

Abstract
We recently reported that the aldehyde residue of an abasic (Ap) site in duplex DNA can generate an interstrand cross-link via reaction with a guanine residue on the opposing strand. This finding is intriguing because the highly deleterious nature of interstrand cross-links suggests that even small amounts of Ap-derived cross-links could make a significant contribution to the biological consequences stemming from the generation of Ap sites in cellular DNA. Incubation of 21-bp duplexes containing a central 5′-CAp sequence under conditions of reductive amination (NaCNBH3, pH 5.2) generated much higher yields of cross-linked DNA than reported previously. At pH 7, in the absence of reducing agents, these Ap-containing duplexes also produced cross-linked duplexes that were readily detected on denaturing polyacrylamide gels. Cross-link formation was not highly sensitive to reaction conditions and, once formed, the cross-link was stable to a variety of work-up conditions. Results of multiple experiments including MALDI-TOF mass spectrometry, gel mobility, methoxyamine capping of the Ap aldehyde, inosine-for-guanine replacement, hydroxyl radical footprinting, and LCMS/MS were consistent with a cross-linking mechanism involving reversible reaction of the Ap aldehyde residue with the N2-amino group of the opposing guanine residue in 5′-CAp sequences to generate hemiaminal, imine, or cyclic hemiaminal cross-links (7-10) that were irreversibly converted under conditions of reductive amination (NaCNBH3/pH 5.2) to a stable amine linkage. Further support for the importance of the exocyclic N2-amino group in this reaction was provided by an experiment showing that installation of a 2-aminopurine-thymine base pair at the cross-linking site produced high yields (15-30%) of a cross-linked duplex at neutral pH, in the absence of NaCNBH3.
Introduction
The glycosidic bonds holding the nucleobases to the 2′-deoxyribose phosphate backbone are weak points in the structure of DNA.1,2 Spontaneous hydrolytic cleavage of this bond at purine residues in genomic DNA has been estimated to generate 10,000 abasic (Ap) sites per cell per day (1, Scheme 1).3-6 In addition, damage inflicted upon the DNA bases by various environmental mutagens and anticancer drugs can increase the rate of spontaneous depurination and, thus, the load of cellular Ap sites.6-15 The action of various DNA-repair glycosylases on damaged nucleobases and misincorporated uracil residues also produces Ap sites in cellular DNA.16,17 Ap sites are mutagenic and, therefore, may contribute to spontaneous and chemically-induced cancers.2,12,18-24 In addition, Ap sites can be cytotoxic.2,18-22,25,26
Scheme 1.

Ap sites in duplex DNA exist as an equilibrium mixture of a ring-closed hemiacetal and ring-opened aldehyde form.27 Aldehydes can form covalent imine adducts with the heterocyclic bases in DNA28-32 and recognition of this fact recently inspired us to explore the ability of Ap sites to generate cross-links in duplex DNA.33 In analogy with interstrand DNA cross-link formation by small molecule agents,34 we anticipated that formation of Ap-derived cross-links in duplex DNA might be most favored at sites where the generation of the linkage induces minimal amounts of distortion.33 Ap-containing duplexes show considerable structural diversity, but often retain B-DNA character.35-38 Molecular models indicated that, in a B-DNA helix, the exocyclic amino group of the guanine residue in a 5′-CAp sequence might be located near the Ap aldehyde (Figure 1). Indeed, we found that interstrand cross-links formed at 5′-CAp sequences in duplex DNA, with the evidence supporting a mechanism involving reaction of the abasic aldehyde group with the N2-amino group of the opposing guanine residue in this sequence (Scheme 2).33
Figure 1.

Molecular model showing the juxtaposition of the N2-amino group of the guanine residue and the abasic site at a 5′-CAp cross-linking site in B-DNA. The image was constructed using Pymol and is based on pdb entry 1dcv.
Scheme 2.

Interstrand cross-links present tremendous challenges to cellular DNA-repair systems.39-42 For example, cross-link repair can be error prone, with the resulting mutations contributing to the etiology of human cancers.43-46 Furthermore, interstrand cross-links can block DNA transcription and replication, thus contributing to cell dysfunction, cell death, senescence, and aging.39-42,47-50 The highly deleterious nature of interstrand DNA-DNA cross-links suggests that even small amounts of Ap-derived crosslinks could contribute significantly to the biological properties of Ap sites in genomic DNA. Therefore, we set out to further characterize formation and properties of the dGAp cross-link in duplex DNA.
Experimental Section
Materials and Methods
Oligonucleotides were purchased from Integrated DNA Technologies. Erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA) hydrochloride was purchased from Tocris Bioscience (Ellisville, MO, USA). Nuclease P1 and phosphodiesterases 1 and 2 were from Sigma-Aldrich (St. Louis, MO, USA). Alkaline phosphatase, proteinase K and other enzymes were from New England Biolabs (Ipswich, MA, USA). [γ−32P]-ATP (6000 Ci/mmol) was purchased from Perkin Elmer. C-18 sep-pak cartridges were purchased from Waters and BS Polyprep columns were obtained from BioRad. Quantification of radioactivity in polyacrylamide gels was carried out using a Personal Molecular Imager (BIORAD) with Quantity One software (v.4.6.5). All other reagents were purchased from Sigma-Aldrich.
Representative procedure for cross-link formation under conditions of reductive amination
Single-stranded 2′-deoxyoligonucleotides were 5′-labeled using standard procedures.51 Labeled DNA was annealed51 with its complimentary strand and treated with the enzyme UDG (50 units/mL, final concentration) to generate Ap sites. The enzyme was removed by phenol-chloroform extraction.51 In individual cross-linking reactions, the Ap-containing double-stranded DNA was incubated in sodium acetate buffer (750 mM, pH 5.2) containing NaCNBH3 (250 mM) at 37 °C for 24 h unless otherwise specified. The DNA was ethanol precipitated from the reaction mixture,51 resuspended in formamide loading buffer,51 loaded onto a 20% denaturing polyacrylamide gel, and the gel electrophoresed for 4 h at 1000 V. The amount of radiolabeled DNA in each band on the gel was measured by phosphorimager analysis. The time course for the formation of the native cross-link was carried out by incubating a solution containing labeled DNA (approximately 200,000 cpm), sodium acetate buffer (750 mM, pH 5.2) containing NaCNBH3 (250 mM) at 37 °C. At specified time points, aliquots (20 μL) were removed and frozen at −20 °C, followed by ethanol precipitation and gel analysis as described above.
Representative procedure for cross-link formation at pH 7 in the absence of reducing agent
Duplex DNA containing a 32P-labeled Ap-containing strand, prepared as described above, was incubated in a buffer composed of HEPES (50 mM, pH 7) containing NaCl (100 mM) at 37 °C for 72 h unless otherwise specified. The DNA was ethanol precipitated, resuspended in formamide loading buffer, loaded onto a 20% denaturing polyacrylamide gel, and the gel electrophoresed for 4 h at 1000 V. Control experiments involving direct loading of cross-linking reactions showed that ethanol precipitation does not markedly alter the yield of the slow-migrating, cross-link band. The amount of radiolabeled DNA in each band on the gel was measured by phosphorimager analysis.
