

Abstract
Symmetric dipyrrylketones 1a,b were synthesized in two steps from the corresponding α-free pyrroles, by reaction with thiophosgene followed by oxidative hydrolysis under basic conditions. The dipyrrylketones produced the corresponding 5-chloro-dipyrrinium salts or 5-ethoxy-dipyrrins on reaction with phosgene or Meerwein’s salt, respectively. Boron complexation of the dipyrrins afforded the corresponding 8-functionalized BODIPYs (borondipyrromethenes) in high yields. The 5-chloro-dipyrrinium salts reacted with methoxide or ethoxide ions to produce monopyrrole esters, presumably via a 5,5-dialkoxy-dipyrromethane intermediate. In contrast, 8-chloro-BODIPYs underwent a variety of nucleophilic substitutions of the chloro group in the presence of alkoxide ions, Grignard reagents, and thiols. In the presence of excess alkoxide or Grignard reagent, at room temperature or above, substitution at the boron center also occurred. The 8-chloro-BODIPY was a particularly useful reagent for the preparation of 8-aryl-, 8-alkyl-, and 8-vinyl-substituted BODIPYs in very high yields, using Pd0-catalyzed Stille cross-coupling reactions. The X-ray structures of eleven BODIPYs and two pyrroles are presented, and the spectroscopic properties of the synthesized BODIPYs are discussed.
Keywords: bodipy, dipyrrin, dipyrrylketone, fluorescence
Introduction
Dipyrrylketones (see Figure 1, A) have received little attention in polypyrrole chemistry. Dipyrrylketones are actually bis-vinylogous amides [νmax (C=O) ca. 1575 cm−1] that do not readily react as normal ketones. For instance, dipyrrylketones are not usually reduced by borohydride.[1] However, upon protonation they experience a significant and completely reversible redshift in their optical spectra [λmax(CH2Cl2) from 340 to 428 nm], clearly demonstrating the presence of the 5-hydroxy-dipyrrylmethene salt (Figure 1, B).
Figure 1.

Optical spectra, in dichloromethane, of dipyrrylketone (A) and dipyrrinium salt (B) after treatment with HCl (g).
Various syntheses of dipyrrylketones are well documented. The most efficient synthesis of unsymmetrical dipyrrylketones involves a modified Vilsmeier–Haack reaction that uses a 2-dimethylamidopyrrole with phosphoryl chloride, and a 2-unsubstituted pyrrole as a nucleophile.[2] Unsymmetrical dipyrrylketones can also be obtained by treatment of a pyrrole-2-acyl chloride with a pyrrole-Grignard derivative. Symmetrical dipyrroketones can be accessed by phosgenation of a pyrrole-Grignard reagent or by oxidative hydrolysis of the corresponding thioketones.[3] Another approach to symmetrical dipyrrylketones involves direct oxidation of the 5-meso-carbon of a dipyrrylmethane by using PbO2/Pb(OAc)4 or Br2/SOCl2.[4] 1,9-Diformyldipyrrylketones have been used in a MacDonald-type porphyrin synthesis to give oxyporphyrins (oxophlorins).[5] The dipyrrylketone system is also present in b-oxobilanes,[6] which are open-chain tetrapyrroles that can be cyclized with trimethyl orthoformate to afford oxophlorins, and subsequently, porphyrins.[1]
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacenes,[7, 8] known as BODIPY dyes, have been recognized as one of the most versatile fluorophores in the last three decades because of their sharp fluorescence emissions, high quantum yields, and relatively high chemical and thermal stabilities.[9, 10] BODIPY dyes are, therefore, intensively employed as biological labeling reagents[11, 12] in protein and DNA research. BODIPYs are normally synthesized from the corresponding 2,2′-dipyrrins, also known as dipyrromethenes, upon complexation with boron, normally accomplished by the use of BF3·OEt2 in the presence of a tertiary amine, such as NEt3, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), or N,N-diisopropylethylamine (DIEA). The synthesis of the dipyrrin precursors usually follows a route involving acid-catalyzed condensation of two pyrroles with a carbon-linking unit; symmetric systems are normally obtained by condensation of pyrrole with an aldehyde,[13] acyl chloride,[14] or anhydride,[15] whereas unsymmetrical systems are normally obtained by the condensation of a 2-formyl- or 2-acyl-pyrrole with an α-free pyrrole.[16] Using these approaches, a variety of 8-substituted BODIPYs have been reported. An alternative synthetic route[17] to symmetric BODIPYs is from dipyrrylketones, such as 1, which can be obtained by reaction of a pyrrole with thiophosgene, followed by oxidative hydrolysis. Dipyrrylketones are shown to be valuable intermediates in the synthesis of 5-chloro-2,2′-dipyrrins and their boron complexes, which can undergo further reactions with nucleophiles. Herein, we report our results in the synthesis of 5-chloro-2,2′-dipyrrins and 8-chloro-BODIPYs from dipyrrylketones, their reactions with nucleophiles, and their use in Pd0-catalyzed cross-coupling reactions. This work advances previous knowledge[17] by reporting new chemistry of 5-chloro-dipyrrins (including a novel fragmentation reaction), describing reactions of 8-chloro-BODIPYs with carbon nucleophiles, and reporting highly efficient organotin reactions.
Results and Discussion
Symmetric dipyrrylketones 1a,b were obtained in two steps (Scheme 1) by treatment of the corresponding α-free pyrrole 2a,b with thiophosgene, followed by oxidative hydrolysis of the resulting dipyrrylthioketone.[3, 18, 19] Alternatively, the dipyrrylketones could be obtained in similar overall yields by reaction of a 2-N,N-dimethylamidopyrrole with excess phosphoryl chloride, followed by treatment with pyrroles unsubstituted at the 2-position, such as 2a,b, and then 10% aqueous Na2CO3. The dipyrrylketones were characterized by FTIR because they show a characteristic band for the amide-like 5-carbonyl group at approximately ν=1575 cm−1 in chloroform, and by HRMS (ESI), and 1H- and 13C NMR spectroscopies. Dipyrrylketones 1a,b were 5-chlorinated[20] in 91–93% yields by reaction with phosgene in dichloromethane or chloroform, to give the corresponding 5-chloro-dipyrrin salts 3a,b. Other chlorinating agents, including POCl3 and (COCl)2, also gave the desired products, although in lower yields. The 5-chloro-dipyrrins 3a and 3b showed characteristic UV/Vis absorptions at λmax=485 and 505 nm in dichloromethane, respectively, and were also characterized by their HRMS (ESI) and 1H and 13C NMR spectra. Complexation of the 5-chloro-dipyrrins 3a,b with boron, by using BF3·OEt2 under basic conditions, gave the corresponding BODIPYs 4a,b in 94–96% yield. Compounds 4a and 4b were characterized by strong UV/Vis absorptions at 503 and 527 nm and brighter fluorescence emissions at 512 and 542 nm, respectively. Single crystals suitable for X-ray structure analysis were obtained for both 8-chloro-BODIPYs 4a,b (Figure 2). The non-hydrogen atoms, except for fluorine in 4a, are fairly coplanar, with a mean deviation of 0.052 Å, and the BF2 plane is almost orthogonal, with a dihedral angle of 89.88(2)°. Compound 4b has three molecules in the asymmetric unit, two of which have the ethyl groups relatively syn, and one has an anti ethyl group. Analogous dihedral angles in the three molecules are in the range 88.45(2)–89.37(2)°. BODIPYs 5a,b were also obtained from dipyrrylketones 1a,b upon reaction with Meerwein’s salt (triethyloxonium tetrafluoroborate).[21] Treatment of 1a and 1b with Meerwein’s salt in chloroform, at room temperature, gave orange-red colored solutions, indicating the formation of the fully conjugated 5-ethoxy-dipyrrins. After aqueous workup, the crude residues were complexed with boron by using similar conditions to those reported above, to produce BODIPYs 5a,b in 71–82% overall yields (Scheme 1). BODIPYs 5a,b could also be obtained from 4a,b, but in lower yield (46–52 %), by nucleophilic addition/elimination of the 8-chloro group, by using sodium ethoxide in ethanol (see below). BODIPYs 5a,b showed intense UV/Vis absorptions at 487 and 508 nm, respectively, and characteristic resonances for the ethoxy group in their 1H NMR spectra at approximately δ =4.12 (q) and 1.53 (t) ppm. The molecular X-ray structures for both BODIPYs 5a,b were also obtained (Figure 2). Molecule 5a almost has Cs symmetry, with the BF2 plane and dipyrryl plane forming a dihedral angle of 89.37(4)°, and the atoms of the OEt group lying at mean of 0.012 Å from the BF2 plane; 5b has a similar structure, with a dihedral angle of 88.68(1)°, but the dipyrryl ethyl groups are anti to each other.
