

Abstract
Flavodoxin (Fld) conformational changes, thermal stability, and cofactor binding were studied using circular dichroism (CD), isothermal titration calorimetry (ITC), and limited proteolysis. Thermodynamics of apo and holo-Fld folding were examined to discern the features of this important electron transfer protein and to provide data on apo-Fld. With the exception of fluorescence and UV-vis binding experiments with its cofactor flavin mononucleotide (FMN), apo-Fld is almost completely uncharacterized in Escherichia coli. Fld is more structured when the FMN cofactor is bound; the association is tight and driven by enthalpy of binding. Surface plasmon resonance binding experiments were carried out under anaerobic conditions for both apo- and holo-Fld and demonstrate the importance of structure and conformation for interaction with binding partners. Holo-Fld is capable of associating with NADP+-dependent flavodoxin oxidoreductase (FNR) and pyruvate formate-lyase activating enzyme (PFL-AE) whereas there is no detectable interaction between apo-Fld and either protein. Limited proteolysis experiments were analyzed by LC-MS to identify the regions in Fld that are involved in conformations changes upon cofactor binding. Docking software was used to model the Fld/PFL-AE complex to understand the interactions between these two proteins and gain insight into electron transfer reactions from Fld to PFL-AE.
Keywords: flavodoxin, NADP+ oxidoreductase, pyruvate formate-lyase activating enzyme, protein-protein interactions, surface plasmon resonance (SPR), circular dichroism (CD), Isothermal titration calorimetry (ITC)
1.0 Introduction
Flavodoxins are small acidic proteins that utilize a flavin mononucleotide cofactor (FMN) for electron transfer reactions. Flavodoxins are widespread in bacteria and are present in some red and green algae [1-3]. In higher eukaryotes, proteins with sequence homology to flavodoxin and its reductase are fused to multidomain proteins as in methionine synthase reductase, which reductively activates methionine synthase [4-6]. Crystal structures are available for holo-Fld from Escherichia coli, however there is no structure for apo-Fld [7]. Fld binds the cofactor FMN with an equilibrium constant of 1 nM and a 1:1 stoichiometry, using multiple hydrogen bonds, salt bridges, and stacking interactions between the isoalloxazine ring and aromatic residues [7]. These interactions are extremely important for determining the reduction potentials of the FMN cofactor once bound to the protein [8, 9]. Flavodoxins are ideal electron donors for a number of biological redox reactions, including those catalyzed by the radical S-adenosylmethionine (SAM) superfamily enzymes due to their low reduction potentials. Several studies have shown that flavodoxin (Fld) is capable of activating pyruvate formate-lyase activating enzyme and anaerobic ribonucleotide reductase activating enzyme (PFL-AE and RNR-AE) as well as biotin synthase (BioB), presumably by reducing the [4Fe-4S] cluster that binds and reductively cleaves SAM [10-12]. The details of these interactions, however, are not well understood.
In addition to interacting with PFL-AE, RNR-AE, and BioB, Escherichia coli Fld serves as an electron transfer partner with NADP+-dependent flavodoxin reductase (FNR), methionine synthase, ferredoxin, pyruvate dependent flavodoxin oxidoreductase, and probably other unknown proteins, and thus plays integral roles in metabolism [10]. Residues involved in the binding site interface of Fld with SAM binding domain of methionine synthase and FNR have been mapped onto the crystal structure of E. coli Fld, showing several residues in common, all located near the FMN binding site [13]. These results suggest that both methionine synthase and FNR bind the same face of Fld and thus presumably bind sequentially [13]. Further support for sequential binding to a single face of Fld comes from the observation of competitive binding between FNR and methionine synthase in spectrophotometric assays [14].
Pyruvate formate-lyase activating enzyme (PFL-AE) is a 28 kDa monomer that contains a [4Fe-4S] cluster and utilizes S-adenosylmethionine (SAM) to activate pyruvate formate-lyase (PFL), a central enzyme in anaerobic glycolysis [15]. The SAM cofactor coordinates the unique iron of the [4Fe-4S] cluster via amino and carboxylate moieties [16, 17]. In the reduced [4Fe-4S]+ state, the cluster donates an electron to SAM, thus promoting homolytic cleavage of the S-C(5′) bond to generate methionine and a 5′-deoxyadenosyl radical; this adenosyl radical abstracts a hydrogen atom from G734 of PFL to generate the active enzyme [15, 18, 19]. Knappe and coworkers have shown that Fld can be used in the activation of PFL, presumably as an electron donor for PFL-AE [20]. When the crystal structure of PFL-AE was solved, a conserved region opposite the PFL binding site was proposed to act as the Fld binding site on PFL-AE [21].
In this work we provide insights into the apo- and holo- forms of E. coli Fld, and their interactions with protein partners, PFL-AE and FNR. Fld unfolding studies were performed with apo- and holo-Fld to probe differences in protein stability and folding thermodynamics as a result of cofactor binding. Limited proteolysis experiments analyzed using LC-MS were used to determine the regions of Fld that were involved in conformational changes. We utilize surface plasmon resonance under anaerobic conditions to examine the interactions between the air-sensitive PFL-AE and Fld and FNR. Findings in the present study indicate that Fld cofactor binding increases protein stability and results in conformational changes that are required for protein-protein interactions.
2.0 Materials and Methods
2.1 Cloning, Expression, and Purification of Fld and FNR
PFU-Turbo DNA polymerase and restriction enzymes NcoI and BamHI were purchased from New England Biolabs. FMN was purchased from MP Biomedicals, LLC, and was used without further purification. Fld (fldA) and FNR (fpr) were separately cloned using PCR to amplify the fldA and fpr genes from E. coli strain B genomic DNA. The PCR products were separately inserted into pET-14b using BamHI and NcoI restriction sites that were incorporated into the ends of the PCR products. The fldA and fpr genes were completely sequenced at Nevada Genomics Center. The purified DNA was used to transform the BL21(DE3)pLysS expression cell line. For protein expression, cells were grown in LB media at 37 °C in Fernbach flasks with agitation at 250 rpm. When cultures reached an O.D. of 0.8, protein production was induced by addition of IPTG to 100 μM final concentration. Cells were harvested three hours later by centrifugation at 6,000 rpm for 10 minutes.
Cells were lysed by chemical lysis (∼ 23 grams of cells in 50 mL of lysis buffer: 20 mM HEPES, 1% w/v Triton X-100, 5% w/v glycerol, 10 mM MgCl2 pH 7.2 with 9 mg PMSF dissolved in 100 μL methanol, 8 mg lysozyme, 1 mg of DNAse and RNAse; cells were lysed for one hour followed by centrifugation at 18,000 rpm for 30 minutes) and Fld and FNR were purified using ammonium sulfate precipitation. Fld and FNR precipitated out in the 60 - 100% fraction and were subsequently purified using a Superdex-75 size exclusion column (Figure S1). Fractions of the highest purity with the most intense color were pooled and concentrated and then flash frozen and stored at -80 °C (Figure S2). Holo-Fld was quantified using a previously determined extinction coefficient of ε467 nm = 8,250 M-1 cm-1 [22].
