deuterium at C2 showed no incorporation of deuterium into dAdo.27 When SAM deuterated at the methyl position (d3-SAM) was used in the reaction, however, the C2 of A2503 was doubly deuterated while dAdo was mono-deuterated.27 These results led to a proposal in which the dAdo radical abstracts a hydrogen atom from the methyl group of the second molecule of SAM, resulting in a mono deuterated dAdo molecule and a SAM-methyl radical.27 This SAM-methyl radical was then proposed to attach to C2 of A2503 with a hydride shift from C2 to the added methyl group giving a doubly deuterated product.27
However, another report published at the same time indicated that the hydrogen abstracted was neither from the substrate A2503 nor SAM but rather from a modified cysteine residue.26 Use of d3-SAM in single-turnover conditions showed no deuterium incorporation into A2503, and when RlmN was expressed and purified from methionine auxotrophic E. coli supplemented with d3-Met, which also contains deuterium at the methyl position, deuterium was detected in the methylated adenosine substrate as well as dAdo.26 This indicated that the hydrogen abstracted probably came from a protein bound methyl group, and mass spectrometry on wild-type RlmN revealed that Cys355 contains the methyl modification.26 The proposed mechanism based on these results begins with the methylation of Cys355 by the first SAM molecule through a nucleophilic substitution typical of other SAM methylations. The second molecule of SAM is reductively cleaved by the reduced Fe-S cluster to produce the dAdo radical which abstracts one of the hydrogens from the methyl group just added to Cys355. This new methyl radical is what then attacks C2 on A2503; the product is finally released when Cys118, as a thiolate, attacks Cys355 forming a disulfide bond (Figure 2, left).26
Figure 2.

Left: Mechanism for the second step in methylthiolation as proposed by Grove et al.45 The methylated cysteine (S355) was generated by SAM dependent methylation in the first step. Right: Structure of RlmN as determined by x-ray crystallography with the N-terminal domain in dark purple and the C-terminal extension in blue (PDB ID 3RFA).
This mechanism is further supported by additional results from Grove et. al.45; they demonstrated that all five cysteines, the three cluster ligands as well as two additional cysteines, are necessary for enzyme functionality. Interestingly, RlmN lacking a [4Fe-4S] cluster does not contain the methylated cysteine modification, which is only restored with the addition of SAM as well as the reconstitution of the cluster. This indicates that the cluster participates in both reductive SAM cleavage as well as the methylation of Cys355.45
The proposed mechanism of Cfr is similar to RlmN26, which is not surprising as Cfr appears to have evolved from RlmN46. However, the ability of Cfr to methylate not only C8, the preferential site, but also C2 of A2503 suggests that there is a difference if not in mechanism then at least in substrate recognition and binding.
The structure of RlmN was just recently solved using x-ray crystallography and showed that, similar to other radical SAM enzymes, RlmN contains a partial (α/β)6 TIM barrel with the [4Fe-4S] cluster ligated at the carboxy end of the barrel (Figure 2, right). In addition to this core radical SAM domain, RlmN contains an N-terminal domain of sixty residues in four short α-helices arranged in a variation of the HhH2 fold, which is usually involved in substrate recognition of nucleic acids47, 48; it is attached to the core by three β strands (β’1-3), which expand the barrel laterally. On the C-terminal end another extension consists of a β strand (β7), which reaches across the barrel opening and terminates in an α helix. These two additional β strand extensions are mechanistically important; the β7 strand contains the catalytically active Cys355 residue, which is methylated in the crystal structure, and positions it within 3.3 Å of the SAM methyl group and the β’1-3 strand contains the other cysteine, Cys118, which is thought to be necessary for completion of the catalytic cycle by releasing the newly methylated substrate.14 The structure of RlmN thus provides further support for the mechanism proposed by Grove et al. as illustrated in Figure 2.14, 26, 45
Radical SAM methylthiolation
There are only five known naturally occurring methylthio modifications, which occur on different components of the translation machinery. Four of these occur on tRNA, all on adenosine, while the fifth occurs on a small ribosomal protein (S12) at an aspartate residue. The function of the methylthio groups is not completely known, however some insight can be gained from the positioning of the modifications within the translation machinery. The tRNA contains the methylthio group at the 3’ nucleotide (A-37) immediately following the anticodon and the modification on aspartate 89 of S12 projects into the acceptor site of the ribosome; these locations for the modifications implicate a role in codon-anticodon stability and translational efficiency and fidelity.49-53