Scheme 1.

Synthesis of 5-chloro-dipyrrins 3 and BODIPYs 4 and 5.
Figure 2.

Molecular structures of some BODIPY products.
The reactions of 5-chloro-dipyrrinium 3a and BODIPY 4a in the presence of nucleophiles were further investigated. In the presence of sodium methoxide in methanol, THF, or chloroform, compound 3a, surprisingly, produced the monopyrrole methyl ester 6 in 81% yield (Scheme 1). If sodium ethoxide was used, the corresponding ethyl ester 7 was obtained (66% yield). The reaction was followed by UV/Vis spectroscopy until complete disappearance of the absorption band of the 5-chloro-dipyrrin occurred. Pyrroles 6 and 7 were fully characterized, including by X-ray crystallography (see the Supporting Information). Two oxygen atoms are added in this unprecedented dipyrrole cleavage reaction to form 6 or 7, therefore, it is suggested that intermediate 5,5-dimethoxydipyrromethane 8 is involved (Scheme 2), and this intermediate undergoes a base-catalyzed version of the known pyrrole deacylation reaction to give 6 or 7 and the corresponding 2-unsubstituted pyrrole.[22] Weaker nucleophiles, including KCN and nBu4NF, did not produce any substitution product. However, the 5-chlorodipyrrin 3a reacted with ethyl magnesium bromide in THF, at room temperature or under reflux, to give a mixture of the desired 5-ethyl-dipyrrin 9 and the reduced meso-free compound 10 (Scheme 3), which could not be separated by chromatography. Complexation of the mixture with boron by using BF3·OEt2 in DIEA produced the corresponding BODIPYs in 20 and 53% yields, respectively, after separation. BODIPY 11 was obtained in higher yield (75%) by reaction of 4a with ethyl magnesium bromide in THF at −78°C; in this case only 10% of the 8-unsubstituted BODIPY was isolated. When the reaction was performed at 0 °C and the mixture was warmed up to room temperature,[23–25] the triethyl substituted BODIPY, 12, was the only product obtained, in 83% yield. Both BODIPYs 11 and 12 had characteristic UV/Vis absorptions, both at 499 nm, and m/z at 276.1713 and 296.2507, respectively. The 1H NMR spectra of BODIPYs 11 and 12 showed the characteristic 8-ethyl protons centered at δ=3.01 (q) and 1.31 (t) ppm, respectively; the B–ethyl protons of BODIPY 12 appeared as broad signals at δ=0.79 and 0.30 ppm.
Scheme 2.

Proposed mechanism for the conversion of 3a into 6 and 7.
Scheme 3.

Reactions of chlorinated 3a and 4a with Grignard reagents.
Because the 8-chloro-BODIPY 4a gave higher yields for the substitution products compared with dipyrrin 3a, owing to the higher stability of the BODIPY core and easier product purification, additional substitution reactions were investigated for 4a, as shown in Scheme 4 and Table 1. Reaction of BODIPY 4a with sodium methoxide in methanol at room temperature produced the corresponding substitution product 13 in 67% yield, along with dimethoxy (21%) and trimethoxy (very minor) side products, in which one or both the fluorine atoms were also replaced.[26] When the reaction was performed at 80°C trimethoxy-BODIPY 15 was the major product, isolated in 65% yield along with the dimethoxy side product in 16% yield. BODIPY 14 was prepared in 81% yield by reaction of 4a with sodium phenoxide at room temperature in THF. Both BODIPYs 5a and 13 showed significantly blueshifted UV/Vis absorptions, both centered at 487 nm, and for 14 at 491 nm, compared with the starting BODIPY 4a. However, substitution of the fluorine atoms, as in BODIPY 15, had no significant effect on the λmax value, as previously observed. The 1H NMR spectra of BODIPYs 13 and 15 showed the 8-OMe protons at δ=3.98 and 3.97 ppm, respectively, whereas the B-OMe protons of 15 appeared at δ=2.89 ppm. Two sulfur nucleophiles were also reacted with BODIPY 4a, in THF and in the presence of K2CO3 as the base (Scheme 4). 2-Mercaptobenzothiazole gave BODIPY 16 in 56% yield, whereas 1-mercaptomethyl-p-carborane[27] gave 17 in 92% yield. BODIPYs 16 and 17 showed redshifted UV/Vis absorptions at 539 and 529 nm, respectively. The 1H NMR spectra of these BODIPYs showed the aromatic protons of 16 at δ=7.28–7.98 ppm and the SCH2 protons of 17 at δ=2.95 ppm. Both BODIPYs 16 and 17 were characterized by X-ray crystallography (Figure 2). In BODIPY 16, the dipyrryl and BF2 planes form a dihedral angle of 88.03(7)°, whereas the mercaptobenzothiazole plane forms a dihedral angle of 77.03(2)° with the dipyrryl unit. In BODIPY 17, the dipyrryl and BF2 planes form a dihedral angle of 88.8 (1)°, the linkage between dipyrryl and para-carborane is extended, with a C-S-CH2-C torsion angle of 175.3(6)°. The best plane of the CSCC linkage forms a dihedral angle of 83.8(2)° with the dipyrryl plane.
Scheme 4.

Nucleophilic substitutions/eliminations of BODIPY 4a.
Table 1.
Stille coupling reactions of 8-chloro-BODIPY 4a.
| |||
|---|---|---|---|
| BODIPY | Organotin | R | Yield [%] |
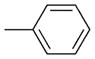
| 18 | Bu3SnPh |
|
93 |
| 19 | Me4Sn | —CH3 | 97 |
| 20 | Bu3Sn(CH=CH2) |
|
97 |
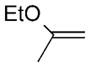
| 21 | Bu3Sn(EtOC=CH2) |
|
93 |
| 22 | Bu3Sn(C≡CH) | —C≡CH | 31 |
| 23 | Bu3Sn-C≡C–Si(Me)3 | —C≡C–Si(Me)3 | 71 |
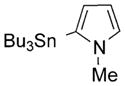
| 24 |
|
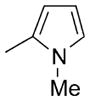
|
95 |
| 25 |
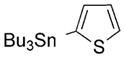
|
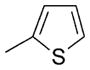
|
99 |
Though 8-chloro-BODIPYs react efficiently in nucleophilic addition–elimination reactions with strong nucleophiles (Scheme 4 and [17]), the 8-chloro-BODIPY 4a was unreactive in the presence of weaker nucleophiles, such as KCN and nBu4NF.
Pd0-catalyzed cross-coupling procedures on BODIPYs have been explored, including the Suzuki,[28] Stille,[29] Heck,[30] and Sonogoshira[31] reactions. These transformations have been performed either directly on the BODIPY core[32–35] or on a substituent,[36] and have facilitated the functionalization of BODIPYs, especially coupling reactions of 3,5- and 2,6-dihalogenated-BODIPYs. In comparison with the pyrrolic positions, the meso-carbon is more electrophilic, as suggested by Mulliken charge distribution and reactivity studies.[37] Recently, various Pd0-catalyzed cross-coupling reactions were performed on 8-halo-BODIPYs[17] with arylboronic acids or aryltin reagents under Suzuki or Stille conditions, and with phenylacetylene under Sonogashira conditions. The products from these reactions were obtained in 36–76% yields, with the lowest yield obtained for the 8-chloro-BODIPY under Suzuki conditions. Concurrently, we were also investigating the Pd0-catalyzed coupling reactions of 8-chloro-BODIPY 4a and the effect of the 1,7-methyl groups on the cross-coupling reactions. A Suzuki reaction[38] between 8-chloro-BODIPY 4a and phenylboronic acid using [Pd(PPh3)4] as the catalyst and K2CO3 as the base in 1,2-dimethoxyethane (DME), at reflux for 24 h, produced the 8-phenyl-BODIPY 18 in 47% yield. A side product from this reaction was the corresponding meso-unsubstituted BODIPY, along with other pyrrolic byproducts from loss of the BF2 moiety under the relatively harsh conditions. BODIPY 18 was synthesized in higher yield (see Table 1) under milder Stille cross-coupling conditions[39] by using tributylphenylstannane and [Pd(PPh3)4], without any base. The reaction mixture was heated at reflux in toluene and TLC analysis indicated that the reaction was complete within five hours, having proceeded in almost quantitative yield. Therefore, we investigated additional Pd0-catalyzed Stille cross-coupling reactions of 8-chloro-BODIPY 4a (Table 1). Various organotin reagents bearing alkyl, alkenyl, alkynyl, and heterocyclic aromatic groups were used in the cross-coupling reactions. All of the coupling reactions produced the corresponding 8-functionalized BODIPYs in almost quantitative yields, except for the reaction with tributylethynyltin, which produced BODIPY 22 in only 31% yield.[40] However, use of the trimethylsilyl-protected organotin reagent afforded a 71% yield of the trimethylsilylalkyne substituted BODIPY 23. The yields for the alkynyl derivatives 22 and 23 are lower than for all other coupling products (Table 1), particularly for 22, probably because of the higher reactivity of the unprotected meso-alkynyl group.