Hydroxyl radical footprinting of cross-linked duplexes
We employed literature protocols to footprint the cross-linked duplexes.52,53 In these experiments, the strand opposing the Ap-containing oligonucleotide was 5′-labeled using standard procedures.51 Labeled DNA was annealed with the uracil-containing complement and treated with UDG to generate the abasic site as described above. The Ap-containing double-stranded DNA (∼500,000 cpm) was incubated in sodium acetate buffer (750 mM, pH 5.2) containing NaCNBH3 (250 mM) at 37 °C for 24 h (in the case of duplex F) or incubated in a buffer composed of HEPES (50 mM, pH 7) containing NaCl (100 mM) at 37 °C for 72 h (in the case of duplex I). The DNA was ethanol precipitated, suspended in formamide loading buffer, and separated on a 0.4 mm thick 20% denaturing polyacrylamide gel. The slow-migrating cross-linked duplex band was visualized using X-ray film, the band cut out of the gel, the gel slice crushed, and the gel pieces vortexed in elution buffer (NaCl 200 mM; EDTA, 1 mM) at room temperature for at least 1 h. The mixture was filtered through a poly-prep column to remove gel fragments and the filtrate desalted using a C18 Sep-pak (100 mg size). The resulting solution was evaporated using a Speed-Vac concentrator, the residue redissolved in water (24 μL), split evenly into three microcentrifuge tubes, and diluted with 2× oxidation buffer (10 μL of a solution composed of sodium phosphate, 20 mM, pH 7.2; NaCl, 20 mM; sodium ascorbate, 2 mM; H2O2, 1 mM). To this mixture was added a solution of iron-EDTA (2 μL, EDTA, 70 mM; (NH4)2Fe(SO4)2•6H2O, 70 mM) to start the reaction, the mixture vortexed briefly, and incubated at room temperature for 1, 2, and 3 min before addition of thiourea stop solution (10 μL of a 100 mM solution in water). Hydroxyl radical footprinting reactions, Maxam-Gilbert G, and Maxam-Gilbert A+G reactions were performed on the labeled single-strand to generate marker lanes.54 The resulting DNA fragments were analyzed using gel electrophoresis as described above.
Preparation of reduced cross-linked duplexes for mass spectrometric analysis
An unlabeled version of DNA duplex A, prepared as described above, was incubated in sodium acetate (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C for 24 h. The DNA was ethanol precipitated, resuspended in formamide loading buffer, and the DNA fragments separated on a 20% denaturing polyacrylamide gel. DNA bands were visualized with UV-shadowing. The reduced crosslink band was cut out of the gel, the gel slice crushed, and the solution vortexed in elution buffer (NaCl, 1 M; EDTA, 500 mM, pH 8) at room temperature for at least 1 h. The solution was filtered through a Poly-prep column, the filtrate desalted using a C18 Sep-pak (100 mg size), and the DNA ethanol precipitated. MALDI-TOF mass spectrometric analyses were carried out on an AB Sciex Voyager DE™ Pro mass spectrometer (Framingham, MA). The aqueous sample solution was combined in a 1/1/1 (V/V/V) ratio with triammonium citrate (0.1 M, aq) and 2′,4′,6′-trihydroxyacetophenone (0.1 M in acetonitrile) for application to a polished stainless steel target for analysis. Negative ion spectra were acquired over a mass range of 5-14 kDa at a 25 kV accelerating potential in the linear delayed-extraction mode using an oligonucleotide of known mass as an external calibrant.55,56
Enzymatic digestion of cross-linked duplex J
The cross-linked duplex J was digested using a 4-enzyme cocktail following the conditions described previously.57 Nuclease P1 (1 unit), phosphodiesterase 2 (0.005 unit), EHNA (2 nmol) and a 5-μL solution containing 300 mM sodium acetate (pH 5.6) and 10 mM zinc chloride were added to 6 pmol of duplex J (final volume, 50 μL). In this context, EHNA served as an inhibitor for deamination of 2′-deoxyadenosine (dA) to 2μ-deoxyinosine (dI) induced by residual adenine deaminase.57 The resulting mixture was incubated at 37 °C for 2 h. To the digestion mixture were then added alkaline phosphatase (1 unit), phosphodiesterase 1 (0.005 unit) and 10 μL of 0.5 M Tris-HCl buffer (pH 8.9). The digestion was continued at 37 °C for 1 h. For digestion with nuclease P1 only, 1 unit of nuclease P1 and a 5-μL solution containing 300 mM sodium acetate (pH 5.6) and 10 mM zinc chloride were added to 6 pmol of duplex J containing the dG-AP crosslink in a final volume of 50 μL. The resulting mixture was incubated at 37 °C for 2 h.
LC-MS/MS and LC-MS/MS/MS Analysis
The LC-MSn experiments were conducted using an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific). Briefly, a 0.5×250 mm or 0.5×150 mm Zorbax SB-C18 column (particle size, 5 μm, Agilent) was used for the separation, and the flow rate was 8.0 μL/min. For the samples arising from digestion with a cocktail of 4 enzymes, a solution of 0.1% (v/v) formic acid in water (A) and a solution of 0.1% (v/v) formic acid in methanol (B) were used as mobile phases. A gradient of 5 min 0% B, 5 min 0-20% B, 40 min 20-50% B, 10 min 50% B, 1 min 50-0% B was employed for the separation. For the samples from nuclease P1 digestion, the mobile phases were 400 mM 1,1,1,3,3,3-hexafluoroisopropanol (HFIP, pH adjusted to 7.0 with triethylamine) (C) and methanol (D). A gradient of 5 min 0% D, 40 min 0-80% D, and 1 min 80-0% D, was employed.
Results and Discussion
Cross-link formation in Ap-containing duplexes under conditions of reductive amination
In our initial report,33 interstrand DNA-DNA cross-links were generated by incubation of 15 base pair (bp), Ap-site-containing duplexes in pH 5.5 buffer (MES, 50 mM) at 30 °C in the presence of the reducing agent sodium cyanoborohydride (NaCNBH3). These conditions of reductive amination58 were designed to facilitate detection of the cross-link via irreversible reduction of the anticipated imine intermediate 8 to give a stable amine linkage (Scheme 2).59-61 In the current work, we examined crosslink formation in several 21-30 bp 2′-deoxyoligonucleotide duplexes, with the goal of more fully characterizing the formation, structure, and properties of the dG-Ap cross-link formed at 5′-CAp sites in duplex DNA.