The first methylthiotransferase (MTTase) to be characterized was MiaB54, which converts i6A to ms2i6A; the time between the characterization of MiaB and the next MTTase was rather long, however study of this subclass of radical SAM enzymes has gained momentum in the past few years. Since the initial report on MiaB, two other MTTases have been identified. RimO is responsible for modifying the ribosomal protein55, and MtaB converts t6A to ms2t6A.56 What is interesting about this subclass of radical SAM enzymes is the bifunctional nature of MTTases, combining sulfur insertion with methylation.57 Also, in addition to the radical SAM Fe-S cluster coordinated by the CX3CX2C motif, MTTases contain a second [4Fe-4S] cluster coordinated by three other conserved cysteines that is essential for function.11
Because of the two different functions a single MTTase carries out, it has been difficult to ascertain a complete mechanism. However, what has been gleaned from biochemical studies has provided answers to some aspects of the methylthiolation process. Like radical SAM methyltransferases, MTTases require two molecules of SAM per turnover, one for dAdo radical production and one to provide the methyl group. The observed products from these two uses of SAM are methionine and dAdo from SAM cleavage and SAH from methylation. It is thought, although not biochemically shown for certain, that the second Fe-S cluster is the source of the inserted sulfur, as is also thought to be the case in other radical SAM enzymes that catalyze sulfur insertion.9-13
These observations coupled with an understanding of radical SAM reactions in general have led to mechanistic proposals for MTTases (Figure 3, left). Two of these mechanisms, using RimO as a model, involve a first step of reductive cleavage of SAM to form the dAdo radical, which then abstracts a hydrogen atom from the β-carbon of Asp89 on S12.12 The substrate radical then attacks a μ-sulfido bridging ligand of the non-radical SAM Fe-S cluster. At this point the two mechanisms diverge; in one pathway, which is believed to be the more likely, the second molecule of SAM methylates the sulfur to which the Asp residue is attached causing a release of the methylthiolated Asp from the cluster.12 In the alternate pathway the Asp bound cluster degrades forming a thiolated Asp intermediate.12 It is this intermediate that is then methylated by the second molecule of SAM. A third possibility exists in which a sulfur on the secondary Fe-S cluster is methylated by one SAM molecule. The methylated sulfur would then be attacked by the Asp radical generated by the other molecule of SAM, releasing the final product.12
Figure 3.

Left: Two proposed mechanisms for radical SAM methylthiolation.12 Right: Structure of apo-RimO as determined by x-ray crystallography, with only the radical SAM (purple and maroon) and TRAM (blue) domains visible (PDB ID 2QGQ).
In order to carry out their reactions MTTases contain three domains, an N-terminal UPF0004 domain, a central radical SAM domain, and a C-terminal TRAM domain.55 The UPF domain contains three of the conserved cysteines that in MiaB ligate one of the Fe-S clusters.11 This domain is also most commonly found with the other two present in MTTases. The radical SAM domain contains the typical CX3CX2C motif ligating the other Fe-S cluster. The TRAM domain, which is only found in radical SAM enzymes of the MTTase subclass, is thought to be involved in substrate recognition.55
The structures of two of these domains, radical SAM and TRAM, were recently revealed in a crystal structure of RimO from Thermatoga maritima (Figure 3, right).15 The structure was obtained for the apo-enzyme crystals prepared with the use of subtilisin, which presumably cleaved the UPF domain from the rest of the protein; however, from the position of the N-terminus of the radical SAM domain, it can be postulated that the UPF domain can interact with the radical SAM Fe-S cluster as modeled from other radical SAM structures.15 The radical SAM domain includes a partial (α/β)6 TIM barrel, and a superposition of RimO with the radical SAM enzyme MoaA (molybdenum cofactor biosynthesis) allowed the likely positions of the radical SAM Fe-S cluster as well as SAM to be determined.15 The location of the TRAM domain is at the distal edge of the concave surface of the radical SAM domain allowing access of the macromolecular substrates to the active site while still closely interacting with them. In RimO the putative surface of substrate binding is negatively charged, complementing the positively charged substrate, the ribosomal S12 protein.15 While these results give an insight into MTTases, a better understanding of the mechanism and the role of the second Fe-S cluster will be possible once the structure of the holo-enzyme has been determined.