The structures of BODIPYs 18–25 were confirmed by 1H and 13C NMR spectroscopy, HRMS (ESI-TOF) mass spectrometry, UV/Vis spectroscopy, and X-ray crystallography (Figure 2); BODIPYs 12, 15 and the mono MeO–B–F compound were also investigated by using 11B NMR spectroscopy. The 1H NMR spectrum of 8-phenyl-BODIPY 18 showed aromatic protons between δ= 7.27–7.49 ppm, consistent with literature reports,[41] and a m/z peak at 324.1717 in the mass spectrum. The meso-methyl group of BODIPY 19 appeared at δ=2.40 ppm in its 1H NMR spectrum and the product showed a m/z peck at 262.1556 in the mass spectrum. For BODIPYs 20 and 21, the 1H NMR spectrum showed the alkene protons at δ=5.56 (d), 5.69 (d), and 6.74 (dd) ppm or as two doublets at δ=4.38(d) and 4.50(d) ppm, respectively. The ethoxy group of 21 produced signals at δ=3.89 (q) and 1.39 (t) ppm in the 1H NMR spectrum and the mass spectra for BODIPYs 20 and 21 showed m/z at 274.1557 and 318.1817, respectively. BODIPY 22 showed a typical peak at δ=3.92 ppm for the terminal alkynyl proton (absent in 23) in its 1H NMR spectrum, and m/z at 272.1380. The meso-rings of BODIPYs 24 and 25 showed peaks between δ=6.06–6.77 and 6.98–7.51 ppm, respectively, and the N-CH3 of 24 showed a peak at δ=3.40 ppm in the 1H NMR spectra. These two BODIPYs were also characterized by m/z at 327.1799 and 352.1094, respectively. The X-ray crystal structures of BODIPYs 18, 19, 20, 24, and 25 were obtained by slow diffusion of hexane into a dichloromethane solution, and are shown in Figure 2. BODIPY 18 deviates from its potential Cs symmetry because the BF2 and phenyl planes are twisted in opposite directions, away from orthogonality with the dipyrryl plane. The BF2 moiety forms a dihedral angle of 88.13(5)° and the phenyl moiety a dihedral angle of 82.40(3)°. BODIPY 19 is unusual in that its dipyrryl group lies in a crystallographic mirror and its BF2 group lies across it; thus, they are exactly orthogonal. The X-ray structure for BODIPY 20 was also obtained (Figure 2). Its dipyrryl and BF2 planes form a dihedral angle of 88.82(2)°. The ethene substituent is not quite orthogonal to the dipyrryl nor coplanar with the BF2 moiety, a C–C–C=C torsion angle about the bond being 72.65(10)°. BODIPY 24 deviates only slightly from Cs symmetry, with the BF2 plane forming a dihedral angle of 89.51(9)° with the dipyrryl plane and the methylpyrrole plane forming an angle of 85.77(4)° with it. In Figure 2, only one of the major orientations of the disordered thiophene is shown for BODIPY 25; it forms a dihedral angle of 84.31(8)° with dipyrryl, and the BF2 plane forms a dihedral angle of 88.56(4)° with the dipyrryl plane.
The spectroscopic properties of the BODIPYs were investigated and their UV/Vis absorption and steady-state fluorescence emission spectra in dichloromethane were recorded. As an example, Figure 3 shows the normalized absorption and fluorescence spectra of BODIPYs 4a, 5a, and 22 (see the Supporting Information for all other BODIPY spectra). Table 2 summarizes the spectroscopic data obtained for the BODIPYs, including their maximum absorption and fluorescence wavelengths, molar extinction coefficients, and fluorescence quantum yields in dichloromethane. As expected, literature compounds 18 and 19 showed absorption and emission profiles similar to those previously reported.[42] The new BODIPYs, 4a and 4b, show spectra of comparable shape and characteristics to other BODIPYs described in the literature, with a strong absorption band corresponding to the S0–S1 (π–π*) transitions centered at 503 and 527 nm, respectively. The approximate 25 nm redshift observed in the absorption and emission maxima is due to the increased alkyl substitution of the BODIPY core (i.e., the 2,6-diethyl substituents in 4b and 5b).[41] The replacement of the 8-chloro group on BODIPY 4a with an alkyl (11 and 19), alkoxy (5a and 13), or phenoxy (14) moiety caused blueshifts of the absorption and emission maxima in the order of 16–19 nm for the alkoxy-BODIPYs, 12–13 nm for the aryloxy-BODIPY and 4–7 nm for the alkyl-BODIPYs. On the other hand, substitution with a phenyl (18) or vinyl (20) group had no noticeable effect on the absorption and emission wavelengths, whereas ≈10 nm redshifts were observed for BODIPYs 21, 24, and 25, and the largest redshifts were obtained for the meso-thio (16) and meso-alkynyl (22) BODIPYs (≈40 nm). The fluorescence quantum yields for BODIPYs 4a and 4b were found to be 0.51 and 0.33, respectively, owing to the heavy-atom effect of the chlorine and introduction of the two ethyl groups in 4b. In accordance with the literature,[17] replacement of the meso-chloro with an alkoxy or phenoxy group caused a significant increase in quantum yield, particularly for BODIPYs 5a and 14 (Φf≈1.0), whereas substitution with a meso-thiol had the opposite effect, Φf of 0.04 and 0.09 were found for BODIPYs 16 and 17, respectively. Substitution of the 8-chloro group with an alkyl (methyl and ethyl) or phenyl group also increased the quantum yields to Φf≈0.8 (for 11 and 19) and 0.62 for 18. However, the quantum yields for the meso-alkenyl, alkynyl, pyrrolyl, and thienyl-BODIPYs 20–25 were appreciably lower, particularly for 20, the fluorescence of which was essentially quenched, due to the greater freedom of rotation for these smaller groups increasing the energy lost to non-radiative decay.[43] Boron substitution had no significant effect on the absorption and emission wavelengths, but did influence the fluorescence quantum yield. Although boron substitution with two methoxy groups significantly increased the quantum yield of BODIPY 15 (Φf=1.0) relative to 13 (Φf= 0.57), substitution with two ethyl groups reduced the quantum yield of BODIPY 12 (Φf=0.32) relative to 11 (Φf=0.83).
Figure 3.

Normalized UV/Vis absorption (solid line) and fluorescence emission spectra (dashed line) of selected BODIPYs 5a, 4a, and 22 (left to right), in dichloromethane.
Table 2.
Spectral properties of BODIPYs in dichloromethane solution at room temperature. The fluorescence quantum yields (λexc=490 nm) were calculated by using, rhodamine 6G (0.80 in ethanol) as the reference.
| BODIPY | Absorbance λmax [nm] | Emission λmax [nm] | Φf | Log ε [M−1cm−1] |
|---|---|---|---|---|
| 4a | 503 | 512 | 0.51 | 4.79 |
| 4b | 527 | 542 | 0.33 | 4.50 |
| 5a | 487 | 493 | 0.99 | 4.77 |
| 5b | 508 | 518 | 0.67 | 4.44 |
| 11 | 499 | 505 | 0.83 | 4.75 |
| 12 | 499 | 507 | 0.32 | 4.20 |
| 13 | 487 | 493 | 0.57 | 4.95 |
| 14 | 491 | 499 | 0.99 | 4.71 |
| 15 | 487 | 496 | 1.00 | 4.67 |
| 16 | 539 | 553 | 0.04 | 4.69 |
| 17 | 529 | 542 | 0.09 | 4.23 |
| 18 | 501 | 511 | 0.62 | 4.76 |
| 19 | 497 | 508 | 0.77 | 4.82 |
| 20 | 505 | 511 | 0.00 | 4.67 |
| 21 | 513 | 522 | 0.14 | 4.61 |
| 22 | 544 | 556 | 0.29 | 4.57 |
| 23 | 545 | 556 | 0.13 | 4.63 |
| 24 | 513 | 521 | 0.10 | 4.73 |
| 25 | 514 | 521 | 0.05 | 4.87 |
Conclusion
The synthesis of BODIPY dyes from dipyrrylketone precursors has been explored. New BODIPY compounds were obtained that are usually very difficult to synthesize by conventional methods. The transformation of dipyrrylketones into 5-chlorodipyrrins was studied, and further reactions involving electrophilic, nucleophilic addition/elimination, and coupling reactions were reported. In this context an unexpected cleavage reaction of 5-chloro-dipyrrins to give monopyrrole-2-carboxylates was discovered. The use 5-chloro-dipyrrins represents an alternative method with broad applicability for the modification of BODIPYs, especially at their 8-meso-carbon. Variations in the photophysical properties of the various new BODIPYs are discussed and interpreted, emphasizing the significant effect caused by meso-substitution. Finally, the X-ray structures of eleven BODIPYs are reported and analyzed within the context of BODIPY solid-state structural characteristics.