Apo-Fld was prepared using trichloroacetic acid precipitation as described previously [23]. This technique has been used in several previous studies to prepare apo-Fld [7, 23-26]. Apo-Fld was quantified using ε280nm = 30,900 M-1 cm-1, which was calculated using the protparam tool in ExPASy (http://www.expasy.org). The concentration of the FMN cofactor was determined using ε445nm = 12,500 M-1 cm-1 [27]. To ensure that the apo-Fld was capable of cofactor binding, a single titration of concentrated apo-Fld was added to a UV-vis cuvette containing FMN to show the 20 nm shift in λmax from 445 nm to 465 nm, characteristic of holo-Fld formation (Figure S3) [22]. PFL-AE was grown and purified as described previously [28]. An extinction coefficient of ε280nm = 39,400 M-1 cm-1 was calculated using ExPASy and correlated well with results from Bradford protein assays [29] after applying a previously-determined correction factor of 0.65 [30].
2.2 CD Spectroscopy
Secondary structure and thermal unfolding experiments were carried out using circular dichroism spectroscopy (CD). Spectra were measured with a Jasco-710 spectropolarimeter using either 0.1 mm or 1 cm pathlength cuvettes. Far-UV measurements were in the range of 195 - 260 nm and near-UV measurements were in the range of 240 - 800 nm. Sensitivity was 100 millidegrees, data pitch was 0.1 nm, using a continuous scan mode with a speed of 100 nm/minute, response was 4 seconds, bandwidth was 1.0 nm, with an accumulation of 3 scans. The buffer used for secondary structure and thermal unfolding experiments was 20 mM HEPES, 50 mM NaCl, pH 8.0. Protein concentrations of 30 μM were used for far-UV measurements and 30 μM or 75 μM for near-UV and visible region measurements. The Fld unfolding curves were independent of protein concentration under the conditions studied. Temperature was maintained for thermal unfolding experiments using an endocal refrigerated circulating water bath from NESLAB model RTE-5. Temperature was increased or decreased at a rate of 0.5 K/minute for unfolding or folding, respectively.
Circular dichroism binding studies were performed on a Jasco-810 spectropolarimeter using a 1 cm cuvette. Scans were taken from 240 - 800 nm using the same parameters as above. The buffer used for CD binding experiments was 20 mM HEPES, 10 mM NaCl, pH 7.4. In these experiments FNR was held constant at 30 μM while Fld was titrated into the FNR. Spectra were also measured for the Fld titration into buffer alone and the contribution of both proteins was subtracted out of the mixture to create difference spectra to visualize changes in visible and near-UV regions as a result of binding. All CD experiments were run in triplicate.
2.3 Analysis of Thermal Unfolding Data
Thermal denaturation of Fld was assessed at pH 8.0 using far-UV (222 nm), near-UV (296 nm), and visible regions (496 nm) of circular dichroism. Thermal unfolding curves were first fit to a two state unfolding model to obtain initial thermodynamic parameters of unfolding (Equations 1 and 2). The spectroscopic signals of the native (SN) and unfolded (SU) states with respective slopes mN and mU, are presumed to vary linearly with temperature. The change in enthalpy, heat capacity, and thermal melting point of the transition correspond to ΔHm, ΔCp, and Tm, respectively. The ΔCp for Fld unfolding was not reported since it has been determined to be unreliable [31, 32]. Apo-Fld and holo-Fld unfolding curves were subsequently fit to a three state unfolding model using a global fit (Equation 3). ΔG for the three state unfolding model follows expressions analogous to Equation 2. The signal of the intermediate (SI) and its associated slope (mI) at T = 0 K are also presumed to vary linearly in relation to temperature. ΔG1 and ΔG2 correspond to differences in free energy between the native/intermediate and intermediate/unfolded equilibria, respectively. Thermodynamic parameters of unfolding are reported with subscripts NI, IU, and NU corresponding to the transition from native to intermediate, intermediate to unfolded, and native to unfolded Fld; NU is reported as the sum of NI and IU.
2.4 Isothermal Titration Calorimetry
Experiments were performed on a VP-ITC (Microcal LLC, Northampton, MA) using 25 μM apo-Fld in the cell with 600 μM FMN cofactor in the syringe. Experimental parameters were as follows: 26 injections, reference power of 10 μcal/second, initial delay of 300 seconds, stir speed of 510 rpm, feedback mode/gain was high and automatic with fast equilibration. Injection duration was 24 seconds, spacing was 300 seconds, and filter period was 2 seconds. All experiments were performed in triplicate at either 25 °C or 37 °C. All Binding isotherm data was analyzed using a best fit to a single-site binding model by Marquardt nonlinear least-squares analysis (Origin 5.0) with VP-ITC software.
2.5 Surface Plasmon Resonance Binding Studies
Surface plasmon resonance experiments were performed under anaerobic conditions in a Coy chamber (Coy Laboratories, Grass Lake, MI) using a Biacore X-100 with the plus package. FNR or PFL-AE was immobilized to a CM5 sensor chip using thiol coupling as recommended by the manufacturer. Apo-Fld, holo-Fld, or FNR was injected as the analyte at a flow rate of 30 μL/minute using single cycle kinetics. Contact time was 60 seconds, dissociation time was 120 seconds, and regeneration time was 120 seconds. The running buffer was 20 mM HEPES, 10 mM NaCl, pH 7.4 and the regeneration buffer was 20 mM HEPES, 500 mM KCl, 0.005 % polysorbate 20, 200 mM imidazole, pH 7.4, all experiments were performed in triplicate at 25 °C. All data was fit using affinity mode in the data analysis software on the Biacore X-100 plus package.
2.6 Limited Proteolysis of Fld
Samples of apo- and holo-Fld were prepared under aerobic conditions in a total volume of 20 μL each. All samples contained 450 μM apo-Fld in 20 mM HEPES, 250 mM NaCl, pH 8.0. Holo-Fld samples were prepared for comparison to apo-Fld by the addition of a 1:1 ratio of the FMN cofactor. Trypsin was added in a 1:500 ratio (mg/mL) to apo- and holo-Fld and each sample was digested for 10 minutes. Samples were then boiled for 10 minutes and flash frozen using liquid N2 and stored until LC-MS analysis using an Agilent 1100 HPLC-ESI-MS using a linear gradient of 100 % H20/0.1 % FA, to 95 % acetonitrile/0.1 % FA for 0 - 7 minutes and held at 95 % acetonitrile/0.1 % FA, from 7 - 8 minutes, followed by re-equilibration with 100 % H20/0.1 % FA for 8 - 10 minutes using a reverse phase Phenomenex Jupiter Proteo C12 column at 25 °C (50 mm L × 1 mm inner diameter, pore size 4 μm, 90 Å pore size) at a flow rate of 400 μL/minute. Peptides eluted at 18 % and 55 % acetonitrile/0.1 % FA. Mass spectrometry was preformed using a Bruker MicroTOF mass spectrometer with an ESI source. Source parameters: drying gas 7 L/minute, nebulizer 4 bar, capillary voltage 4500 V., spectra were collected in positive mode from 300 - 3000 m/z at a rate of 1 Hz. All samples were analyzed at the Montana State University mass spectrometry core facility.
2.7 Docking of Fld and PFL-AE
A sequence alignment was performed using Clustal Omega to determine the residues involved in the Fld binding site on PFL-AE (http://www.clustal.org/). The amino acid sequences for PFL-AE from the following organisms were aligned and the FASTA ID is listed: Escherichia coli (P0A9N6), Clostridium pasteurianum (Q46267), Haemophilus influenzae (P43751), Listeria innocua (P0A443), Staphylococcus aureus (Q2FK43), Staphylococcus epidermidis (Q5HKI0), Streptococcus mutans (O68575), Clostridium novyi (A0Q1M3), and Shewanella sp. (A0KVG9). The residues with 100% sequence conservation were then mapped onto the structure of PFL-AE and the structure was examined to determine which residues were likely to be involved in the interface between PFL-AE and Fld. Previous work has been done to determine the residues involved in Fld binding with FNR and the SAM binding domain of methionine synthase [13]. The residues thought to be involved in the binding site for Fld and PFL-AE were used to construct an in silico rigid body complex between the two proteins using ZDOCK software [33].