We generated duplexes A-I (Scheme 2), containing authentic Ap sites by treatment of the corresponding 2′-deoxyuridine-containing 2′-deoxyoligonucleotide duplexes with uracil DNA glycosylase (UDG).62-64 Prior to annealing of the duplex and treatment with UDG, the uracil-containing strand of the duplexes was 5′-32P-labeled using standard methods to allow detection of the products following separation on 20% denaturing polyacrylamide gels (PAGE).51
As expected, the uracil-containing oligonucleotides treated with UDG (50 U/mL, 45-90 min, 37 °C) were smoothly cleaved by piperidine workup (1 M, 95 °C, 25 min) to generate two faster-migrating bands on the gel, which we assigned as 4 (minor band) and 5 (major band), indicating that the Ap-sites were generated in ≥90% yields (Lanes 3, 8, 13, and 18, Figure 2). The Ap-containing duplex A was incubated in a solution containing sodium acetate (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C. The DNA was then ethanol precipitated,51 redissolved in formamide loading buffer51 without heating, and the products resolved using denaturing gel electrophoresis to reveal a slow-migrating band in the region where the cross-linked duplex was expected to migrate (Lane 4, Figure 2).33 Interestingly, the yield of the slow-migrating band generated by duplex A was substantially greater (18.2 ± 0.6%) than that obtained from duplex J in our previous studies. In our earlier work, the 15 bp duplex J generated 2-3% yields of cross-linked duplex under similar conditions.33 Possible reasons for the differences in yield are offered in the Conclusions below. Given the strikingly high yields of the slow-migrating band generated by duplex A compared to the previously studied duplex J, we felt it was important to carefully characterize this putative interstrand DNA-DNA cross-link.
Figure 2.

Interstrand cross-link formation in duplexes A-D under conditions of reductive amination. Duplex A, lanes 1-5, B, lanes 6-10, C, lanes 11-15, and D, lanes 16-20. The uracil-containing precursor 2′-deoxyoligonucleotides appear in lanes 1, 6, 11, and 16. The abasic-site-containing duplexes without incubation appear in lanes 2, 7, 12, and 17. The abasic-site-containing duplexes cleaved by treatment with piperidine (1 M, 95 °C, for 25 min) appear in lanes 3, 8, 13, and 18). The cross-linking reactions involving incubation of the abasic-site-containing duplex in sodium acetate buffer (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C appear in lanes 4, 9, 14, and 19. The abasic-site-containing duplexes in sodium acetate buffer (750 mM, pH 5.2), NaCNBH3 (250 mM), and CH3ONH2 hydrochloride (2 mM) at 37 °C appear in lanes 5, 10, 15, and 20. The 32P-labeled 2′-deoxyoligonucleotides were resolved on a sequencing gel and the radioactivity in each band quantitatively measured by phosphorimager analysis.
Consistent with the involvement of the Ap aldehyde group in formation of the slow-migrating band from duplex A, addition of methoxyamine (CH3ONH2, 2 mM) at the beginning of the assay significantly inhibited formation of the slow-migrating band (lane 5, Figure 2). Methoxyamine efficiently “caps” the Ap aldehyde via formation of a stable oxime (6, Scheme 2) that prevents both cross-link formation and strand cleavage.65,66 We also examined a duplex in which the strand complementary to the Ap-containing oligonucleotide was extended on the 3′-end to generate an overhang (duplex B, Scheme 2). Importantly, the mobility of the slow-migrating band generated from this duplex (lane 9, Figure 2) was retarded relative to that produced by duplex A (lane 4, Figure 2). This provided evidence that the slow-migrating band contained both the 32P-labeled strand and its complement. Similar to the result observed for duplex A, generation of the slow-migrating band from duplex B was blocked by the presence of methoxyamine in the assay mixture (lane 10, Figure 2). In addition, we constructed a duplex in which the Ap-containing strand was extended on the 3′-end to generate an overhang (duplex C, Scheme 2). Again, the slow-migrating band generated from this duplex was gel-shifted relative to that produced by duplex A (lane 14, Figure 2). This indicated that the slow-migrating band contained the full-length Ap-containing strand that had not undergone cleavage at the Ap site to yield truncated products such as 4 or 5 (Scheme 2) that have shed the 3′-end of the oligonucleotide. A substrate bearing 3′-overhangs on both ends (duplex D) generated a slow-migrating band whose gel mobility was retarded even further relative to duplexes B and C (lane 19, Figure 2). Overall, these results provided evidence that the slow-migrating bands observed in Figure 2 are cross-linked duplexes that contain both full-length strands of the starting duplex and whose formation is dependent upon the aldehyde group of the Ap site. A time course experiment indicated that substantial yields of cross-link are established well inside of 24 h, with an apparent half-time to completion of approximately 4.5 h (Figure 3).
Figure 3.

Time course for the formation of cross-linked duplex under conditions of reductive amination. Duplex A was incubated in sodium acetate buffer (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C and at 0, 1, 2, 4, 6, 8, 12, and 23 h aliquots were removed from the reaction and frozen prior to sequencing gel analysis (lanes 5-12). The lower bands correspond to the full length labeled 2′-deoxyoligonucleotides and the upper band cross-linked DNA. Lane 1 is the 32P-labeled uracil-containing precursor 2′-deoxyoligonucleotide, lane 2 is the 32P-labeled abasic-site-containing duplex without incubation, and lane 3 is the 32P-labeled abasic-site-containing duplexes cleaved by treatment with piperidine (1 M, 95 °C, for 25 min). Lane 4 is the abasic-site-containing duplex incubated in pH 7 buffer for 24 h at 37 °C. The 32P-labeled 2′-deoxyoligonucleotides were resolved on a sequencing gel and the radioactivity in each band quantitatively measured by phosphorimager analysis. Figure 1 shows a full image of the gel. This and subsequent gel images display only the region of the gel where full length and cross-linked DNA appear.
Evidence that formation of the reduced cross-link involves reaction of the Ap site with the opposing guanine residue in the 5′-CAp sequence: Incorporation of the non-natural nucleobase inosine and hydroxyl radical footprinting of the cross-linked duplex
In this section, we describe two experiments that shed light on the sites of cross-link attachment in the duplexes described above. First, we constructed duplex E (Scheme 2) in which the guanine residue on the strand opposing the Ap site in the 5′-CAp sequence was replaced with the nonnatural nucleobase inosine. The substitution of an inosine residue for guanine at the putative cross-linking site amounts to the targeted deletion of a single NH2 group in the 21 bp duplex A (the structure of inosine is shown in Scheme 2). Duplexes A and E were incubated in pH 5.2 sodium acetate buffer (750 mM) containing NaCNBH3 (250 mM) at 37 °C. In the event, we found that replacement of the guanine residue in duplex A with inosine in duplex E prevented cross-link formation (Figure 4). This result provided evidence for the involvement of the N2-amino of the guanine residue in cross-link formation at the 5′-CAp sequences in duplexes A-D. Interestingly, in the case of the inosine-containing duplex E, a faint band migrating more slowly than the dG-Ap cross-link can be observed (lane 4, Figure 4). This suggested that a low yield of some alternative cross-linked structure was generated when the N2-amino group of dG was not poised to capture the Ap aldehyde; however, this minor product was not further characterized.
Figure 4.