Experimental Section
General
Commercially available reagents were purchased from Sigma–Aldrich Co., and were used without further purification. All air and moisture sensitive reactions were performed under an argon atmosphere Melting points were measured on an Electrothermal MEL-TEMP instrument. Analytical thin-layer chromatography (TLC) was performed on polyester backed TLC plates 254 (precoated, 200 μm, Sorbent Technologies). Column chromatography was performed on silica gel (Sorbent Technologies, 60 Å, 40–63 μm) 1H-, 13C- and 11B NMR spectra were recorded, by using a Bruker AV-400 spectrometer, in CDCl3 (7.26 ppm, 1H and 77.0 ppm, 13C, and BF3.Et2O at 0.0 ppm, 11B). Chemical shifts are given in parts per millions (ppm) relative to tetramethylsilane (TMS, 0 ppm); multiplicities are indicated as br (broad), s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). All spectra were recorded at room temperature. Mass spectra were obtained at the LSU Department of Chemistry Mass Spectrometry Facility using an Applied Biosystems QSTAR XL ESI-TOF.
1,3,7,9-Tetramethyldipyrryl-5-ketone 1a
A solution of thiophosgene (1.15 g, 10.0 mmol) in toluene (10 mL) was added dropwise to a solution of 2,4-dimethylpyrrole (2.0 g, 21.0 mmol) in dry ether (30 mL) at 0 °C. The mixture was stirred for about 1 h (until TLC analysis showed the starting material had disappeared). Then, methanol (30 mL) was added and the reaction mixture was stirred for another 30 min. The solvents were removed in vacuo and the residue was purified on a silica-gel column eluted with toluene/chloroform (4:1). The thioketone product was obtained as an orange solid. (980 mg, 40%); m.p. 180–181 °C; 1H NMR (400 MHz; CDCl3): δ=8.96 (s, 2H), 5.91 (s, 2H), 2.27 (s, 6H), 2.07 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=175.30, 133.30, 127.98, 127.56, 111.74, 13.09, 12.65 ppm; HRMS (ESI): m/z calcd for C13H16N2S: 233.1107 [M+H+]; found: 233.1106. To the thioketone (232 mg, 1.0 mmol) in EtOH (95 %, 100 mL) was added KOH (1.0 g), and to this mixture was added aqueous H2O2 (5 %, 10 mL) dropwise in an ice bath. Then, the solution was heated in a steam bath for about 5 min. The solution was cooled to room temperature and water (300 mL) was added and the solution was extracted with chloroform three times. The organic layers were collected and washed with water, brine and then dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by sublimation to give the product dipyrrylketone as a pale yellow solid. (170 mg, 78%); m.p. 232–233°C; 1H NMR (400 MHz; CDCl3): δ=8.63 (s, 2H), 5.85 (s, 2H), 2.28 (s, 6H), 2.19 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=175.30, 133.30, 127.98, 127.56, 111.74, 13.09, 12.65 ppm; IR ν̃max (C=O) (Nujol)=1573 cm−1; UV/Vis (CH2Cl2): λmax (ε)=295 (8600), 341 nm (14 600); HRMS (ESI): m/z calcd for C13H16N2O: 217.1335 [M+H+]; found: 217.1334.
2,8-Diethyl-1,3,7,9-tetramethyl-5-dipyrrylketone 1b
Method A
4-Ethyl-3,5-dimethylpyrrole-2-dimethylamide (8.0 g. 41.2 mmol) was dissolved in excess POCl3 and warmed at 50°C for 15 min before being evaporated to dryness. The resulting complex (λmax=380 nm) was taken up in CH2Cl2 (20 mL). 3-Ethyl-2,4-dimethylpyrrole (5.6 g, 45.5 mmol) was then added and the mixture was heated under reflux for 90 min. The brown solution (λmax=408 nm) was stirred vigorously with 10% aq Na2CO3 (100 mL) and heated under reflux for 2 h. The dipyrrylketone product, precipitated from solution as a yellow solid, was filtered off and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (50 mL) and the combined organic extracts were washed with water and dried over anhydrous MgSO4 before evaporation to small volume. The yellow solid that precipitated was combined with the earlier solid and was recrystallized from CH2Cl2 to give 1b (10.1 g, 89%) as pale yellow needles.
Method B
The synthesis of 1b followed the procedure reported above for 1a. Thioketone yield: (1.30 g, 43%); m.p. 172–174°C; 1H NMR (400 MHz; CDCl3): δ=8.95 (s, 2H), 2.37 (q, J=7.5 Hz, 4H), 2.23 (s, 6H), 2.00 (s, 6H), 1.05 ppm (t, J=7.5 Hz, 6H); 13C NMR (100 MHz; CDCl3): δ=189.39, 136.19, 134.67, 126.67, 125.56, 17.47, 14.99, 11.73, 10.88 ppm; HRMS (ESI): m/z calcd for C17H25N2S: 289.1733 [M+H+]; found: 289.1739. Dipyrrylketone 1b yield: (224 mg, 82%); m.p. 205–207 °C (literature[41] 207°C); 1H NMR (400 MHz; CDCl3): δ=8.61 (s, 2H), 2.40 (q, J=7.5 Hz, 4H), 2.23 (s, 6H), 2.14 (s, 6H), 1.07 ppm (t, J=7.5 Hz, 6H); 13C NMR (100 MHz; CDCl3): δ=175.23, 129.65, 127.24, 125.27, 124.25, 17.37, 15.20, 11.41, 10.70 ppm; IR ν̃max (C=O) (CHCl3 or Nujol)=1575 cm−1; UV/Vis (CHCl3): λmax (ε)=288 (11700), 357 nm (25 000); HRMS (ESI): m/z calcd for C17H25N2O: 273.1967 [M+H+]; found: 273.2013.
5-Chloro-1,3,7,9-tetramethyldipyrromethene hydrochloride 3a
Dipyrrylketone 1a (314 mg, 1.45 mmol) was dissolved in CHCl3 and phosgene in toluene solution was added before stirring at room temperature for 1 h. When the reaction was complete according to UV/Vis spectroscopy and TLC analysis, nitrogen gas was passed through solution to purge it of excess phosgene, which was directed to a NaHCO3 solution trap. Solvent was then removed in vacuo and the red solid was recrystallized from CHCl3/petroleum ether to give 3a as a red solid (356 mg, 91%); m.p.> 260°C; 1H NMR (400 MHz; CDCl3): δ=12.93 (s, 2H), 6.23 (s, 2H), 2.59 (s, 6H), 2.34 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=153.70, 144.35, 139.63, 128.18, 120.71, 16.27, 14.38 ppm; UV/Vis (CH2Cl2): λmax (ε)= 485 nm (23 500); HRMS (ESI): m/z calcd for C13H16ClN2: 235.0997; found: 235.0992.
5-Chloro-2,8-diethyl-1,3,7,9-tetramethyldipyrromethene hydrochloride 3b
Yield: (439 mg, 93%); m.p.>300°C; 1H NMR (400 MHz; CDCl3): δ=12.50 (s, 2H), 2.35 (s, 6H), 2.15 (s, 6H), 2.40 (q, J=7.6 Hz, 4H), 1.05 ppm (t, J=7.6 Hz, 6H); 13C NMR (100 MHz; CDCl3): δ=152.41, 138.81, 137.68, 133.13, 128.08, 17.44, 14.28, 12.72, 12.63 ppm; UV/Vis (CH2Cl2): λmax (ε)=509 nm (57 500); HRMS (ESI): m/z calcd for C17H24ClN2: 291.1623; found: 291.1635; elemental analysis calcd (%) for C17H24Cl2N2: C 62.4, H 7.3, N 8.6; found: C 62.7, H 7.5, N 8.8.
BODIPY 4a
Chloro-dipyrrin hydrochloride 3a (235 mg, 0.87 mmol) was dissolved in CHCl3 and DIEA (8 mmol, 9.0 equiv) was added before the mixture was stirred for 30 min. Then, BF3·OEt2 (14 mmol, 16 equiv) was added. The resulting mixture was stirred overnight at room temperature. The solution was washed with saturated aq NaHCO3, brine and then dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by chromatography on silica gel (eluted with CH2Cl2/hexane) to give the BODIPY 4a (230 mg, 94%); m.p. 235–237°C; 1H NMR (400 MHz; CDCl3): δ= 6.09 (s, 2H), 2.52 (s, 6H), 2.45 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=155.26, 142.84, 136.54, 129.72, 121.36, 16.68, 14.54 ppm; UV/Vis (CH2Cl2): λmax (ε)=503 nm (61 700); HRMS (ESI): m/z calcd for C13H15ClF2N2: 282.1016; found: 282.1010.