Conclusions
Methyltransferases and methylthiotransferases are two subclasses of an extensive superfamily of radical SAM enzymes. These two, however, are unique and puzzling in their ability to use two molecules of SAM in two distinct capacities: as a methyl group donor and as the source of the dAdo radical. Because of the dual roles of SAM, two adenosyl products, SAH and dAdo, are formed. Deoxyadenosine is known to inhibit radical SAM enzymes58, and SAH, while not directly studied in the radical SAM MTases, would likely result in a similar inhibition as this is the case with MTase enzymes that utilize a direct SN2 methyl substitution.59 Further, evidence suggests that in vivo ratios of SAM to SAH, sometimes referred to as the methylation index, affects the activities of MTases.60, 61 An interesting and as-yet unexplored question is how the methylation index affects the activities of radical SAM enzymes, particularly those involved in methylation and methylthiolation.
For the MTases it is interesting that the dAdo radical abstracts a hydrogen atom from the methyl group of the second SAM molecule after the methyl has been added to a conserved cysteine. Another fascinating implication that deserves further investigation is the role of the radical SAM [4Fe-4S] cluster in methylation of the cysteine residue. To date no other radical SAM enzymes show a secondary role for their dAdo radical producing clusters. The dual role for the radical SAM cluster adds yet another complexity to the ability of radical SAM enzymes to control difficult and sensitive radical chemistry.
The presence of two Fe-S clusters in the MTTases is not surprising, as radical SAM enzymes involved in sulfur insertion all have additional clusters that are thought to be the source of the sulfur. A mechanistic anomaly within this subgroup concerns tRNA modification: direct hydrogen atom abstraction from the substrate would be from an sp2 hybridized carbon for which there is no precedence in the radical SAM family. Further mechanistic studies using labeled substrates as well as a crystal structure of a holo-MTTase could help to illuminate how MTTases use the second Fe-S cluster as well as whether the sp2 hydrogen is in fact the one abstracted. Further insights into the biological significance of the methylthio modifications that these enzymes impart are being revealed. Just recently the eukaryotic MTTase Cdkal1 was linked to the development of type 2 diabetes. Cdkal1 synthesizes ms2t6A in tRNALys (UUU); mice pancreatic cells that were deficient in this enzyme misread the Lys codon in proinsulin, which resulted in a reduction of glucose-stimulated proinsulin synthesis.62
Radical SAM enzymes catalyze a remarkably diverse range of chemical reactions, all of which are initiated by the reductive homolytic cleavage of an S-C bond of SAM, in order to generate a carbon-centered radical intermediate. Most radical SAM enzymes characterized to date appear to cleave only the S-C(5’) bond to generate the 5’-dAdo radical intermediate, although two enzymes have been recently shown to catalyze reductive cleavage of the S-C(γ) bond to generate the alternate 3-amino-3-carboxypropyl (ACP) radical.31, 32 Although reductive homolytic cleavage of the third S-C bond of SAM to generate a methyl radical has not been demonstrated, the MTases and MTTases discussed in this review do cleave (presumably in a nucleophilic, heterolytic manner) the S-C(methyl) bond of SAM to provide the methyl group required for the reaction.44, 57 These enzymes thus represent fascinating examples of the dual use of SAM (as a methyl donor and a radical precursor) in a single enzyme.
Acknowledgements
Research on radical SAM enzymes in the Broderick lab is supported by the National Institutes of Health grant GM54608 and the Department of Energy Grant DE-FG02-10ER16194. The Astrobiology Biogeocatalysis Research Center is supported by NASA (NNA08C-N85A).