Cross-link formation was abrogated by replacement of the opposing guanine residue in the 5′-CAp sequence with an inosine residue. The lower bands correspond to the 32P-labeled full length labeled 2′-deoxyoligonucleotides and the upper band cross-linked DNA. Lane 1 is the abasic-site-containing 2′-deoxyoligonucleotide duplex without incubation. Lane 2 is the abasic-site-containing 2′-deoxyoligonucleotide duplex A cleaved by treatment with piperidine (1 M, 95 °C, 25 min). Lane 3 is duplex A incubated in sodium acetate buffer (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C. Lane 4 is duplex E incubated in sodium acetate buffer (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C. The 32P-labeled 2′-deoxyoligonucleotides were resolved on a sequencing gel and the radioactivity in each band quantitatively measured by phosphorimager analysis. The image shows the region of the gel where full-length oligonucleotide and cross-link migrate.
We also carried out a hydroxyl radical footprinting experiment to determine the base to which the Ap residue was attached on the opposing strand of the cross-linked duplex F. For this experiment, the complement to the Ap-containing strand was 5′-32P-labeled. Under conditions of reductive amination described above, duplex F generated a good yield of the slow-migrating band analogous to the yields obtained from duplexes A-D. As a technical note, longer duplexes were employed in the hydroxyl radical footprinting experiments to move bands near the cross-linking site out of a “salt flare” zone in the gel so that they could be clearly visualized. The slow-migrating band was excised from the gel, subjected to cleavage by H2O2-iron-EDTA,67 and the resulting mixture of labeled DNA fragments resolved on a sequencing gel. In this type of experiment, the presence of a cross-link results in an interruption of the “ladder” of DNA fragments at the site of cross-link attachment.52,53,68,69 Here, we observed a clear interruption in the fragment ladder at G4 of the labeled strand (Figures S1 and S2; numbering of the guanine residues in duplex F is shown in Scheme 2). This result provided additional evidence that the cross-link extends from the Ap site to the opposing guanine residue in the central 5′-CAp sequence of this duplex (Scheme 2).
Analysis of the reduced cross-link via MALDI mass spectrometry
Using conditions of reductive amination, we prepared a version of duplex A lacking the 5′-32P group on the Ap-containing strand. Preparative gel electrophoresis was used to separate the cross-linked material from the native single strands. DNA in the gel was visualized by UV-shadowing, and the slow-moving, cross-linked band was excised from the gel. DNA was eluted from the gel, desalted using a C18 cartridge, and ethanol precipitated. MALDI-TOF mass spectrometric analysis of the DNA obtained in this manner gave a strong signal that closely matched that expected for a reduced cross-link structure such as 10 in Scheme 2 (m/z for [M–H]− observed: 12,720.8; calcd: 12,721.5; relative mass error 55 ppm, Figure S3).
Formation of the native (unreduced) dG-Ap cross-link in neutral aqueous solution
We examined formation of the dG-Ap cross-link at physiological pH in the absence of added reducing agent. In these experiments, duplex A was incubated in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C, ethanol precipitated,51 redissolved in formamide loading buffer51 without warming, and analyzed by denaturing gel electrophoresis. A slow-migrating band was observed in the region where the cross-linked duplex migrates (Lane 4, Figure 5). Consistent with the involvement of the Ap aldehyde group in formation of this slow-migrating band, addition of methoxyamine (CH3ONH2, 20 mM) at the beginning of the assay blocked formation of the slow-migrating band (lane 5, Figure 5). Similar to the results described above for the reduced cross-link, replacement of the guanine residue in the central 5′-CAp sequence with an inosine residue abrogated formation of the major slow-migrating band. Together, these findings provided evidence that the slow-migrating band generated in this experiment was an unreduced dG-Ap interstrand cross-link. The native (unreduced) cross-link could be resolved from the reduced cross-link on a 0.4 mm thick, 20% denaturing polyacrylamide gel if the cross-linked material was run at least 12 cm from the well, with the reduced cross-link migrating slightly faster than the unreduced material (Figure 6). A time course for the reaction revealed that the unreduced cross-link formed rapidly. Indeed, an easily detectable amount of the cross-link formed during the 1 h incubation in which the Ap-site was generated by the action UDG on the uracil-containing 2′-deoxyoligonucleotide duplex. Further incubation in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C, revealed that the apparent half-time to completion of the cross-linking reaction was less than 24 h (Figure S4).
Figure 5.

Detection of native (unreduced) dG-Ap cross-link duplex A and evidence that replacement of the opposing guanine residue in the 5′-CAp sequence of duplex A with inosine in duplex E abrogates cross-link formation. The lower bands correspond to the full-length 32P-labeled 2′-deoxyoligonucleotides and the upper band cross-linked DNA. Lane 1 is the uracil-containing precursor of 2′-deoxyoligonucleotide duplex A. Lane 2 is the abasic-site-containing 2′-deoxyoligonucleotide duplex A without incubation. Lane 3 is the abasic-site-containing 2′-deoxyoligonucleotide duplex A cleaved by treatment with piperidine (1 M, 95 °C, 25 min). Lane 4 is duplex A incubated in with CH3ONH2 (2 mM) in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 5 is duplex A incubated in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 6 is the abasic-site-containing 2′-deoxyoligonucleotide duplex E without incubation. Lane 7 is the abasic-site-containing 2′-deoxyoligonucleotide duplex E cleaved by treatment with piperidine (1 M, 95 °C, 25 min). Lane 8 is duplex E incubated in with CH3ONH2 (2 mM) in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 9 is duplex E incubated in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. The 32P-labeled 2′-deoxyoligonucleotides were resolved on a sequencing gel and the radioactivity in each band quantitatively measured by phosphorimager analysis. The image shows the region of the gel where full-length oligonucleotide and cross-link migrate.
Figure 6.
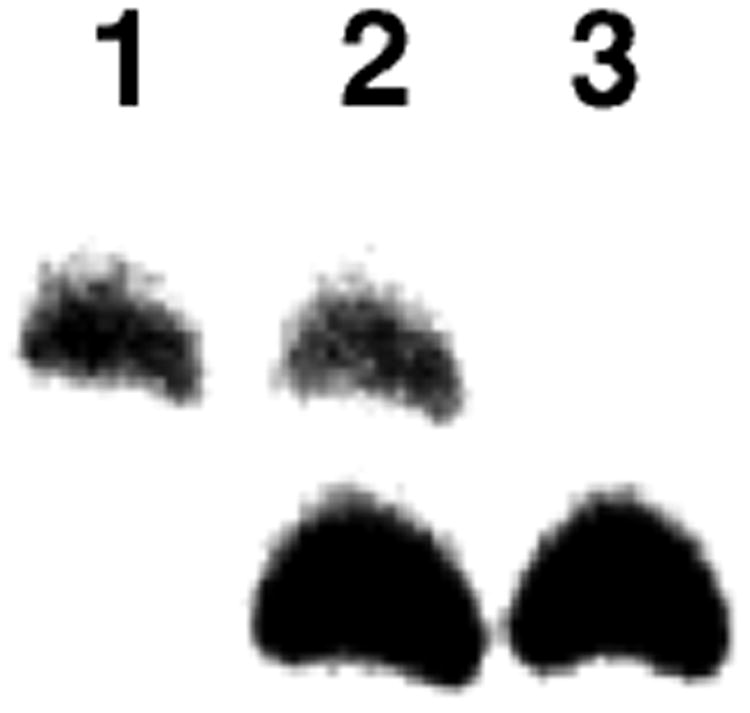
The reduced and unreduced dG-Ap cross-linked duplexes can be resolved by gel electrophoresis. Lane 1 is the native (unreduced) cross-link generated by incubation of duplex A in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 3 is the reduced cross-link generated by incubation of duplex A in sodium acetate buffer (750 mM, pH 5.2) and NaCNBH3 (250 mM) at 37 °C. Lane 2 is a mixture of the reduced and native cross-links. The image shows the region of the gel where the cross-link migrates.