3.0 Results
3.1 Secondary Structure of E. coli Fld
The secondary structure of Fld was examined by far-UV circular dichroism to determine if FMN cofactor binding to the apo-Fld resulted in changes in secondary structure. For these experiments, FMN was added to apo-Fld to generate the holo-Fld. Native apo-Fld was compared to native holo-Fld and our data shows that cofactor binding does not appear to change the secondary structure of the protein (Figure 1). Apo- and holo-Fld both contain features at 220 and 208 nm, which correspond to alpha helical structure, suggesting that they are both well folded. After the native apo- and holo-Fld proteins were thermally unfolded, far-UV CD spectra were compared to show that unfolded apo- and holo-Fld are very similar and exhibit features at 203 nm characteristic of an unfolded protein [34].
Figure 1.
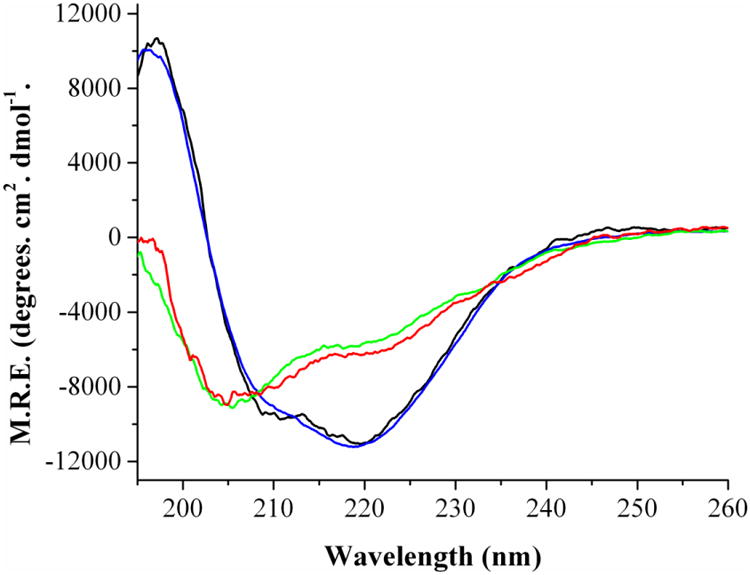
Secondary Structure of E. coli Fld as determined by far-UV CD. Black: Native apo-Fld at 24 °C, Blue: Native holo-Fld at 24 °C, Red: Unfolded apo-Fld at 86 °C, Green: Unfolded holo-Fld at 94 °C. Protein concentrations were 30 μM, 0.1 mm cuvette, 195-260 nm, Sensitivity = 100 millidegrees, data pitch = 0.1 nm, accumulation of 4 scans.
3.2 Cofactor Binding Increases the Thermal Stability of Fld
The thermal stability of Fld was measured using circular dichroism in the far-UV and near-UV regions (Figure 2). Two thermal transitions were evident for apo-Fld unfolding, the first one was in the near-UV region at 323.6 ± 0.7 K and the second was in the far-UV region at 326.3 ± 0.5 K (Figure 2A). The superposition test was used to show that Fld unfolds via a three state mechanism based on non-superimposable unfolding curves where an intermediate accumulates in the transition region [35-37] and thermodynamics of unfolding have been determined for each transition (Table 1). The enthalpy of unfolding from the native state to the completely unfolded state (ΔHm NU) of the apo-protein was determined to be 77 ± 4.6 kcal/mol•K, which agrees well with the previously determined values for apo-Fld from D. desulfuricans and Anabaena [27, 38]. Refolding of apo-Fld was also examined using circular dichroism by cooling unfolded samples to determine whether signal intensity can be restored. Both unfolding and refolding curves were superimposable and the signal intensity was completely restored upon sample cooling, therefore the three state unfolding process for apo-Fld is reversible under the conditions studied.
Figure 2.
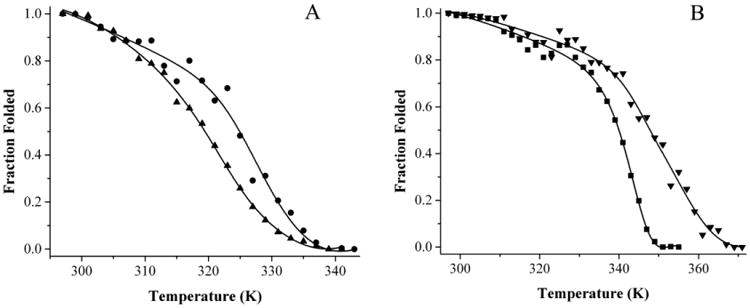
Thermal unfolding curves for holo-Fld as monitored in the near-UV, and far-UV regions by CD. A) Apo-Fld near-UV (▲) and far-UV (●) unfolding curves. B) Holo-Fld near-UV (■) and far-UV (▼) unfolding curves. The unfolding curves were globally fit to a three state folding model.
Table 1.
Thermodynamics of apo- and holo-Fld unfolding. Data was obtained from fitting Fld thermal unfolding transitions to a three state unfolding equation (Equation 1).
| Apo-Fld | Holo-Fld | ||
|---|---|---|---|
| Hm NI (kcal/mol) | 46.1 ± 4.1 | Hm NI (kcal/mol) | 65.5 ± 1.7 |
| Tm NI (K) | 323.6 ± 0.7 | Tm NI (K) | 345.9 ± 0.4 |
| Hm IU (kcal/mol) | 30.9 ± 2.1 | Hm IU (kcal/mol) | 39.6 ± 2.4 |
| Tm IU (K) | 326.3 ± 0.5 | Tm IU (K) | 352.8 ± 0.4 |
The thermal unfolding of holo-Fld produces two different melting transitions, one at 345.9 ± 0.4 K observed in the near-UV and visible regions of the CD spectrum, and the second at 352.8 ± 0.4 K observed in the far-UV region (Figure 2B). The near-UV and visible region unfolding curves overlay well, whereas the far-UV unfolding curve is not super-imposable with these, indicating that holo-Fld, much like apo-Fld unfolds via a three state mechanism that involves an intermediate. The ΔHm NU of the holo-protein is 105.1 ± 3 kcal/mol•K, indicating that the holo-protein is more stable and structured than the apo-protein; the thermodynamics of both transitions have been determined (Table 1). This value agrees well with that determined for D. desulfuricans holo-Fld [27]. After holo-Fld was unfolded, the samples were slowly cooled to monitor refolding of the protein. Based on far-UV, near-UV and visible region data, signal intensity can be restored and protein folding is a reversible equilibrium process provided that the sample is not heated much above the thermal melting point, similar to observations for D. desulfuricans Fld [27]. Both apo- and holo-Fld unfold to the same extent albeit at different temperatures and have features at 203 nm similar to other unfolded proteins (Figure 1) [34].
When unfolding curves are compared between apo and holo-Fld, it is obvious that there is an increase in stability upon cofactor binding. Increased unfolding enthalpy and thermal melting points suggest that the native and intermediate states of apo-Fld are both stabilized upon interaction with the FMN cofactor. Thermal transitions are independent of protein concentration under these conditions and increased thermal melting points for both holo-Fld unfolding transitions suggest that the FMN cofactor is bound during both unfolding transitions.