BODIPY 4b
BODIPY 4b was prepared, using the same procedure as described above, from 5-chloro-dipyrrin hydrochloride 3b (325 mg, 96%); m.p. 173–175 °C; 1H NMR (400 MHz; CDCl3): δ=2.50 (s, 6H), 2.41 (m, 10H), 1.05 ppm (t, J=7.6 Hz, 6H); 13C NMR (100 MHz; CDCl3): δ=153.36, 138.10, 135.16, 133.20, 129.16, 17.11, 14.75, 13.82, 12.49 ppm; UV/Vis (CH2Cl2): λmax (ε)=527 nm (31 600); HRMS (ESI): m/z calcd for C17H22BClFN2: 319.1549 [M+−F]; found: 319.1550.
BODIPY 5a
Method A
Dipyrrylketone 1a (60 mg, 0.27 mmol) was dissolved in dry CHCl3 (20 mL) and triethyloxonium tetrafluoroborate (105.1 mg, 0.54 mmol) was added. The solution turned red with time and UV/Vis spectroscopy showed a sharp peak at 438 nm. The mixture was stirred overnight and then washed with water, brine and finally dried over anhydrous Na2SO4. After evaporation, the residue was dissolved in CHCl3 (30 mL) and DIEA (1.62 mmol, 6 equiv) was added and the solution was stirred for about 30 min before addition of BF3·OEt2 (2.7 mmol, 10 equiv); the solution was then stirred overnight. UV/Vis spectroscopy showed a sharp peak at 487 nm. Then, the mixture was washed with aq NaHCO3, brine and then dried over anhydrous Na2SO4. The residue was purified by silica-gel column chromatography (eluted with CH2Cl2/hexane) to give BODIPY 5a as an orange red solid (64 mg, 82%).
Method B
BODIPY 4a (56.4 mg, 0.20 mmol) was dissolved in MeOH in an ice bath and NaOEt (20.4 mg, 0.3 mmol) was added. The mixture was stirred for 45 min and was monitored by TLC analysis and UV/Vis spectroscopy. The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by chromatography (eluted with CH2Cl2/hexane) to give an orange solid (26.8 mg, 46%); m.p. 126–128 °C; 1H NMR (400 MHz; CDCl3): δ=6.02 (s, 2H), 4.12 (q, J=7.1 Hz, 2H), 2.51 (s, 6H), 2.38 (s, 6H), 1.53 ppm (t, J=7.1 Hz, 3H); 13C NMR (100 MHz; CDCl3): δ= 159.12, 154.13, 139.13, 127.94, 119.57, 74.14, 15.31, 14.45, 13.75 ppm; UV/Vis (CH2Cl2): λmax (ε)=487 nm (58 900); HRMS (ESI): m/z calcd for C15H19BF2N2O: 292.1668; found: 292.1654.
BODIPY 5b
Method A: Yield: (66 mg, 71%); Method B: Yield: (36.2 mg, 52%); m.p. 135–137 °C; 1H NMR (400 MHz; CDCl3): δ=4.09 (q, J=7.1 Hz, 2H), 2.48 (s, 6H), 2.48 (q, J=7.5 Hz, 4H), 2.32 (s, 6H), 1.55 (t, J= 7.1 Hz, 3H), 1.06 ppm (t, J=7.5 Hz, 6H); 13C NMR (100 MHz; CDCl3): δ=158.04, 152.19, 134.73, 131.53, 73.88, 17.04, 15.29, 14.68, 12.41, 11.22 ppm; UV/Vis (CH2Cl2): λmax (ε)=508 nm (27 500); HRMS (ESI): m/z calcd for C19H27BFN2O (M+−F): 329.2200; found: 329.2167.
Methyl 3,5-dimethylpyrrole-2-carboxylate 6
Dipyrromethene 3a (47.0 mg, 0.17 mmol) was dissolved in CHCl3 and 0.5 M NaOMe in MeOH (1.0 mL, 0.5 mmol) was added at room temperature; the mixture was stirred overnight and the reaction was monitored by TLC analysis and spectrophotometry. The solution was washed with water, brine and then dried over anhydrous Na2SO4. The solvent was removed and the product was purified by silica-gel column chromatography (eluted with MeOH/CH2Cl2); Yield: 21 mg, 80.8%; m.p. 93–95°C (literature[44] 98–99°C); 1H NMR (400 MHz; CDCl3): δ=9.27 (s, 1H), 5.79 (s, 1H), 3.83 (s, 3H), 2.30 (s, 3H), 2.25 ppm (s, 3H); 13C NMR (100 MHz; CDCl3): δ=163.43, 132.97, 129.14, 117.53, 111.34, 50.92, 12.96, 12.82 ppm; HRMS (ESI): m/z calcd for C8H12NO2: 154.0868 [M+H+]; found: 154.0870 (see the Supporting Information for X-ray structure).
Ethyl 3,5-dimethylpyrrole-2-carboxylate 7
This compound was prepared as described for pyrrole 6, except that sodium ethoxide was used as the base in ethanol. Yield: 19.0 mg, 66.2%; m.p. 118–119°C (literature[45] 125°C); 1H NMR (400 MHz, CDCl3): δ=8.68 (s, 1H), 5.79 (s, 1H), 4.31 (q, J=7.1 Hz, 2H), 2.30 (s, 3H), 2.24 (s, 3H), 1.34 ppm (t, J=7.1 Hz, 3H); 13C NMR (100 MHz; CDCl3): δ=161.69, 132.28, 129.02, 117.78, 111.35, 59.68, 14.57, 13.07, 12.77 ppm; HRMS (ESI): m/z calcd for C9H14NO2: 168.1019 [M+H+]; found: 168.1016 (see the Supporting Information for X-ray structure).
BODIPY 11
Method A
5-Chloro-dipyrrin 3a (100 mg, 0.37 mmol) was dissolved in CHCl3 (40 mL) and 1.0 M EtMgBr in THF (1.30 mL, 1.30 mmol) was added dropwise at 0 °C; the reaction was left stirring for 1 h at 0 °C and then for another 2 h at room temperature. Then, the solution was poured into ice water to destroy excess EtMgBr, and extracted with CHCl3. The organic layers were combined and washed with water, brine and then dried over anhydrous Na2SO4. After evaporation, the residue (a mixture of compounds 9 and 10) in CHCl3 (30 mL) and DIEA (0.45 mL, 7 equiv, 2.58 mmol) in an ice bath was treated with BF3·OEt2 (0.53 mL, 12.0 equiv, 4.3 mmol) and the mixture was stirred overnight. The solvent was removed and the residue was purified on a silica-gel column (eluted with CH2Cl2/hexane) to give BODIPY 11. Yield: 20.1 mg, 19.6%.
Method B
ChloroBODIPY 4a (56.4 mg, 0.2 mmol) was dissolved in anhydrous THF and 1.0 M EtMgBr in THF (0.5 mL, 0.5 mmol) was added dropwise to the solution at −70°C with vigorous stirring. When the reaction was complete, according to TLC analysis and spectrophotometry, the solution was poured into ice water to destroy the excess Grignard reagent. Then the mixture was extracted three times with CHCl3, the organic phases were combined and washed with water, brine and then dried over anhydrous Na2SO4. The solvent was evaporated and the residue was purified by silicagel column chromatography (eluted with CH2Cl2/hexane) to give the product as an orange-red solid (40 mg, 75%); m.p. 214–216°C; 1H NMR (400 MHz; CDCl3): δ=6.05 (s, 2H), 3.01 (q, J=7.5 Hz, 2H), 2.52 (s, 6H), 2.44 (s, 6H), 1.32 ppm (t, J=7.5 Hz, 3H); 13C NMR (100 MHz; CDCl3): δ=153.87, 147.87, 140.30, 131.20, 121.55, 21.29, 16.30, 15.45, 14.45 ppm; HRMS (ESI): m/z calcd for C15H19BF2N2: 276.1718; found: 276.1713; UV/Vis (CH2Cl2): λmax (ε) 499 nm (56 200). The meso-unsubstituted BODIPY was also eluted from the column (6.0 mg, 10%); m.p. 185–187°C; 1H NMR (400 MHz; CDCl3): δ=7.04 (s, 1H), 6.05 (s, 2H), 2.53 (s, 6H), 2.25 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=156.75, 141.20, 133.41, 120.07, 119.02, 14.67, 11.28 ppm; UV/Vis (CH2Cl2): λmax (ε)=507 nm (19 600); HRMS (ESI): m/z calcd for C13H15BF2N2: 248.1405; found: 248.1404.