Installation of a 2-aminopurine-thymine base pair at the cross-linking site gives excellent yields of cross-linked duplexes at neutral pH in the absence of reducing agent
The evidence presented above suggested that the exocyclic N2-amino group of guanine was central to formation of the dG-Ap cross-link at 5′-CAp sequences. In the 2-aminopurine-thymine base pair, the exocyclic N2-amino group of the 2-aminopurine residue resides in a location similar to that of the N2-amino group of the guanine residue in a typical G-C base pair (Figure 7). Furthermore, work from Greenberg and coworkers provided evidence that 2-aminopurine residues in DNA are more reactive than guanine residues toward an aldehyde residue in a DNA duplex.70 For these reasons, we felt it would be interesting to examine whether 2-aminopurine could substitute for guanine in the cross-linking reaction examined here. We prepared two duplexes containing a 2-aminopurine-thymine base pair adjacent to an Ap site. Duplex G contained an adenine opposing the Ap site and duplex H contained a guanine residue opposing the Ap site. Upon incubation in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C duplexes G and H generated remarkable yields (15.1 ± 0.5 and 31.4 ± 0.1, respectively) of a slow-migrating band that co-migrated with the dG-Ap cross-linked duplex (Figure 8, lanes 5 and 9). In both cases, the presence of methoxyamine blocked formation of the slow-migrating band, indicating the involvement of the Ap aldehyde residue in the formation of this product. Footprinting of the cross-linked duplex I with H2O2-iron-EDTA67 confirmed that the cross-link attachment was located at the 2-aminopurine residue (Figures S5 and S6). Curiously, both duplexes G and H generated secondary bands that migrated more slowly than the major cross-link species (Figure 8). These could be anomeric isomers of the 2-aminopurine cross-link or cross-links derived from reactions of other neighboring bases with the Ap aldehyde; however, the identity of these minor products remains uncertain at this time.
Figure 7.

The 2-aminopurine-thymine base pair places an exocyclic amino group in the same location as a typical G-C base pair.
Figure 8.

Evidence for cross-link formation in 2-aminopurine-containing 2′-oligodeoxyribonucleotide duplexes G and H. The lower band corresponds to full-length labeled 2′-deoxyoligonucleotide and the upper band cross-linked DNA. The lower bands correspond to the full length labeled 2′-deoxyoligonucleotides and the upper band cross-linked DNA. Lane 1 is the uracil-containing precursor of 2′-deoxyoligonucleotide duplex G. Lane 2 is the abasic-site-containing 2′-deoxyoligonucleotide duplex G without incubation. Lane 3 is the abasic-site-containing 2′-deoxyoligonucleotide duplex G cleaved by treatment with piperidine (1 M, 95 °C, 25 min). Lane 4 is duplex G incubated with CH3ONH2 (2 mM) in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 5 is duplex G incubated in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 6 is the abasic-site-containing 2′-deoxyoligonucleotide duplex H without incubation. Lane 7 is the abasic-site-containing 2′-deoxyoligonucleotide duplex H cleaved by treatment with piperidine (1 M, 95 °C, 25 min). Lane 8 is duplex H incubated in with CH3ONH2 (2 mM) in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. Lane 9 is duplex H incubated in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. The 32P-labeled 2′-deoxyoligonucleotides were resolved on a sequencing gel and the radioactivity in each band quantitatively measured by phosphorimager analysis. The image shows the region of the gel where full-length oligonucleotide and cross-link migrate.
Formation and stability of the native and reduced cross-links under various conditions of pH, heat, and treatment with nucleophiles
Following cross-link formation, we subjected the native and reduced cross-linked duplexes to a variety of work-up conditions and then assessed the amount of remaining cross-link by denaturing gel electrophoresis. Cross-link yields in the reactions subjected to work-up conditions were normalized against a standard cross-linking reaction without post-reaction treatment run side-by-side on the same gel. We found that the yields of native cross-link were diminished by heating at 60 °C, 90 °C, or piperidine workup (100 mM, 60 °C) all for 15 min (Figure S7). On the other hand, adjustment of the pH to 3, followed by incubation for 15 min at 24 °C had no significant effect on yield of the native cross-link. Adjustment of the pH to 10, followed by incubation for 15 min at 24 °C increased cross-link yield somewhat. This result may mesh with a chemical precedent showing that the equilibrium formation of aminoglycosides from glucose and aryl amines is favored at pH 11.71 Post-formation treatment of the unreduced cross-link with methoxyamine (20 mM, 15 min) or hydrazine (20 mM, 15 min) did not substantially diminish the cross-link yield (Figure S7). Given that methoxyamine strongly blocks cross-link formation (presumably via oxime formation with the Ap aldehyde residue), the inability of this reagent to promote dissociation of the cross-link is striking. Treatment with N,N-dimethylethylenediamine led to a small increase in the cross-link yield. The yields of the reduced cross-link were not significantly altered by the various post-formation treatment conditions explored here. This is consistent with the anticipated stability of the alkylamine attachment proposed for the reduced cross-link (10, Scheme 2). Importantly, this showed that reductive amination affords a chemically stable cross-link that may be used in biochemical and biological studies.
Imine formation is a notoriously complex process that is affected by various catalysts and conditions.72 Therefore, we sought to ensure that the cross-linking process examined here was robust and occurs under a wide variety of conditions. We found that the presence of physiologically-relevant concentrations of the biological thiol glutathione (1 mM) during cross-link formation had no substantial effect on the yield of the unreduced cross-link (Figure S7). Similarly, dithiothreitol had no significant effect. With two exceptions, cross-link yields were not dramatically altered when the reaction was carried out in buffers other than the HEPES buffer that was used in our standard reactions or when salt conditions were varied (Figure S7, Panel B). The exceptions were the buffers MES (50 mM, pH 5.0) and Bis-tris (50 mM, pH 7)-DTPA (10 mM) that gave significantly enhanced yields of the cross-link. Additives such as adenine, diaminopurine, and 4-chloroaniline that have shown the ability to catalyze cross-link formation in other systems52,72,73 did not enhance formation of the dG-Ap cross-link. Similarly, the amino acids lysine and arginine had little or no effect on the cross-link yields (Figure S7, Panel B).