3.3 Thermodynamics of Binding of FMN to apo-Fld
FMN binding to apo-Fld was measured using isothermal titration calorimetry at 25 °C and 37 °C to determine the affinity and thermodynamics of interaction (Figure 3). At 25 °C the KD was 5.6 ± 1.2 nM. UV-vis and fluorescence binding assays have been performed previously on E. coli apo-Fld, yielding equilibrium constants of 1 nM, which is in agreement with our ITC data [7]. The ΔH of binding was -21.6 ± 0.6 kcal/mole and ΔS = -34.4 ± 1.6 cal/mol•K with a stoichiometry of 0.93 ± 0.01 and a ΔG = -11.3 ± 0.1 kcal/mol. At 37 °C apo-Fld cofactor binding affinity was slightly weaker exhibiting a KD = 8.2 ± 0.8 nM and a stoichiometry of 0.92 ± 0.08. The ΔH of binding was -31.9 ± 3.3 kcal/mol with a ΔS = -65.6 ± 10.4 cal/mol•K and a ΔG = -11.5 ± 0.1 kcal/mol and a ΔCp = -802 ± 200 cal/mol•K. It is clear from this data that FMN binding to apo-Fld is driven by enthalpy and that there is an unfavorable contribution from entropy of binding. To ensure that FMN was indeed binding apo-Fld, a control experiment was performed using UV-vis in the visible region where FMN was added to buffer and a scan was measured followed by the addition of apo-Fld (Figure S3). This data clearly shows a 20 nm shift in λmax from 445 nm to 465 nm characteristic of apo-Fld binding the FMN cofactor resulting in holo-protein formation [22].
Figure 3.
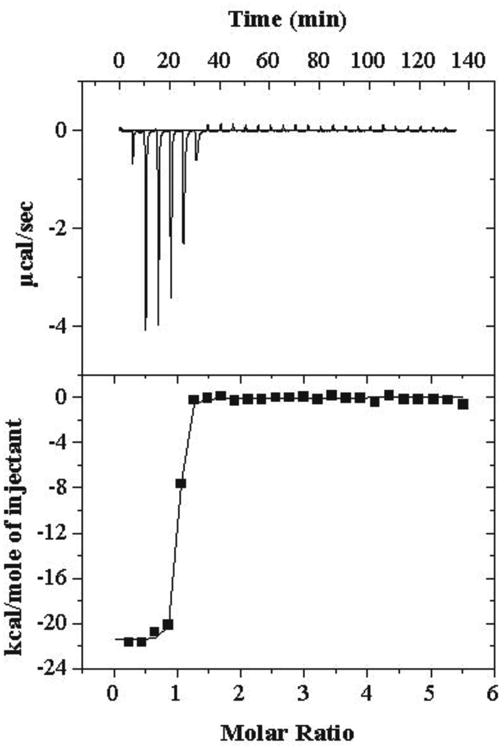
Isothermal titration calorimetry of apo-Fld binding FMN at 37 °C. Injections (26 at 10 μL each and the first one at 2 μL) of FMN (600 μM stock concentration) were added to 25 μM apo-Fld in the cell. The reference power was 10 μcal/second, initial delay of 300 seconds, stir speed of 510 rpm, feedback mode/gain was set to high and automatic with fast equilibration, duration of 24 seconds, spacing of 300 seconds, with a filter period of 2 seconds
3.4 Binding Interactions Perturb FNR CD Spectra
FNR is a NADP+-dependent reductant for Fld in E. coli. Previous work on FNR has provided evidence that upon interaction with Fld or ferredoxin, the visible absorbance is perturbed and has been attributed to changes in the electronic environment of the FAD cofactor [39-41]. In Figure 4 we show the CD spectra of holo-Fld alone (green trace), FNR alone (blue trace), and a spectral addition of these two spectra (red trace). We also show the CD spectrum obtained for a 1:1 mixture of Fld and FNR (black trace). It is clear from comparison of the black and red traces that the physical mixture of Fld and FNR produces a different CD spectrum than that obtained by spectral addition of the data for the isolated proteins, indicating that protein-protein interactions are perturbing the CD spectra of one or both proteins in the mixture. As another way to illustrate these effects, we titrated Fld into a solution of FNR and monitored changes by CD spectroscopy; the contribution of Fld and FNR was subtracted out from each titration spectrum in order to emphasize the changes in FNR upon Fld binding. As expected, both near-UV and visible regions monitored by CD exhibited changes characteristic of binding (Figure 4), indicating that when Fld and FNR interact, aromatic residues and the FAD cofactor experience a change in environment. These changes were determined to be proportional to the amount of titrated flavodoxin in the mixture. The difference spectrum shows decreased intensity and a red shift in the near-UV region as well as intensity changes in the visible region (Figure 4 inset). It is not known whether conformational changes cause the electronic environment of the FAD cofactor to be perturbed or if the close proximity of cofactors during binding causes this phenomenon.
Figure 4.
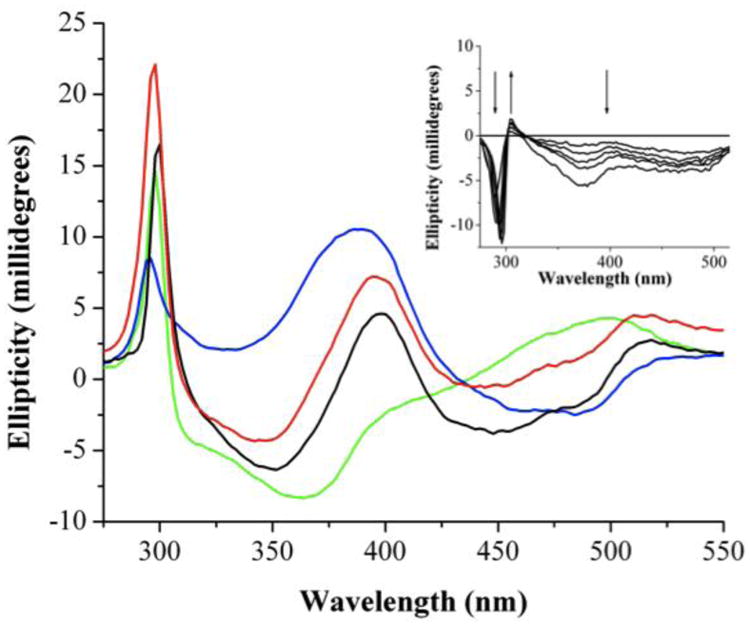
Binding of holo-Fld to FNR as monitored by CD spectroscopy. Blue: 30 μM FNR; Green: 30 μM Fld; Red: spectral addition of FNR and Fld; Black: Mixture of 30 μM FNR and 30 μM Fld. Inset shows difference spectra corresponding to a titration of (0 μM, 7.5 μM, 15 μM, 22.5 μM, 30 μM, 60 μM) Fld into FNR (30 μM) to show changes in the near-UV and visible regions as a result of Fld binding FNR.
3.5 Fld Binding to FNR and PFL-AE
FNR and Fld have been shown to be capable of supplying low-potential electrons for radical SAM enzymes such as PFL-AE, with the electrons originating from NADPH [10, 12, 42, 43]. In order to probe the interactions between Fld and its partners FNR and PFL-AE, we have utilized surface plasmon resonance studies under anaerobic conditions; anaerobic conditions were required in order to maintain the integrity of the [4Fe-4S] cluster of PFL-AE. A KD of 3.7 ± 1.6 μM was determined for holo-Fld binding with FNR; this agrees well with the previously determined value of 2.0 ± 1 μM [39]. Interestingly, apo-Fld was found to be inert as a binding partner for FNR. Holo-Fld interacts with PFL-AE with a KD of 23.3 ± 3.8 μM (Figure 5). When apo-Fld was substituted for holo-Fld, there was no interaction between these two proteins. The observation that holo-Fld binds to both FNR and PFL-AE with micromolar affinity, while binding of apo-Fld cannot be detected for either protein, suggests that Fld cofactor binding produces a conformational change in Fld rendering it capable of interacting with binding partners.