Notes and references
- 1.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shepard EM, Broderick JB. In: Comprehensive Natural Products II: Chemistry and Biology. Mander L, Liu H-W, editors. Vol. 8. Elsevier; Oxford: 2010. pp. 625–661. [Google Scholar]
- 3.Frey PA, Hegeman AD, Ruzicka FJ. Crit. Rev. Biochem. Mol. Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 4.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. J. Am. Chem. Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 5.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J. Am. Chem. Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 6.Henshaw TF, Cheek J, Broderick JB. J. Am. Chem. Soc. 2000;122:8331–8332. [Google Scholar]
- 7.Vey JL, Drennan CL. Chem. Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dowling DP, Vey JL, Croft AK, Drennan CL. Biochim. Biophys. Acta. 2012 doi: 10.1016/j.bbapap.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2001;40:8343–8351. doi: 10.1021/bi0104625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cicchillo RM, Lee K-H, Baleanu-Gogonea C, Nesbitt NM, Krebs C, Booker SJ. Biochemistry. 2004;43:11770–11781. doi: 10.1021/bi0488505. [DOI] [PubMed] [Google Scholar]
- 11.Hernández HL, Pierrel F, Elleingand E, Garcia-Serres R, Huynh BH, Johnson MK, Fontecave M, Atta M. Biochemistry. 2007;46:5140–5147. doi: 10.1021/bi7000449. [DOI] [PubMed] [Google Scholar]
- 12.Lee K-H, Saleh L, Anton BP, Madinger CL, Benner JS, Iwig DF, Roberts RJ, Krebs C, Booker SJ. Biochemistry. 2009;48:10162–10174. doi: 10.1021/bi900939w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arragain S, Handelman SK, Forouhar F, Wei F-Y, Tomizawa K, Hunt JF, Douki T, Fontecave M, Mulliez E, Atta M. J. Biol. Chem. 2010;285:28425–28433. doi: 10.1074/jbc.M110.106831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boal AK, Grove TL, McLaughlin MI, Yennawar NH, Booker SJ, Rosenzweig AC. Science. 2011 doi: 10.1126/science.1205358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arragain S, Garcia-Serres R, Blondin G, Douki T, Clemancey M, Latour J-M, Forouhar F, Neely H, Montelione GT, Hunt JF, Mulliez E, Fontecave M, Atta M. J. Biol. Chem. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guianvarc'h D, Florentin D, Bui BTS, Nunzi F, Marquet A. Biochem. Biophys. Res. Commun. 1997;236:402–406. doi: 10.1006/bbrc.1997.6952. [DOI] [PubMed] [Google Scholar]
- 17.Knappe J, Neugebauer FA, Blaschkowski HP, Gänzler M. Proc. Natl. Acad. Sci. U.S.A. 1984;81:1332–1335. doi: 10.1073/pnas.81.5.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruszczycky MW, Choi S.-h., Liu H.-w. J. Am. Chem. Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjdia A, Leprince J, Sandström C, Vaudry H, Berteau O. J. Am. Chem. Soc. 2009;131:8348–8349. doi: 10.1021/ja901571p. [DOI] [PubMed] [Google Scholar]
- 20.Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J. Am. Chem. Soc. 2007;129:15147–15155. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- 21.Cheek J, Broderick JB. J. Am. Chem. Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 22.Frey PA, Rothe M, Wagner AFV, Knappe J. J. Biol. Chem. 1994;269:12432–12437. [PubMed] [Google Scholar]
- 23.Escalettes F, Florentin D, Bui BTS, Lesage D, Marquet A. J. Am. Chem. Soc. 1999;121:3571–3578. [Google Scholar]
- 24.Kilgore JL, Aberhart DJ. J. Chem. Soc., Perkin Trans. 1991;1:79–84. [Google Scholar]
- 25.Gambarelli S, Luttringer F, Padovani D, Mulliez E, Fontecave M. Chembiochem : a European journal of chemical biology. 2005;6:1960–1962. doi: 10.1002/cbic.200500182. [DOI] [PubMed] [Google Scholar]
- 26.Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. Science. 2011;332:604–607. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- 27.Yan F, Fujimori DG. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3930–3934. doi: 10.1073/pnas.1017781108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnusson OT, Reed GH, Frey PA. J. Am. Chem. Soc. 1999;121:9764–9765. [Google Scholar]
- 29.Magnusson OT, Reed GH, Frey PA. Biochemistry. 2001;40:7773–7782. doi: 10.1021/bi0104569. [DOI] [PubMed] [Google Scholar]
- 30.Zhu X, Dzikovski B, Su X, Torelli AT, Zhang Y, Ealick SE, Freed JH, Lin H. Mol. Biosyst. 2011;7:74–81. doi: 10.1039/c0mb00076k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, Lin H. Nature. 2010;465:891–896. doi: 10.1038/nature09138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demick JM, Lanzilotta WN. Biochemistry. 2011;50:440–442. doi: 10.1021/bi101255e. [DOI] [PubMed] [Google Scholar]