Characterization of the dG-Ap cross-links using LC-MS
We carried out mass spectrometric experiments to characterize the unreduced dG-Ap cross-link in duplex DNA. Unlabeled, cross-linked duplex was isolated using preparative gel electrophoresis as described above. In the first experiment, we digested DNA containing the dG-Ap cross-link (Duplex J, Scheme 2) with nuclease P1. The digestion mixture was subjected to LC-MS/MS analysis on an LTQ linear ion trap mass spectrometer, using previously reported experimental conditions.74-76 The LC-MS/MS result revealed the presence of a product whose mass is consistent with the partially digested “tetramer” cross-link shown in Figure 9 (m/z 583.5, [M-2H]2−). Elimination of 2′-deoxyadenosine-5′-phosphate (pdA) to give the singly-deprotonated ion of m/z 837, is consistent with the presence of 3′-flanking dA residues in this tetrameric dG-Ap cross-link. Partial digestion of this cross-link is consistent with our previous data showing that mitomycin C and psoralen-derived cross-links are resistant to P1 cleavage on the 3′-side of the cross-link attachments.74-76
Figure 9.

LC-ESI-MS and MS/MS for the analysis of the nuclease P1 digestion mixture of duplex J with a dG-Ap cross-link. Shown in (a) is the selected-ion chromatogram (SIC) for monitoring the loss of a 2′-deoxyadenosine-5′-phosphate from the [M - 2H]2− ion of the tetramer shown (i.e., the m/z 583.5→ 837 transition). Inset gives the higher resolution “ultra-zoom” ESI-MS for the [M - 2H]2− ion of the tetramer. Displayed in (b) is the tandem mass spectrum for the [M - 2H]2− ion of the tetramer.
In the second experiment, treatment of the cross-linked duplex J with a four-enzyme cocktail including nuclease P1, alkaline phosphatase, and phosphodiesterases I and II, resulted in release of the fully digested dG-Ap cross-link (Figure 10). LC-MS/MS analysis revealed two major peaks eluting at 39 and 50 min in the selected-ion chromatogram (SIC) for the m/z 384→268→240 transitions, which monitor sequential neutral losses of 2-deoxyribose and CO from the cross-link. The presence of two peaks in the chromatogram is consistent with the expectation that the unreduced, digested crosslink will exist as an isomeric mixture of the α- and β-anomers of both the pyranose and furanose forms of the 2-deoxyribose sugar adduct (11 and 12, Scheme 3). The α- and β-anomers of the pyranose form are expected to predominate.77
Figure 10.

LC-ESI-MS/MS/MS for the analysis of the 4-enzyme digestion mixture of duplex J with a dG-Ap cross-link. Shown in (a) is the selected-ion chromatogram (SIC) for monitoring the m/z 384→268→240 transition (proposed structures for fragment ions in this fragmentation pathway are shown on the right). Depicted in (b) is MS/MS/MS arising from the fragmentation of the ion of m/z 268 observed in MS/MS from the cleavage of the [M + H]+ ion of the completely digested crosslink remnant.
Scheme 3.

Conclusion
The experiments described here examined the formation and properties of interstrand dG-Ap cross-links formed at 5′-CAp sequences in duplex DNA. Other groups have characterized related cross-links generated by oxidized Ap sites in DNA;52,69,78 however, it is important to emphasize that the structure and reactivity of the oxidized Ap sites are distinct from that of the “native” Ap site that is the focus of our work.
We found that incubation of DNA duplexes containing an abasic site in a 5′-CAp sequence under physiologically-relevant conditions in the absence of reducing agents yields easily detectable amounts of a slow-moving cross-linked duplex within 1 h as monitored by denaturing polyacrylamide gel electrophoresis. Consistent with the notion that the slow-moving band on the gel was an interstrand cross-link resulting from the reaction of the Ap aldehyde group with the opposing guanine residue, formation of this product was inhibited by methoxyamine capping of the Ap site and by an inosine-for-guanine replacement at the 5′-CAp site. LC-MS/MS analysis of digested cross-linked duplex J revealed that the cross-link involves a covalent linkage between the Ap site and the opposing guanine residue. Once formed, the native (unreduced) cross-link is stable to a variety of conditions (Figure S7) and we hypothesize that the equilibrium formation of the cyclic hemiaminal 9 is favored and renders this cross-link more stable than a typical imine or hemiaminal linkage. Cross-link formation was not highly sensitive to identity of the buffer, salt concentration, or the presence of the biological thiol glutathione (1 mM). The reversible nature of the chemistry examined here may allow dissociation of the cross-link during sample manipulation and electrophoresis. Thus, the yields of unreduced cross-link observed in these experiments presumably represent a lower limit of the amount of cross-link present in the reactions. This work provides the first clear evidence that Ap-derived cross-links can form under physiologically-relevant conditions. Moreover, the stability of the cross-link hints at the potential for these lesions to block DNA transcription and replication.
Incubation of duplexes containing an 5′-CAp sequence in the presence of the reducing agent NaCNBH3 in pH 5.2 buffer generated good yields (∼20%) of a slow-moving band on denaturing polyacrylamide gels. The results of MALDI-TOF mass spectrometry, and gel mobility experiments provided evidence that the slow-moving band generated under these conditions of reductive amination did, indeed, contain both full-length strands of the starting duplex. The cross-link generated under reducing conditions was structurally distinct from the native cross-link, as it could be resolved from the native unreduced cross-link on denaturing gels. Methoxyamine capping, inosine-for-guanine replacement, and hydroxyl radical footprinting further provided evidence that cross-link formation involved attachment of the Ap aldehyde group to the opposing guanine residue at the 5′-CAp sequences. The cross-link generated under reducing conditions was stable against all of the workup conditions examined here (Figure S7, panel A).
Overall, the evidence is consistent with the chemical mechanism for cross-link formation at 5′-CAp sequences illustrated in Scheme 2, in which reaction of the Ap aldehyde residue with the N2-amino group of the opposing guanine residue yields hemiaminal, imine, or cyclic hemiaminal linkages (structures 7-10). Under conditions of reductive amination (NaCNBH3/pH 5.2), equilibrium amounts of the imine can be irreversibly converted to the amine linkage. Indeed, both MALDI-TOF-MS data and the improved stability of cross-link generated under reducing conditions were consistent with the anticipated alkylamine structure for this cross-link (10, Scheme 2). Le Chatelier's principle tells us that the reduced cross-link has the potential to accumulate beyond the equilibrium levels of its precursors 9 and 10. The system is somewhat complex, however, and formation of the reduced cross-link likely must compete against irreversible reduction of the abasic aldehyde to the alcohol.