Figure 5.
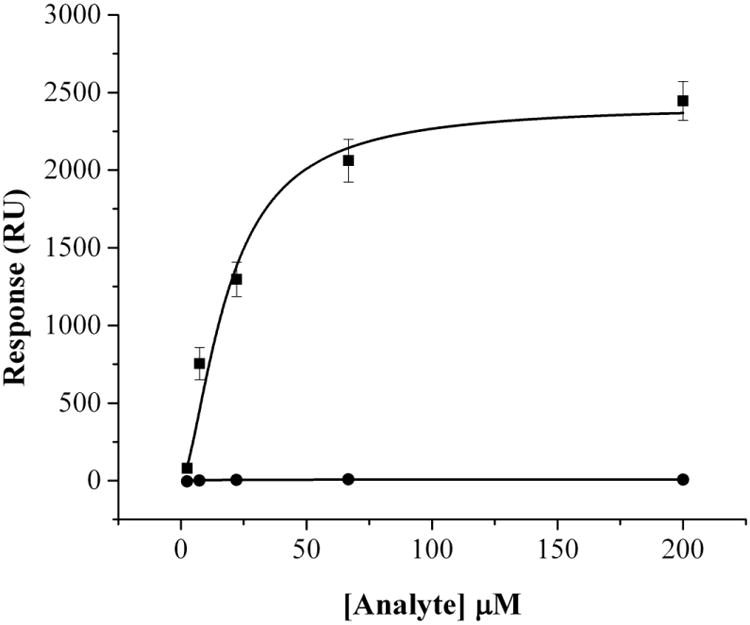
Binding of PFL-AE to apo- and holo-Fld using surface plasmon resonance under anaerobic conditions. Single cycle kinetics mode was used at 25 °C and PFL-AE was covalently attached to a CM5 sensor chip using thiol coupling. Either apo-Fld (●) or holo-Fld (■) was titrated over PFL-AE at concentrations of 2.47, 7.41, 22.2, 66.7, and 200 μM. The running buffer used was 20 mM HEPES, 10 mM KCl, pH 7.4 with a flow rate of 30 μL/minute used in assays with a contact time of 60 seconds and dissociation time of 120 seconds. The regeneration buffer was 20 mM HEPES, 500 mM KCl, 0.005 % Polysorbate 20, 200 mM imidazole, pH 7.4 and the regeneration time was 120 seconds.
3.6 Fld Conformational Changes Upon FMN Cofactor Binding
Fld was examined using limited proteolysis to determine which regions of the protein change in conformation as a result of FMN cofactor binding. Samples were digested using trypsin and peptides were analyzed by LC-MS and identified using Mascot. Peptides that were unique to apo- and holo-Fld samples were considered to be involved in apo-Fld conformational changes induced by FMN cofactor binding. Unique peptides observed in the holo-Fld sample correspond to D29-K38 and S39-R112 (Table S1). The apo-Fld sample contained unique peptides corresponding to S39-K81, G133-K159, and G113-K162. Cleavage was only observed at K81 in apo-Fld, indicating that this region is more open and accessible to proteolysis (Figure 6). Fld cleavage was observed at structured and unstructured regions, suggesting that Fld peptides are unstable after the protein is nicked and readily unfold, facilitating cleavage at structured regions [44].
Figure 6.
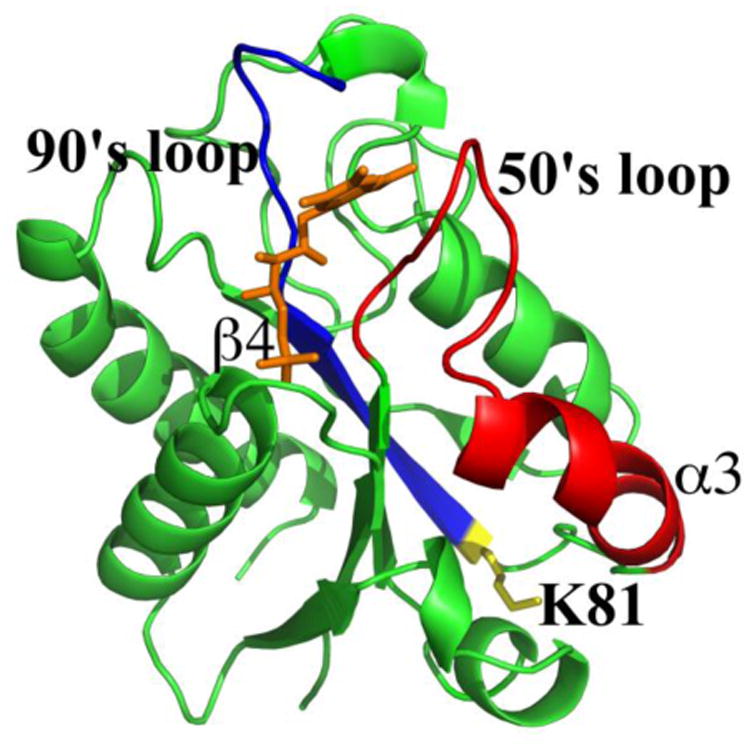
Conformational changes in Fld induced by FMN cofactor binding. Apo- and holo-Fld was digested with trypsin and peptides were analyzed by LC-MS. Blue: 90's loop of Fld connected to β-sheet 4, Red: 50's loop of Fld connected to α-helix 3. The side chain is shown for the unique apo-Fld cleavage point at K81 and it is colored yellow and labeled. The FMN cofactor is colored orange.
3.7 Docking of Fld with PFL-AE
When the structure of PFL-AE was solved, a sequence alignment was performed that showed an area of 100 % sequence conservation, which was proposed to be the Fld binding site [21]. We have reproduced and analyzed this alignment in order to determine the residues that were likely to be involved in the interface between PFL-AE and Fld (Figure 7 A). PFL-AE residues Q27, G28, C29, R32, H37, N38, D40, T41, W42, E79, Q83, F86, and K208 were 100 % sequence conserved and confined to a region where the [4Fe-4S] cluster is close to the surface of the enzyme. Previous studies suggest that electrostatic interactions are important for Fld binding and that Fld binding partners contain positively charged residues at the binding site interface [45, 46]. However, hydrophobic residues are thought to contribute the bulk of binding energy to Fld protein-protein interactions [13, 47]. Electrostatic mapping results show that there is only one location where there is a large positively charged patch on the surface of PFL-AE and this positively charged area largely overlays the region of 100 % conservation. When hydrophobic residues were mapped on PFL-AE, there was a large hydrophobic patch that coincided with the 100 % conserved residues and positively charges residues. We used these findings in conjunction with results from a previous study that identified Fld residues involved in interactions with FNR and the SAM binding domain from methionine synthase [13] to produce a docked structure of Fld and PFL-AE using ZDOCK software (Figure 7 B). The Fld binding site on PFL-AE is the only location other than the PFL binding site where the [4Fe-4S] cluster is close to the surface of the enzyme, which would be necessary for efficient electron transfer from Fld to PFL-AE. There are also aromatic residues between the FMN cofactor in Fld and the [4Fe-4S] cluster in PFL-AE that may be involved in the electron transfer pathway. In the Fld/PFL-AE complex, FMN is located 10.7 Å from the [4Fe-4S] cluster in PFL-AE (Figure 7 C). In the Fld crystal structure, W57 stacks with the FMN cofactor 3.8 Å away. In Fld, W57 is within 3.3 Å of W42 from PFL-AE forming a bridge between the two cofactors. In PFL-AE, W42 is located 3.1 Å from the [4Fe-4S] cluster. Indeed, tryptophan and tyrosine are greatly enriched at binding site interfaces and tryptophan can act an electron donor/acceptor between cofactors and significantly increase long distance electron transfer rates [48, 49].