BODIPY 12
1.0 M Ethyl magnesium bromide (1.0 mL, 1.0 mmol) in THF was added to chloroBODIPY 4a (56.4 mg, 0.2 mmol) in THF (20 mL) at room temperature and stirred for 1 h; the reaction was quenched by addition of aq NH4Cl and then extracted with CHCl3. The organic extract was washed with water, brine and then dried over anhydrous Na2SO4. BODIPY 12 was isolated as the sole product by using silica-gel column chromatography (eluted with CH2Cl2/hexane). Yield: 49 mg, 83 %; m.p. 135–137 °C; 1H NMR (400 MHz; CDCl3): δ=6.08 (s, 2H), 3.07 (q, J=7.5 Hz, 2H), 2.46 (s, 6H), 2.44 (s, 6H), 1.31 (t, J=7.5 Hz, 3H), 0.79 ppm (m, 4H), 0.30 (m, 6H); 13C NMR (100 MHz; CDCl3): δ=150.08, 135.52, 131.77, 122.06, 21.34 16.97, 16.39, 15.93, 9.00, 1.02 ppm; 11B NMR (128 MHz; CDCl3): δ= 1.37 ppm; UV/Vis (CH2Cl2): λmax (ε)=499 nm (15 800); HRMS (ESI): m/z calcd for C19H30BN2: 296.2533; found: 296.2507.
BODIPY 13
8-Chloro-BODIPY 4a (56.4 mg, 0.2 mmol) was dissolved in MeOH (30 mL) in an ice bath and NaOMe (16.5 mg, 0.3 mmol) was added; the mixture was stirred for 45 min and the reaction was monitored by TLC analysis and spectrophotometry. The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by silica-gel column chromatography (eluted with CH2Cl2/hexane). The major fraction was the desired BODIPY product 13. Yield: 37.1 mg, 67%; m.p. 160–161°C; 1H NMR (400 MHz; CDCl3): δ=6.03 (s, 2H), 3.98 (s, 3H), 2.51 (s, 6H), 2.40 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=159.95, 154.76, 138.33, 128.30, 119.56, 64.32, 49.12, 14.47, 13.76 ppm; UV/Vis (CH2Cl2): λmax (ε)= 487 nm (89 100); HRMS (ESI): m/z calcd for C14H18BF2N2O: 278.1511; found: 278.1504. The monomethoxyboron-substituted BODIPY was also separated as a minor fraction from the column (Yield: 12.0 mg, 21%); 1H NMR (400 MHz; CDCl3): δ=6.02 (s, 2H), 3.97 (s, 3H), 2.89 (s, 3H), 2.51 (s, 6H), 2.37 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=159.95, 154.76, 138.33, 128.30, 119.56, 64.24, 49.12, 14.47, 13.76 ppm; 11B NMR (128 MHz; CDCl3): δ=1.58 ppm; HRMS (ESI): m/z calcd for C14H17BFN2O: 259.1418 [M+−OMe]; found: 259.1392. A small amount of the dimethoxyboron BODIPY 15 was also isolated from the column, but in insufficient quantity to fully characterize.
BODIPY 14
8-Chloro-BODIPY 4a (56.4 mg, 0.2 mmol) was dissolved in THF (30 mL) in an ice bath and sodium phenoxide (34.8 mg, 0.3 mmol) was added; the mixture was stirred for 2 h at room temperature and monitored by TLC analysis and spectrophotometry. The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by silica-gel column chromatography (eluted with CH2Cl2/hexane) to give an orange solid (55 mg, 81%); m.p. 188–190 °C; 1H NMR (400 MHz; CDCl3): δ=7.36–7.25 (m, 2H), 7.08–7.06 (m, 1H), 7.01–6.99 (m, 2H), 5.99 (s, 2H), 2.55 (s, 6H), 2.04 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=157.52, 155.78, 151.59, 140.42, 130.08, 127.62, 122.84, 119.99, 114.70, 14.60, 14.14 ppm; UV/Vis (CH2Cl2): λmax (ε)=491 nm (51 300); HRMS (ESI): m/z calcd for C19H20BF2N2O: 340.1668; found: 340.1662.
BODIPY 15
8-Chloro-BODIPY 4a (56.4 mg, 0.2 mmol) was dissolved in MeOH (30 mL) and NaOMe (54.0 mg, 0.75 mmol) was added before the solution was heated at reflux temperature, under argon for 12 h. The solvent was removed and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was separated by silica-gel chromatography (eluted with MeOH/CH2Cl2). Yield: 38 mg, 65%; m.p. 93–95°C; 1H NMR (400 MHz; CDCl3): δ=6.01 (s, 2H), 3.97 (s, 3H), 2.86 (s, 6H), 2.48 (s, 6H), 2.41 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ= 159.92, 154.87, 137.38, 129.02, 119.33, 64.13, 49.04, 14.41, 13.81 ppm; 11B NMR (128 MHz; CDCl3): δ=2.39 ppm; UV/Vis (CH2Cl2): λmax (ε)=487 nm (46 800); HRMS (ESI): m/z calcd for C16H23BN2NaO3: 324.173 [M++Na]; found: 324.1719. The monomethoxyboron-substituted BODIPY (see above) was also separated as a minor fraction from the column (Yield: 9.3 mg, 16 %).
BODIPY 16
A solution of 8-chloro-BODIPY 4a (56.4 mg, 0.2 mmol), 2-mercaptobenzothiozole (40.0 mg, 0.24 mmol), and K2CO3 (41.5 mg, 0.3 mmol) in THF (30 mL) was stirred at room temperature for 12 h or until the starting material was almost completely consumed. The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by silica-gel column chromatography (eluted with CH2Cl2/hexane). Yield: 46.1 mg, 56%; m.p. 208–210°C. 1H NMR (400 MHz; CDCl3): δ=7.90 (dt, J=8.4, 0.9 Hz, 1H), 7.68 (m, 1H), 7.44 (ddd, J=8.4, 7.3, 1.3 Hz, 1H), 7.31 (ddd, J= 8.3, 7.3, 1.2 Hz, 1H), 6.10 (s, 2H), 2.58 (s, 6H), 2.46 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=166.56, 158.25, 153.59, 144.75, 135.37, 134.42, 128.70, 126.46, 124.76, 123.07, 122.00, 121.08, 17.26, 14.97 ppm; UV/Vis (CH2Cl2): λmax (ε)=539 nm (49 000); HRMS (ESI): m/z calcd for C20H18BFN3S: 394.1019 [M−F]; found: 394.1010.
BODIPY 17
A solution of 8-chloro-BODIPY 4a (56.4 mg, 0.2 mmol), p-carborane thiol (45.6 mg, 0.24 mmol), and K2CO3 (41.5 mg, 3.0 mmol) in THF (30 mL) was stirred at room temperature for 12 h or until the starting material was consumed, The solvent was removed in vacuo and the residue was taken up in CHCl3 and washed with water, brine and then dried over anhydrous Na2SO4. The product was purified by column chromatography (eluted with CH2Cl2/hexane) to give a red solid (80 mg, 92%); m.p. 150–152°C; 1H NMR (400 MHz; CDCl3): δ=6.06 (s, 2H), 2.95 (s, 2H), 2.51 (s, 3H), 2.49 (s, 3H), 2.17–1.68 ppm (m, 11H); 13C NMR (100 MHz; CDCl3): δ=156.14, 143.51, 136.84, 134.23, 122.50, 59.04, 46.04, 31.91, 16.91, 14.68 ppm; UV/Vis (CH2Cl2): λmax (ε)=529 nm (17 000); HRMS (ESI): m/z calcd for C16H26B11F2N2S: 437.2808; found: 437.3033.
General procedure for Stille coupling reactions
8-Chloro-BODIPY 4a (56.4 mg, 0.2 mmol), [Pd(PPh3)4] (10.0 mg, 5% mmol) and organotin reagent (0.24–0.30 mmol) were added to a round-bottomed flask. The flask was flushed with argon and then anhydrous toluene was added, followed by injection of the Stille reagents. The mixture was refluxed for 5 h, after which time the reaction was complete; the toluene was then removed and the residue was taken up in dichloromethane and washed with water, brine and then dried over anhydrous Na2SO4. The crude product was purified by silica-gel column chromatography (eluting with CH2Cl2/hexane).
BODIPY 18
Yield: 60.1 mg, 93%; m.p. 165–166°C; 1H NMR (400 MHz; CDCl3): δ=7.49–7.47 (m, 3H), 7.29–7.27(m, 2H), 5.98 (s, 2H), 2.56 (s, 6H), 1.37 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ= 154.90, 142.19, 140.52, 131.14, 123.79, 121.11, 17.24, 14.55 ppm; UV/Vis (CH2Cl2): λmax (ε)=501 nm (57 500); HRMS (ESI): m/z calcd for C19H20BF2N2: 324.1718; found: 324.1717.