The yields of the reduced dG-Ap cross-link obtained in the current study were substantially greater than those reported in our previous work.33 The Ap-containing duplexes used here were longer than the 15 bp duplexes employed in the original studies and it is possible that partial melting of the duplexes in our early work limited cross-link yields. In addition, in the 21 bp duplex used here, a thymine rather than an adenine residue rests on the 3′-side of the guanine residue involved in the cross-linking reaction and flanking bases may affect the cross-link yield (K. Johnson and M. Fekry, unpublished observations). Finally, the sodium acetate buffer used in the current reductive amination experiments may catalyze cross-link formation.79-81
We believe that the exocyclic N2-amino group of guanine is the nucleophile that reacts with the Ap aldehyde to generate the dG-Ap cross-link as postulated in Scheme 2. A wealth of precedents show that the N2-amino group of guanine residues in DNA can react with aldehyde groups in small molecules to form covalent adducts.28-32 At the same time, it is important to recognize chemical precedents showing that intramolecular reactions of aldehydes with endocyclic nitrogens can yield covalent adducts.31,82 Thus, the exact chemical structure of the dG-Ap cross-links remains uncertain and efforts are underway to synthesize authentic standards of the putative cross-linked nucleoside analogs of 9 and 10 for use in structure elucidation.
Further support for the importance of the exocyclic N2-amino group at the cross-linking site came from the experiments in which the critical G-C base pair adjacent to the Ap site was replaced with a 2-aminopurine-thymine base pair (in duplexes G and H). In this base pair, the exocyclic amino group of the 2-aminopurine residue is expected to reside in a location similar to the N2-amino group of the guanine residue in a typical G-C base pair (Figure 7).83 Indeed, excellent yields of the slow-migrating, cross-linked DNA band were obtained from duplexes G and H (15.1 ± 0.5 and 31.4 ± 0.1, respectively).
It is interesting to consider how DNA structure and reactivity combine to determine the sites at which significant yields of native (unreduced) Ap-derived crosslinks are formed. This cross-link formation is an equilibrium process and relatively small changes in the free energy of the reaction will engender significant changes in Keq.84 For example, the difference in equilibrium yields of cross-linked duplex described above for duplexes G and H (15% versus 31%, respectively) reflect a free energy difference of only 0.5 kcal/mol between the two systems (ΔΔG = −RTln(K1/K2)). Even the rather large difference in equilibrium cross-link yields observed for duplexes A and H reflects a free energy difference of only about 2 kcal/mol. Such small changes in free energy can easily arise from subtle structural changes in the large macromolecular ensemble of the crosslink. For example, the relatively high cross-link yields obtained for the 2-aminopurine-containing duplexes G and H could arise from the greater conformational flexibility of the 2-aminopurine-T base pair in duplex DNA as compared to a G-C base pair.70,83 Overall, however, the energetic analysis suggests that, within closely related DNA sequences, it may be rather difficult to rationalize or predict the cross-link yields obtained in different sequence contexts unless additional structural information is brought to bear on the problem. The ability of rather subtle changes in local sequence to significantly alter the yields of Ap-derived cross-links is illustrated by the different yields of cross-link generated in duplexes G and H that differ only in the identity of the purine directly opposing the Ap (adenine versus guanine, respectively). The structural reasons for this sequence effect remain uncertain. Considering the separate issue of DNA reactivity, the remarkably high yields of native cross-link generated by 2-aminopurine in duplexes G and H further offer the possibility that different nucleobases possess different inherent abilities to react with the Ap-aldehyde residue. Along these lines, the striking results reported here for 2-aminopurine suggest that adenine residues may be competent in the generation of Ap-derived cross-links (2-aminopurine is a structural isomer of adenine). To the best of our knowledge, however, there exists no quantitative information regarding the relative rates and equilibria associated with the reactions of different nucleobases with aldehydes.
Our observations of Ap-derived interstrand DNA-DNA cross-links could be relevant to the cytotoxic and mutagenic properties of Ap sites in DNA. The highly deleterious nature of cross-links suggests that even small amounts of Ap-derived crosslinks could make a significant contribution to the overall biological effects stemming from the generation of Ap sites in cellular DNA. Some of the cross-linked duplexes reported here may be useful substrates for the study of cross-link repair. Overall, our results provide a firm foundation for future investigations of the structure, occurrence, and biological consequences of Ap-derived cross-links in cellular DNA.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health for partial support of this work (CA83925 and 119131 to K.G. and ES019873 to Y.W. and ES021007 to K.G and Y.W.). We thank Professor Marc Greenberg for helpful discussions during the course of this work. MALDI mass spectrometry was conducted at the Charles W. Gehrke Proteomics Center at the University of Missouri and we thank Beverly DaGue for assistance with these experiments.
Footnotes
Supporting Information Available. Hydroxyl radical footprinting of cross-linked duplexes F and I, MALDI-TOF-MS data for reduced cross-linked duplex A, time course for formation of the native cross-link formation in duplex A, and yields and stability of cross-link under various conditions. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gates KS, Nooner T, Dutta S. Chem Res Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- 2.Gates KS. Chem Res Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amosova O, Coulter R, Fresco JR. Proc Nat Acad Sci USA. 2006;103:4392–4397. doi: 10.1073/pnas.0508499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Nyberg B. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura J, Swenberg JA. Cancer Res. 1999;59:2522–2526. [PubMed] [Google Scholar]
- 6.Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- 7.Park JH, Troxel AB, Harvey RG, Penning TM. Chem Res Toxicol. 2006;19:719–728. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rusyn I, Asakura S, Y L, Kosyk O, Koc H, Nakamura J, Upton PB, Swenberg JA. DNA repair. 2005;4:1099–1110. doi: 10.1016/j.dnarep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 9.Viswesh V, Gates KS, Sun D. Chem Res Toxicol. 2010;23:99–107. doi: 10.1021/tx900301r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torres MC, Rieger RA, Iden CR. Chem Res Toxicol. 1996;9:1313–1318. doi: 10.1021/tx960107t. [DOI] [PubMed] [Google Scholar]