Figure 7.
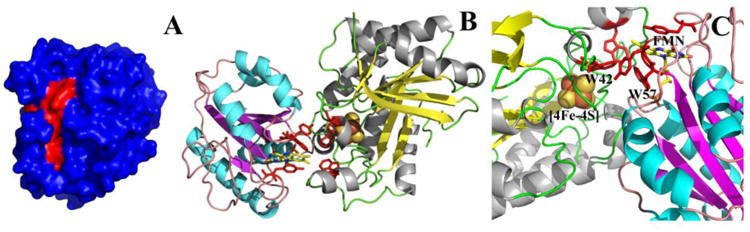
A) Surface representation of PFL-AE with 100 % sequence conserved residues colored red. B) Docked structure of Fld bound to PFL-AE constructed using ZDOCK: Fld (1AHN), PFL-AE (3C8F). C) Zoomed in view of the active sites in the Fld/PFL-AE complex. The side chains are colored red for aromatic residues that form a bridge between cofactors. The FMN cofactor, W57 from flavodoxin, W42 from PFL-AE, and the [4Fe-4S] cluster are all labeled.
4.0 Discussion
In this study, we set out to determine whether cofactor binding induces changes in stability and conformation in apo-Fld, and what effects these structural changes would have on protein-protein interactions. Far-UV CD shows that native apo- and holo-Fld have features that correspond to structured, well-folded proteins and FMN cofactor binding does not appear to alter protein secondary structure. At high temperatures, both apo- and holo-Fld exhibit features corresponding to unfolded proteins. Thermal unfolding studies support the idea that apo-Fld cofactor binding increases protein stability. Both apo-Fld and holo-Fld unfold via a three state unfolding mechanism that involves an intermediate. Fld unfolding studies show that binding of the cofactor produces an increase in the stability of both the native protein as well as the intermediate. Overall, the unfolding of apo-Fld and holo-Fld from E. coli are in very nice agreement with the results obtained for Fld from D. desulfuricans [27].
Isothermal titration calorimetry data clearly shows that Fld cofactor binding is a favorable process. The binding is driven by enthalpy and exhibits an unfavorable entropy contribution that may correspond to a decrease in conformational freedom of the cofactor and cofactor binding loops, much like in H. pylori [24]. The crystal structure from Anabaena PCC 7119 clearly shows that when Fld binds its cofactor, W57 and Y94 swing back into the interior of the protein and hold the isoalloxazine ring in place using stacking interactions [50]. It is likely that Escherichia coli Fld uses the same mechanism for binding FMN.
Association experiments between Fld and FNR show that when the proteins interact, aromatic residues experience a change in environment (based on near-UV CD changes) that may be a result of conformational changes or an altered environment at the interface of the complex. The electronic environment of the FAD cofactor is also perturbed during binding, which may be due to conformational changes that alter the cofactor environment. However, it believed that when these proteins bind, their cofactors are juxtaposed for electron transfer such that the FMN cofactor in Fld fits into a cavity near the FAD cofactor in FNR, possibly contributing to the perturbed visible region spectrum [13]. Surface plasmon resonance experiments demonstrate the functional significance of conformational changes associated with cofactor binding in Fld. Holo-Fld interacts with PFL-AE and FNR, however in absence of the FMN cofactor, Fld is no longer capable of binding either protein. Activity assays demonstrate that Fld is the electron donor for PFL-AE and that FNR reduces Fld [10].
Unique Fld cleavage patterns were observed during limited proteolysis experiments of apo- and holo-Fld analyzed by LC-MS, indicating that conformational changes induced by cofactor binding occur in the unique peptide regions. The crystal structure of E. coli holo-Fld shows that residues from the 50's and 90's loops hydrogen bond with FMN and that W57 and Y94 interact with the isoalloxazine ring of the cofactor via π-stacking interactions [7]. Fld residues involved in binding FNR or the SAM binding domain of methionine synthase were located in the same region identified in cofactor binding in the crystal structure and include S40, Y58, Y59, A62, D68, D93, Y94, A95, F127, K131, and Q149 [13]. Our results show that the regions identified in FMN cofactor binding and protein-protein interactions experience conformational changes, which explain why cofactor binding would affect protein-protein interactions. A unique cleavage site in apo-Fld at K81 provides insight into a possible mechanism of conformational change where FMN binding brings α3 and β4 in closer proximity and alters the PFL-AE and FNR binding site (Figure 6).
The interaction between PFL-AE and Fld has been modeled using ZDOCK software. The rigid body model of the Fld/PFL-AE complex was generated based on bioinformatic data of PFL-AE and previous studies of Fld because experimental data was not available. A distance of 10.7 Å is estimated between the FMN cofactor and the [4Fe-4S] cluster, which puts the two relatively close, however this distance would ensure an outer sphere electron transfer mechanism. The docked structure of the Fld/PFL-AE complex shows Fld bound to the side opposite the PFL binding site on PFL-AE, which is the only other location on PFL-AE where the [4Fe-4S] cluster is located close to the surface of the enzyme. Sequence alignments performed on other radical SAM enzymes have shown that this region is conserved and is thought to be a common binding site for electron donors [51]. Additionally aromatic residues were found between the Fld (W57) and PFL-AE (W42) cofactors that may be involved in electron transfer from the FMN cofactor to the [4Fe-4S] cluster.
Overall our data suggests that cofactor binding in alters Fld structure near the cofactor binding region where protein binding partners interact, explaining why FNR and PFL-AE only bind holo-Fld. Considering this information and studies on FNR and methionine synthase that show competitive binding with Fld and overlapping binding sites, PFL-AE and FNR may bind sequentially as well. In this case, FNR would first bind and reduce Fld before Fld interacts with and presumably reduces PFL-AE. PFL-AE can then activate PFL in the presence of SAM and PFL substrates to produce active PFL.
5.0 Conclusions
Fld cofactor binding is not only required for a functional Fld that is capable of transferring electrons, but it also induces conformational changes that increase protein stability and facilitate protein-protein interactions. Fld interacts with FNR and PFL-AE with low micromolar affinity that would be expected for electron donor and acceptor proteins involved in PFL activation. Bioinformatic data on PFL-AE in conjunction with previous Fld studies suggest that PFL-AE binds Fld at a site that is separate from the PFL binding site, therefore making it possible for Fld to reduce PFL-AE when it is bound to SAM, PFL, and PFL substrates.
Supplementary Material
Limited proteolysis results of apo- and holo-Fld analyzed by LC-MS. Fld peptides that were unique to each sample are labeled with an asterisk. Errors are reported as the difference between the observed mass and the theoretical mass of the peptide in units of Daltons.
FPLC chromatogram for a Fld purification using the Superdex-75 size exclusion column. Blue trace corresponds to Fld absorbance at 280 nm and the red trace corresponds absorbance at 466 nm. Fractions were collected from 650 - 790 mL in 10 mL increments.