BODIPY 19
Yield: 50.7 mg, 97%; m.p. 245–247°C; 1H NMR (400 MHz; CDCl3): δ=6.05 (s, 2H), 2.56 (s, 6H), 2.51 (s, 6H), 2.40 ppm (s, 3H); 13C NMR (100 MHz; CDCl3): δ=153.59, 141.43, 141.01, 132.06, 121.25, 17.31, 16.37, 14.43 ppm; UV/Vis (CH2Cl2): λmax (ε)=487 nm (66 000); HRMS (ESI): m/z calcd for C14H18BF2N2: 262.1562; found: 262.1556.
BODIPY 20
Yield: 53.1 mg, 97%; m.p. 228–230°C; 1H NMR (400 MHz; CDCl3): δ=6.74 (dd, J=14.3, 9.1 Hz, 1H), 6.03 (s, 2H), 5.69 (dd, J=12.0, 2.2 Hz, 1H), 5.56 (d, J=17.5, 1.8 Hz, 1H), 2.53 (s, 6H), 2.23 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=154.90, 142.19, 140.52, 131.14, 123.79, 121.11, 17.24, 14.55 ppm; UV/Vis (CH2Cl2): λmax (ε)=505 nm (46 000); HRMS (ESI): m/z calcd for C15H18BF2N2: 274.1562; found: 274.1557.
BODIPY 21
Yield: 59.1 mg, 93%; m.p. 105–107 °C; 1H NMR (400 MHz; CDCl3): δ=6.03 (s, 2H), 4.46 (d, J=2.9 Hz, 1H), 4.37 (d, J=2.8 Hz, 1H), 3.89 (q, J=7.0 Hz, 2H), 2.52 (s, 6H), 2.23 (s, 6H), 1.39 ppm (t, J=7.0 Hz, 3H); 13C NMR (100 MHz; CDCl3): δ=155.97, 153.56, 142.50, 134.98, 131.29, 88.11, 63.67, 14.65, 14.34 ppm; UV/Vis (CH2Cl2): λmax (ε)=513 nm (40 700); HRMS (ESI): m/z calcd for C17H22BF2N2O: 318.1824; found: 318.1817.
BODIPY 22
Yield: 16.8 mg, 31 %; m.p.>260°C; 1H NMR (400 MHz; CDCl3): δ=6.07 (s, 2H), 3.92 (s, 1H), 2.53 (s, 6H), 2.45 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=155.07, 142.61, 133.28, 121.06, 118.92, 94.42, 79.19, 15.47, 14.63 ppm; UV/Vis (CH2Cl2): λmax (ε)= 544 nm (37 200); HRMS (ESI): m/z calcd for C15H16BF2N2: 272.1405; found: 272.1380.
BODIPY 23
Yield: 21.7 mg, 71.1% (starting with 25 mg, 0.088 mmol of 4a); m.p.≈250°C “(decomposed)”; 1H NMR (400 MHz; CDCl3): δ=6.06 (s, 2H), 2.52 (s, 6H), 2.46 (s, 6H), 0.29 ppm (s, 9H); 13C NMR (100 MHz; CDCl3): δ=154.40, 142.23, 133.05, 120.78, 120.05, 115.01, 110.44, 15.54, 14.61, 0.68 ppm; UV/Vis (CH2Cl2): λmax (ε)=545 nm (42 500); HRMS (ESI): m/z calcd for C18H23BFN2Si: 325.1708 [M+−F]; found: 325.1699.
BODIPY 24
Yield: 62.1 mg, 95%; m.p. 179–181°C; 1H NMR (400 MHz; CDCl3): δ=6.77 (dd, J=2.7, 1.7 Hz, 1H), 6.21 (dd, J=3.6, 2.7 Hz, 1H), 6.07 (dd, J=3.6, 1.7 Hz, 1H), 6.01 (s, 2H), 3.40 (s, 3H), 2.55 (s, 6H), 1.50 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ= 156.14, 143.34, 133.13, 131.73, 124.64, 122.47, 121.01, 109.34, 108.84, 33.84, 14.67, 12.68 ppm; UV/Vis (CH2Cl2): λmax (ε)=513 nm (53 700); HRMS (ESI): m/z calcd for C18H21BF2N2: 327.1827.
BODIPY 25
Yield: 69.8 mg, 99%; m.p. 197–199°C; 1H NMR (400 MHz; CDCl3): δ=7.50 (dd, J=5.0, 1.3 Hz, 1H), 7.13 (dd, J=5.1, 3.5 Hz, 1H), 6.99 (dd, J=3.5, 1.3 Hz, 1H), 6.00 (s, 2H), 2.55 (s, 6H), 1.58 ppm (s, 6H); 13C NMR (100 MHz; CDCl3): δ=156.07, 143.50, 134.64, 133.99, 132.41, 127.81, 127.61, 127.41, 121.50, 14.64, 13.55 ppm; UV/Vis (CH2Cl2): λmax (ε)=514 nm (74 100); HRMS (ESI): m/z calcd for C17H17BF2N2NaS: 352.1102 [M++Na]; found: 352.1094.
Crystal data and refinement
Diffraction data were collected at low temperature (90 or 100 K) on either a Nonius KappaCCD diffractometer equipped with MoKα radiation (λ=0.71073 Å) or a Bruker Kappa Apex-II DUO diffractometer equipped with Mo or CuKα radiation (λ=1.54184 Å). Refinement was by full-matrix least squares using SHELXL, with H atoms in idealized positions. Compound 4b has three independent molecules. Compound 19 lies on a mirror plane in the crystal, and its central Me group is disordered into two conformations. Disorder of the thiophene was present in 25, and the crystal of 17 was a non-merohedral twin.
Crystal data
For 4a: C13H14BClF2N2; monoclinic; P21/c; a= 8.6711(17), b=12.274(3), c=11.850(2) Å; β=97.570(8)°; Z=4; T= 100 K; R=0.040. 4b: C17H22BClF2N2; triclinic; P1; a=8.6228(5), b= 12.4360(7), c=12.4491(6) Å; α=93.806(3), β=107.076(3), γ= 100.514(3)°; Z=3; T=100 K; R=0.052. 5a: C15H19BF2N2O; monoclinic; P21/n; a=10.5564(9), b=11.8910(10), c=12.4508(14) Å; β= 112.982(4)°; Z=4; T=90 K; R=0.036. 5b: C19H27BF2N2O; monoclinic; Cc; a=13.9165(9), b=17.1597(10), c=8.4065(5) Å; β= 114.970(5)°; Z=4; T=100 K; R=0.034. 16: C20H18BF2N3S2; monoclinic; P21/c; a=11.7044(17), b=14.871(2), c=10.9035(17) Å; β= 93.116(10)°; Z=4; T=100 K; R=0.038. 17: C16H27B11F2N2S; monoclinic; P21/c; a=13.8364(9), b=13.3718(11), c=12.0936(12) Å; β= 93.295(6)°; Z=4; T=100 K; R=0.136. 18: C19H19BF2N2; monoclinic; P21/n, a=11.812(3), b=7.0804(17), c=19.362(5) Å; β=99.545(14)°; Z=4; T=100 K, R=0.049. 19: C14H17BF2N2; orthorhombic; Pnma; a=11.3154(16), b=7.0396(10), c=16.013(2) Å; Z=4; T=100 K; R= 0.039. 20: C15H17BF2N2; monoclinic; P21/n; a=10.0991(5), b= 11.7547(6), c=11.9808(7) Å; β=107.886(3)°; Z=4; T=100 K; R= 0.046. 24: C18H20BF2N3; monoclinic; P21; a=7.2814(11), b= 12.4570(19), c=9.2579(15) Å; β=100.858(8)°; Z=2; T=100 K; R= 0.042. 25: C17H17BF2N2S; monoclinic; P21/c; a=6.6072(10), b= 18.543(2), c=12.7436(15) Å; β=92.804(6)°; Z=4; T=90 K; R= 0.034. CCDC-959961–959970, 959972 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Steady-state absorption and fluorescence spectroscopy
The absorption measurements were carried out on an Agilent 8453 spectrophotometer and the steady-state fluorescence spectroscopic studies were performed in dichloromethane on a PTI Quanta-Master4/2006SE spectrofluorimeter. The slit width was 3 nm for both the excitation and emission. For the fluorescence quantum yield measurements, dilute solutions with absorbance between 0.04–0.06 at the excitation wavelength were used. The fluorescence quantum yields of BODIPYs were obtained by comparing the area under the corrected emission spectrum of the test sample with that of rhodamine 6G in ethanol. All spectra were recorded at room temperature by using non-degassed samples, spectroscopic grade solvents and a 10 mm quartz cuvette.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health, grant CA132861.