- 33.Raynaud C, Sarçabal P, Meynial-Salles I, Croux C, Soucaille P. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5010–5015. doi: 10.1073/pnas.0734105100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien JR, Raynaud C, Croux C, Girbal L, Soucaille P, Lanzilotta WN. Biochemistry. 2004;43:4635–4645. doi: 10.1021/bi035930k. [DOI] [PubMed] [Google Scholar]
- 35.Broderick JB. Nature. 2010;465:877–878. doi: 10.1038/465877a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kampmeier JA. Biochemistry. 2010;49:10770–10772. doi: 10.1021/bi101509u. [DOI] [PubMed] [Google Scholar]
- 37.Layer G, Moser J, Heinz DW, Jahn D, Schubert W-D. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hänzelmann P, Schindelin H. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toh S-M, Xiong L, Bae T, Mankin AS. RNA. 2008;14:98–106. doi: 10.1261/rna.814408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giessing AMB, Jensen SS, Rasmussen A, Hansen LH, Gondela A, Long K, Vester B, Kirpekar F. RNA. 2009;15:327–336. doi: 10.1261/rna.1371409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan F, LaMarre JM, Rohrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. J. Am. Chem. Soc. 2010;132:3953–3964. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grove TL, Radle MI, Krebs C, Booker SJ. J. Am. Chem. Soc. 2011;133:19586–19589. doi: 10.1021/ja207327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaminska KH, Purta E, Hansen LH, Bujnicki JM, Vester B, Long KS. Nucleic Acids Res. 2010;38:1652–1663. doi: 10.1093/nar/gkp1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao X, Grishin NV. Nucleic Acids Res. 2000;28:2643–2650. doi: 10.1093/nar/28.14.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller DJ, Ouellette N, Evdokimova E, Savchenko A, Edwards A, Anderson WF. Protein Sci. 2003;12:1432–1442. doi: 10.1110/ps.0302403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Díaz I, Ehrenberg M. J. Mol. Biol. 1991;222:1161–1171. doi: 10.1016/0022-2836(91)90599-2. [DOI] [PubMed] [Google Scholar]
- 50.Gustilo EM, Vendeix FA, Agris PF. Curr. Opin. Microbiol. 2008;11:134–140. doi: 10.1016/j.mib.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaczanowska M, Rydén-Aulin M. Microbiol. Mol. Biol. Rev. 2007;71:477–494. doi: 10.1128/MMBR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konevega AL, Soboleva NG, Makhno VI, Semenkov YP, Wintermeyer W, Rodnina MV, Katunin VI. RNA. 2004;10:90–101. doi: 10.1261/rna.5142404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbonavičius J, Qian Q, Durand JMB, Hagervall TG, Björk GR. EMBO J. 2001;20:4863–4873. doi: 10.1093/emboj/20.17.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierrel F, Björk GR, Fontecave M, Atta M. J. Biol. Chem. 2002;277:13367–13370. doi: 10.1074/jbc.C100609200. [DOI] [PubMed] [Google Scholar]
- 55.Anton BP, Saleh L, Benner JS, Raleigh EA, Kasif S, Roberts RJ. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1826–1831. doi: 10.1073/pnas.0708608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anton BP, Russell SP, Vertrees J, Kasif S, Raleigh EA, Limbach PA, Roberts RJ. Nucleic Acids Res. 2010;38:6195–6205. doi: 10.1093/nar/gkq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pierrel F, Douki T, Fontecave M, Atta M. J. Biol. Chem. 2004;279:47555–47563. doi: 10.1074/jbc.M408562200. [DOI] [PubMed] [Google Scholar]
- 58.Challand MR, Ziegert T, Douglas P, Wood RJ, Kriek M, Shaw NM, Roach PL. FEBS Lett. 2009;583:1358–1362. doi: 10.1016/j.febslet.2009.03.044. [DOI] [PubMed] [Google Scholar]
- 59.Clarke S, Banfield K. In: Homocysteine in Health and Disease. Carmel R, Jacobsen DW, editors. Cambridge University Press; Cambridge, UK: 2001. pp. 63–78. [Google Scholar]
- 60.Zappia V, Zydek-Cwick CR, Schlenk F. J. Biol. Chem. 1969;244:4499–&. [PubMed] [Google Scholar]
- 61.Perna AF, Ingrosso D, Zappia V, Galletti P, Capasso G, De Santo NG. J. Clin. Invest. 1993;91:2497–2503. doi: 10.1172/JCI116485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei F-Y, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, Yamagata K, Suzuki T, Tomizawa K. J. Clin. Invest. 2011;121:3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]