12 % SDS-PAGE gel of a Fld purification. Lane one is the molecular weight marker. Each lane is from one 10 mL fraction starting at 650 mL and going to 790 mL from left to right.
UV-vis data of Fld binding its cofactor FMN. Scans were taken in the visible region at 25 °C, from 300 - 800 nm. The buffer used was 20 mM HEPES, 50 mM NaCl, pH 8.0. Black: 62.2 μM FMN, Red: 64 μM apo-Fld plus 61.6 μM FMN.
Highlights.
E. coli flavodoxin cofactor binding is very favorable and driven by enthalpy.
Cofactor binding increases structure and stability and is required for protein binding.
Flavodoxin interacts with protein partners with low micromolar affinity.
A flavodoxin binding site has been found that may provide insight into enzyme activation.
Acknowledgments
We are grateful to GE Biacore for the Biacore X-100 Inspiration Contest that provided the Biacore X-100 plus machine and materials for the surface plasmon resonance experiments. I would also like to thank Dave Dooley's group at Montana State University and Michele McGuirl's group at the University of Montana for use of their circular dichroism instruments.
This work was supported by NIH grant 2R01GM054608.
6.0 Appendices
6.1 Two state and three state unfolding equations
| (1) |
| (2) |
| (3) |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sancho J. Flavodoxins: sequence, folding, binding, function and beyond. Cell Mol Life Sci. 2006;63:855–864. doi: 10.1007/s00018-005-5514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peleato ML, Ayora S, Inda LA, Gomez-Moreno C. Isolation and characterization of two different flavodoxins from eukaryote Chlorella fusca. Biochem J. 1994;302:807–811. doi: 10.1042/bj3020807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukuyama K, Matsubara H, Rodgers LJ. Crystal structure of oxidized flavodoxin from red algae Chlondrus crispus refined at 1.8 Å resolution. Description of the flavodoxin mononucleotide binding site. J Mol Biol. 1992;225:775–789. doi: 10.1016/0022-2836(92)90400-e. [DOI] [PubMed] [Google Scholar]
- 4.Olteanu H, Banerjee R. Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J Biol Chem. 2001;276:35558–35563. doi: 10.1074/jbc.M103707200. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Q, Modi S, Smith G, Paine M, McDonage PD, Wolf CR, Tew D, Lian Ly, Roberts GCK, Driessen HPC. Crystal structure of the FMN-binding domain of human cytochrome P450 reductase at 1.93 Å resolution. Protein Sci. 1999;8:298–306. doi: 10.1110/ps.8.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paine MJI, Garner AP, Powell D, Sibbald J, Sales M, Pratt N, Smith T, tew DG, Wolf CR. Cloning and characterization of a novel human dual flavin reductase. J Biol Chem. 2000;275:1471–1478. doi: 10.1074/jbc.275.2.1471. [DOI] [PubMed] [Google Scholar]
- 7.Hoover DM, Ludwig ML. A flavodoxin that is required for enzyme activation: the structure of oxidized flavodoxin from Esherichia coli at 1.8 Å resolution. Protein Sci. 1997;6:2525–2537. doi: 10.1002/pro.5560061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Z, Swenson RP. Electrostatic effects of surface acidic amino acid residues on the oxidation-reduction potentials of the flavodoxin from Deslfovibrio vulgaris (Hildenborough) Biochemistry. 1995;34:3183–3192. doi: 10.1021/bi00010a007. [DOI] [PubMed] [Google Scholar]
- 9.Kasim M, Swenson RP. Alanine-scanning of the 50's loop in the Clostridium beijerinckii flavodoxin: evaluation of additivity and the importance of interactions provided by the main chain in the modulation of the oxidation-reduction potentials. Biochemistry. 2001;40:13548–13555. doi: 10.1021/bi011587c. [DOI] [PubMed] [Google Scholar]
- 10.Blaschkowski HP, Neuer G, Ludwig-Festl M, Knappe J. Routes of flavodoxin and ferredoxin reduction in Escherichia coli. CoA-acylating pyruvate: flavodoxin and NADPH: flavodoxin oxidoreductases participating in the activation of pyruvate formate-lyase. Eur J Biochem. 1982;123:563–569. [PubMed] [Google Scholar]
- 11.Bianchi V, Eliasson R, Fontecave M, Mulliez E, Hoover DM, Matthews RG, Reichard P. Flavodoxin is required for the activation of the anaerobic ribonucleotide reductase. Biochem Biophys Res Commun. 1993;197:792–797. doi: 10.1006/bbrc.1993.2548. [DOI] [PubMed] [Google Scholar]
- 12.Ifuku O, Koga N, Haze Si, Kishimoto J, Wachi Y. Flavodoxin is required for conversion of dethiobiotin to biotin in Escherichia coli. Eur J Biochem. 1994;224:173–178. doi: 10.1111/j.1432-1033.1994.tb20009.x. [DOI] [PubMed] [Google Scholar]
- 13.Hall DA, Vander Kooi CW, Stasik CN, Stevens SY, Zuiderweg ER, Matthews RG. Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase. Proc Natl Acad Sci U S A. 2001;98:9521–9526. doi: 10.1073/pnas.171168898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoover DM, Jarrett JT, Sands RH, Dunham WR, Ludwig ML, Matthews RG. Interaction of Escherichia coli colbalamin-dependent methionine synthase and its physiological partner flavodoxin: binding of flavodoxin leads to axial ligand dissociation from the cobalamin cofactor. Biochemisty. 1997;36:127–138. doi: 10.1021/bi961693s. [DOI] [PubMed] [Google Scholar]
- 15.Henshaw TF, Cheek J, Broderick JB. The [Fe-4S]1+ cluster of pyruvate formate-lyase activating enzyme generates the glycyl radical on pyruvate formate-lyase: EPR-detected single turnover. J Am Chem Soc. 2000;122:8331–8332. [Google Scholar]
- 16.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. Coordination of adenosylmethionine to a unique iron site of the [4Fe-4S] of pyruvate formate-lyase activating enzyme: a mössbauer spectroscopic study. J Am Chem Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 17.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 18.Frey M, Rothe MAF, Wagner V, Knappe J. Adenosylmethionine-dependent synthesis of the glycyl radical in pyruvate formate-lyase by abstraction of the glycine C-2 pro-S Hydrogen atom. J Biol Chem. 1994;269:12432–12437. [PubMed] [Google Scholar]
- 19.Volker Wagner AF, Frey M, Meugebauer FA, Schäfer W, Knappe J. The free radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci. 1992;89:996–1000. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knappe J, Schacht J, Möckel W, Höpner T, Vetter H, Jr, Edenharder R. Pyruvate formate-lyase reaction in Escherichia coli. European J Biochem. 1969;11:316–327. doi: 10.1111/j.1432-1033.1969.tb00775.x. [DOI] [PubMed] [Google Scholar]
- 21.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci U S A. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vetter H, Jr, Knappe J. Flavodoxin and ferredoxin of Escherichia coli. H-Z Physiol Chem. 1971;352:433–446. doi: 10.1515/bchm2.1971.352.1.433. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew SG, Massey V. Purification and characterization of flavodoxin from Peptostreptococcus elsdenii. J Biol Chem. 1969;244:794–802. [PubMed] [Google Scholar]