Footnotes
BODIPY=difluoroborondipyrromethene.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201304310 and contains 1H-, 11B-, and 13CNMR spectra, pyrrole X-ray structures and absorption/emission profiles for all BODIPYs.
References
- 1.Smith KM. Quart Rev Chem Soc. 1971;25:31–85. [Google Scholar]
- 2.Ballantine JA, Jackson AH, Kenner GW, McGillivray G. Tetrahedron. 1966;22:241–259. [Google Scholar]
- 3.Goud TV, Tutar A, Biellmann JF. Tetrahedron. 2006;62:5084–5091. [Google Scholar]
- 4.Smith KM, Vicente MGH. Science of Synthesis. Vol. 17.8. Georg Thieme Verlag; Stuttgart: 2004. pp. 1081–1334. [Google Scholar]
- 5.Clezy PS, Liepa AJ, Smythe GA. Aust J Chem. 1970;23:603–608. [Google Scholar]
- 6.Jackson AH, Kenner GW, McGillivray G, Smith KM. J Chem Soc C. 1968:294–302. [Google Scholar]
- 7.Treibs A, Kreuzer FH. Justus Liebigs Ann Chem. 1968;718:208–223. doi: 10.1002/jlac.19687180118. [DOI] [PubMed] [Google Scholar]
- 8.Falk H, Hofer O, Lehner H. Monatsh Chem. 1974;105:169–178. [Google Scholar]
- 9.Loudet A, Burgess K. Chem Rev. 2007;107:4891–4932. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 10.Ulrich G, Ziessel R, Harriman A. Angew Chem. 2008;120:1202–1219. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:1184–1201. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- 11.Karolin J, Johansson LBA, Strandberg L, Ny T. J Am Chem Soc. 1994;116:7801–7806. [Google Scholar]
- 12.Tan K, Jaquinod L, Paolesse R, Nardis S, Di Natale C, Di Carlo A, Prodi L, Montalti M, Zaccheroni N, Smith KM. Tetrahedron. 2004;60:1099–1106. [Google Scholar]
- 13.Lee CH, Lindsey JS. Tetrahedron. 1994;50:11427–11440. doi: 10.1016/j.tet.2008.08.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulrich G, Ziessel R. J Org Chem. 2004;69:2070–2083. doi: 10.1021/jo035825g. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Fan J, Gao X, Wang B, Sun S, Peng X. J Org Chem. 2009;74:7675–7683. doi: 10.1021/jo901149y. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Burgess K. Chem Commun. 2008:4933–4935. doi: 10.1039/b810503k. [DOI] [PubMed] [Google Scholar]
- 17.Leen V, Yuan P, Wang L, Boens N, Dehaen W. Org Lett. 2012;14:6150–6153. doi: 10.1021/ol3028225. [DOI] [PubMed] [Google Scholar]
- 18.Osorio-Martínez CA, Urías-Benavides A, Gómez-Durán CFA, Bañuelos J, Esnal I, López Arbeloa I, Peña-Cabrera E. J Org Chem. 2012;77:5434–5438. doi: 10.1021/jo300724m. [DOI] [PubMed] [Google Scholar]
- 19.Plater MJ, Aiken S, Bourhill G. Tetrahedron. 2002;58:2405–2413. [Google Scholar]
- 20.Fischer H, Orth H. Justus Liebigs Ann Chem. 1933;502:237–264. [Google Scholar]
- 21.Diem MJ, Burow DF, Fry JL. J Org Chem. 1977;42:1801–1802. [Google Scholar]
- 22.Smith KM, Miura M, Tabba HD. J Org Chem. 1983;48:4779–4781. [Google Scholar]
- 23.Li L, Nguyen B, Burgess K. Bioorg Med Chem Lett. 2008;18:3112–3116. doi: 10.1016/j.bmcl.2007.10.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kee HL, Kirmaier C, Yu L, Thamyongkit P, Youngblood WJ, Calder ME, Ramos L, Noll BC, Bocian DF, Scheidt WR, Birge RR, Lindsey JS, Holten D. J Phys Chem B. 2005;109:20433–20433. doi: 10.1021/jp0525078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goze C, Ulrich G, Mallon LJ, Allen BD, Harriman A, Ziessel R. J Am Chem Soc. 2006;128:10231–10239. doi: 10.1021/ja062405a. [DOI] [PubMed] [Google Scholar]
- 26.Gabe Y, Ueno T, Urano Y, Kojima H, Nagano T. Anal Bioanal Chem. 2006;386:621–626. doi: 10.1007/s00216-006-0587-y. [DOI] [PubMed] [Google Scholar]
- 27.Bhupathiraju NVSDK, Vicente MGH. Bioorg Med Chem. 2013;21:485–495. doi: 10.1016/j.bmc.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 29.Stille JK, Lau KSY. J Am Chem Soc. 1976;98:5841–5849. [Google Scholar]
- 30.Heck RF. J Am Chem Soc. 1968;90:5518–5526. [Google Scholar]
- 31.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 32.Rohand T, Qin W, Boens N, Dehaen W. Eur J Org Chem. 2006:4658–4663. [Google Scholar]
- 33.Thivierge C, Bandichhor R, Burgess K. Org Lett. 2007;9:2135–2138. doi: 10.1021/ol0706197. [DOI] [PubMed] [Google Scholar]
- 34.a) Qin W, Rohand T, Dehaen W, Clifford JN, Driesen K, Beljonne D, VanAverbeke B, Van der Auweraer M, Boens N. J Phys Chem A. 2007;111:8588–8597. doi: 10.1021/jp073547+. [DOI] [PubMed] [Google Scholar]; b) Leen V, Braeken E, Luckermans K, Jackers C, Van derAuweraer M, Boens N, Dehaen W. Chem Commun. 2009:4515–4517. doi: 10.1039/b906164a. [DOI] [PubMed] [Google Scholar]
- 35.Jiao L, Yu C, Uppal T, Liu M, Li Y, Zhou Y, Hao E, Hu X, Vicente MGH. Org Biomol Chem. 2010;8:2517–2519. doi: 10.1039/c001068e. [DOI] [PubMed] [Google Scholar]
- 36.Li F, Yang SI, Ciringh Y, Seth J, Martin CH, Singh DL, Kim D, Birge RR, Bocian DF, Holten D, Lindsey JS. J Am Chem Soc. 1998;120:10001–10017. [Google Scholar]
- 37.Ortiz MJ, Agarrabeitia AR, Duran-Sampedro G, Prieto JB, Lopez TA, Massad WA, Montejano HA, Garcia NA, Arbeloa IL. Tetrahedron. 2012;68:1153–1162. [Google Scholar]
- 38.a) Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437–3440. [Google Scholar]; b) Miyaura N, Yano T, Suzuki A. Tetrahedron Lett. 1980;21:2865–2868. [Google Scholar]; c) Miyaura N, Suzuki A. Chem Commun. 1979:866–867. [Google Scholar]; d) Miyaura N, Yanagi T, Suzuki A. Synth Commun. 1981;11:513–519. [Google Scholar]
- 39.a) Stille JK. Angew Chem. 1986;98:504–519. [Google Scholar]; Angew Chem Int Ed. 1986;25:508–524. [Google Scholar]; b) Milstein D, Stille JK. J Am Chem Soc. 1978;100:3636–3638. [Google Scholar]
- 40.Gräf K, Korzdorfer T, Kummel S, Thelakkat M. New J Chem. 2013;37:1417–1426. [Google Scholar]
- 41.Gabe Y, Urano Y, Kikuchi K, Kojima H, Nagano T. J Am Soc Chem. 2004;126:3357–3367. doi: 10.1021/ja037944j. [DOI] [PubMed] [Google Scholar]
- 42.Sun J, Zhong F, Yi X, Zhao J. Inorg Chem. 2013;52:6299–6310. doi: 10.1021/ic302210b. [DOI] [PubMed] [Google Scholar]
- 43.Gibbs JH, Robins LT, Zhou Z, Bobadova-Parvanova P, Cottam M, McCandless GT, Fronczek FR, Vicente MGH. Bioorg Med Chem. 2013;21:5770–5781. doi: 10.1016/j.bmc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boiadjiev SE, Lightner DA. Tetrahedron: Asymmetry. 2002;13:1721–1732. [Google Scholar]
- 45.Fischer H, Orth H. Justus Liebigs Ann Chem. 1931;489:62–86. [Google Scholar]
- 46.Fischer H, Orth H. Die Chemie des Pyrrols. Akademische Verlag; Leipzig: 1934. p. 238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.