- 24.Credmades N, Velazquez-Campoy A, Freire E, Sancho J. The flavodoxin from Helicobacter pylori: structural determinants of thermostability and FMN cofactor binding. Biochemistry. 2008;47:627–639. doi: 10.1021/bi701365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cremades N, Sancho J. Molten globule and native state ensemble of Helicobacter pylori flavodoxin: can crowding, osmolytes or cofactors stabilize the native conformation relative to the molten globule? Biophys J. 2008;95:1913–1927. doi: 10.1529/biophysj.108.130153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Anna JA, Tollin G. Studies of flavin-protein interaction in flavoproteins using protein fluorescence and circular dichroism. Biochemistry. 1972;11:1073–1080. doi: 10.1021/bi00756a020. [DOI] [PubMed] [Google Scholar]
- 27.Muralidhara BK, Wittung-Stafshede P. Thermal unfolding of Apo and Holo Desulfovibrio desulfuricans flavodoxin: cofactor stabilizes folded and intermediate states. Biochemistry. 2004;43:12855–12864. doi: 10.1021/bi048944e. [DOI] [PubMed] [Google Scholar]
- 28.Broderick JB, Henshaw TF, Cheek J, Wojtuszewski K, Smith SR, Trojan MR, McGhan RM, Kopf A, Kibbey M, Broderick WE. Pyruvate formate-lyase-activating enzyme: strictly anaerobic lisolation yields active enzyme containing a [3Fe-4S]+ cluster. Biochem Biophys Res Commun. 2000;269:451–456. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 29.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Broderick JB, Duderstadt RE, Fernandez DC, Wojtuszewski K, Henshaw TF, Johnson MK. Pyruvate formate-lyase activating enzyme is an iron-sulfur protein. J Am Chem Soc. 1997;119:7396–7397. [Google Scholar]
- 31.Privalov PL, Potekhin SA. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 1986;131:4–51. doi: 10.1016/0076-6879(86)31033-4. [DOI] [PubMed] [Google Scholar]
- 32.Privalov PL. Thermodynamics of protein folding. J Chem Thermodyn. 1997;29:447–474. [Google Scholar]
- 33.Chen R, Li L, Weng Z. ZDOCK: an inital-stage protein docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 34.Chen L, Jiang ZG, Khan AA, Chishti AH, McKnight CJ. Demantin exhibits a natively unfolded core domain and an independently folded headpiece domain. Protein Sci. 2009;18:629–636. doi: 10.1002/pro.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irún MP, Garcia-Mira MM, Sanchez-Ruiz MJ, Sanch J. Native hydrogen bonds in a molten globule: The apoflavodoxin thermal intermediate. J Mol Biol. 2001;306:877–888. doi: 10.1006/jmbi.2001.4436. [DOI] [PubMed] [Google Scholar]
- 36.Maldonado S, Jimènez MÁ, Langdon GM, Sancho J. Cooperative Stabilization of a molte globule apoflavodoxin fragment. Biochemistry. 1998;37:10589–10696. doi: 10.1021/bi980368x. [DOI] [PubMed] [Google Scholar]
- 37.Luo Y, Kay MS, Baldwin RL. Cooperativity of folding of the apomyoglobin pH 4 intermediate studied by glycine and proline mutations. Nature Struct Biol. 1997;4:925–930. doi: 10.1038/nsb1197-925. [DOI] [PubMed] [Google Scholar]
- 38.Campos L, Sancho J. Native-specific stabilization of flavodoxin by the FMN cofactor: structural and thermodynamic explanation. Proteins. 2006;63:581–594. doi: 10.1002/prot.20855. [DOI] [PubMed] [Google Scholar]
- 39.Wan JT, Jarrett JT. Electron accetor specificity of ferredoxin (flavodoxin):NADP+ oxidoreductase from Escherichia coli. Arch Biochem Biophys. 2002;406:116–126. doi: 10.1016/s0003-9861(02)00421-6. [DOI] [PubMed] [Google Scholar]
- 40.Jung YS, Roberts VA, Stout DC, Burgess BK. Complex formation between Azotobacter vinelandii ferredoxin I and is physiological electron donor NADPH-ferredoxin reductase. J Biol Chem. 1999;274:2978–2987. doi: 10.1074/jbc.274.5.2978. [DOI] [PubMed] [Google Scholar]
- 41.Lambeth DJ, Seybert DW, Kamin H. Andredoxin reductase andredoxin complex. J Biol Chem. 1980;255:4667–4672. [PubMed] [Google Scholar]
- 42.Sanyal I, Gibson KJ, Flint DH. Escherichia coli biotin synthase: an investigation into the factors required for its activity and its sulfur donor. Arch Biochem Biophys. 1996;326:48–56. doi: 10.1006/abbi.1996.0045. [DOI] [PubMed] [Google Scholar]
- 43.Mulliez E, Padovani D, Atta M, Alcouffe C, Fontecave M. Activation of class III ribonucleotide reductase by flavodoxin: A protein radical-driven electron transfer to the iron-sulfur center. Biochemistry. 2001;40:3730–3736. doi: 10.1021/bi001746c. [DOI] [PubMed] [Google Scholar]
- 44.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 45.Hall DA, Jordan-Starck TC, Loo RO, Ludwig ML, Matthews RG. Interaction of flavodoxin with cobalamin-dependent methionine synthase. Biochemistry. 2000;39:10711–10719. doi: 10.1021/bi001096c. [DOI] [PubMed] [Google Scholar]
- 46.Medina M, Peleato LM, Mendez E, Gomez-Moreno C. Identification of specific carboxyl groups on Anabaena PCC 7119 flavodoxin which are involved in the interaction with ferredoxin-NADP+ reductase. Eur J Biochem. 1992;203:373–379. doi: 10.1111/j.1432-1033.1992.tb16560.x. [DOI] [PubMed] [Google Scholar]
- 47.Nogués I, Martinez-Júlvez M, Navarro JA, Hervás M, Armenteros L, Angel de la Rosa M, Brodie TB, Hurley JK, Tollin G, Gómez-Moreno C, Medina M. Role of hydrophobic interactions in the flavodoxin mediated electron transfer from photosystem I to ferredoxin-NADP+ reductase in Anabaena PCC 7119. Biochemistry. 2003;42:2036–2045. doi: 10.1021/bi0270541. [DOI] [PubMed] [Google Scholar]
- 48.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 49.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlček A, Jr, Richards JH, Winkler JR, Gray HB. Tryptophan-accelerated electron flow through proteins. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 50.Genzor CG, Perales-Alcón A, Sancho J, Romero A. Closure of a tyrosine/tryptophan aromatic gat leads to a compact fold in apo flavodoxin. Nat Struct Biol. 1996;3:329–332. doi: 10.1038/nsb0496-329. [DOI] [PubMed] [Google Scholar]
- 51.Vey JL, Drennan CL. Strutural insights into radical generation by the radical SAM superfamily. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Limited proteolysis results of apo- and holo-Fld analyzed by LC-MS. Fld peptides that were unique to each sample are labeled with an asterisk. Errors are reported as the difference between the observed mass and the theoretical mass of the peptide in units of Daltons.
FPLC chromatogram for a Fld purification using the Superdex-75 size exclusion column. Blue trace corresponds to Fld absorbance at 280 nm and the red trace corresponds absorbance at 466 nm. Fractions were collected from 650 - 790 mL in 10 mL increments.
12 % SDS-PAGE gel of a Fld purification. Lane one is the molecular weight marker. Each lane is from one 10 mL fraction starting at 650 mL and going to 790 mL from left to right.
UV-vis data of Fld binding its cofactor FMN. Scans were taken in the visible region at 25 °C, from 300 - 800 nm. The buffer used was 20 mM HEPES, 50 mM NaCl, pH 8.0. Black: 62.2 μM FMN, Red: 64 μM apo-Fld plus 61.6 μM FMN.