- 11.Masta A, Gray PJ, Phillips DR. Nucleic Acids Res. 1994;22:3880–3886. doi: 10.1093/nar/22.19.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Chandrasena ER, Yuan Y, Peng Kw, van Breemen RB, Thatcher GRJ, Bolton JL. Chem Res Toxicol. 2010;23:1365–1373. doi: 10.1021/tx1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Bont R, van Larebeke N. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- 14.Marnett LJ, Burcham PC. Chem Res Toxicol. 1993;6:771–785. doi: 10.1021/tx00036a005. [DOI] [PubMed] [Google Scholar]
- 15.Gates KS. In: Reviews of Reactive Intermediates. Platz MS, Moss RA, Jones MJ, editors. John Wiley and Sons, Inc.; Hoboken: 2007. pp. 333–378. [Google Scholar]
- 16.Guillet M, Bioteux S. Mol Cell Biol. 2003;23:8386–8394. doi: 10.1128/MCB.23.22.8386-8394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.David SS, Williams SD. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 18.Loeb LA, Preston BD. Ann Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 19.Boiteux S, Guillet M. DNA Repair. 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Lhomme J, Constant JF, Demeunynck M. Biopolymers. 2000;52:65–83. doi: 10.1002/1097-0282(1999)52:2<65::AID-BIP1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 21.Wilson DM, III, Barsky D. Mutation Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 22.Evans AR, Limp-Foster M, Kelley MR. Mutation Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 23.Yu SL, Lee SK, Johnson RE, Prakash L, Prakash S. Mol Cell Biol. 2003;23:382–388. doi: 10.1128/MCB.23.1.382-388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auerbach P, Bennett RAO, Bailey EA, Krokan HE, Demple B. Proc Nat Acad Sci USA. 2005;102:17711–17716. doi: 10.1073/pnas.0504643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaaper RM, Loeb LA. Proc Nat Acad Sci USA. 1981;78:1773–1777. doi: 10.1073/pnas.78.3.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor AF, Weiss B. J Bacteriol. 1982;151:351–357. doi: 10.1128/jb.151.1.351-357.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilde JA, Bolton PH, Mazumdar A, Manoharan M, Gerlt JA. J Am Chem Soc. 1989;111:1894–1896. [Google Scholar]
- 28.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Chem Res Toxicol. 2000;13:1149–1157. doi: 10.1021/tx000118t. [DOI] [PubMed] [Google Scholar]
- 29.Riggins JN, Daniels JS, Rouzer CA, Marnett LJ. J Am Chem Soc. 2004;126:8237–8243. doi: 10.1021/ja040009r. [DOI] [PubMed] [Google Scholar]
- 30.Chaw YFM, Crane LE, Lange P, Shapiro R. Biochemistry. 1980;19:5525–5531. doi: 10.1021/bi00565a010. [DOI] [PubMed] [Google Scholar]
- 31.Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ. Acc Chem Res. 2008;41:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. J. o. t. A. C. S. J Am Chem Soc. 2003;125(1):50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 33.Dutta S, Chowdhury G, Gates KS. J Am Chem Soc. 2007;129:1852–1853. doi: 10.1021/ja067294u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopkins PB, Millard JT, Woo J, Weidner MF, Kirchner JJ, Sigurdsson ST, Raucher S. Tetrahedron. 1991;47:2475–2489. [Google Scholar]
- 35.Hoehn ST, Turner CJ, Stubbe J. Nucleic Acids Res. 2001;29:3413–3423. doi: 10.1093/nar/29.16.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, Dupradeau FY, Case DA, Turner CJ, Stubbe J. Nucleic Acids Res. 2008;36:253–262. doi: 10.1093/nar/gkm622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de los Santos C, El-khateeb M, Rege P, Tian K, Johnson F. Biochemistry. 2004;43:15349–15357. doi: 10.1021/bi048400c. [DOI] [PubMed] [Google Scholar]
- 38.Beger RD, Bolton PH. J Biol Chem. 1998;273:15565–15573. doi: 10.1074/jbc.273.25.15565. [DOI] [PubMed] [Google Scholar]
- 39.Schärer OD. ChemBioChem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 40.Noll DM, Mason TM, Miller PS. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hlavin EM, Smeaton MB, Miller PS. Env Mol Mutagenesis. 2010;51:604–624. doi: 10.1002/em.20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muniandy PA, Liu J, Majumdar A, Liu St, Seidman MM. Crit Rev Biochem Mol Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenberg RB, Alberti M, Hearst JE, Chua MA, Saffran WA. J Biol Chem. 2001;276:31551–31560. doi: 10.1074/jbc.M103588200. [DOI] [PubMed] [Google Scholar]
- 45.Shen X, Li L. Env Mol Mutagenesis. 2010;51:493–499. doi: 10.1002/em.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho TV, Schärer OD. Env Mol Mutagenesis. 2010;51:552–566. doi: 10.1002/em.20573. [DOI] [PubMed] [Google Scholar]
- 47.Rajski SR, Williams RM. Chem Rev. 1998;98:2723–2795. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 48.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GTJ, Meinecke P, Kleijer WJ, Vijg J, Jaspers NGJ, Hoeijmakers JHJ. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 49.Bergstrahl DT, Sekelsky J. Trends in Genetics. 2007;24:70–76. doi: 10.1016/j.tig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Grillari J, Katinger H, Voglauer R. Nucleic Acids Res. 2007;35:7566–7576. doi: 10.1093/nar/gkm1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Lab Manual. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 52.Sczepanski JT, Jacobs AC, Majumdar A, Greenberg MM. J Am Chem Soc. 2009;131:11132–11139. doi: 10.1021/ja903404v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luce RA, Hopkins PB. Methods Enzymol. 2001;340:396–412. doi: 10.1016/s0076-6879(01)40433-2. [DOI] [PubMed] [Google Scholar]
- 54.Maxam AM, Gilbert W. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 55.Lavanant H, Lange C. Rapid Commun Mass Spec. 2002;16:1928–1933. doi: 10.1002/rcm.816. [DOI] [PubMed] [Google Scholar]
- 56.Koomen JM, Russell WK, Tichy SE, Russell DH. J Mass Spectrom. 2002;37 doi: 10.1002/jms.312. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Yuan B, Guerrero C, Bahde R, Gupta S, Wang Y. Anal Chem. 2011;83:2201–2209. doi: 10.1021/ac103099s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Borch RF, Hassid AI. J Org Chem. 1972;37:1673–1674. [Google Scholar]
- 59.Manoharan M, Andrade LK, Cook PD. Org Lett. 1999;1:311–314. doi: 10.1021/ol9906209. [DOI] [PubMed] [Google Scholar]
- 60.Dohno C, Okamoto A, Saito I. J Am Chem Soc. 2005;127:16681–16684. doi: 10.1021/ja054618q. [DOI] [PubMed] [Google Scholar]
- 61.Angelov T, Guainazzi A, Schärer OD. Org Lett. 2009;11:661–664. doi: 10.1021/ol802719a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Varshney U, van de Sande JH. Biochemistry. 1991;30:4055–4061. doi: 10.1021/bi00230a033. [DOI] [PubMed] [Google Scholar]
- 63.Eftedal I, Guddal PH, Slupphaug G, Volden G, Krokan HE. Nucleic Acids Res. 1993;11:2095–2101. doi: 10.1093/nar/21.9.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nilsen H, Yazdankhah SP, Eftedal I, Krokan HE. FEBS Lett. 1995;362:205–209. doi: 10.1016/0014-5793(95)00244-4. [DOI] [PubMed] [Google Scholar]
- 65.Rosa S, Fortini P, Karran P, Bignami M, Dogliotti E. Nucleic Acids Res. 1991;19:5569–5574. doi: 10.1093/nar/19.20.5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coombs MM, Livingston DC. Biochim Biophys Acta. 1969;174:161–173. doi: 10.1016/0005-2787(69)90239-1. [DOI] [PubMed] [Google Scholar]
- 67.Pogozelski WK, McNeese TJ, Tullius TD. J Am Chem Soc. 1995;117:6428–6433. [Google Scholar]
- 68.Huang H, Solomon MS, Hopkins PB. J Am Chem Soc. 1992;114:9240–9241. [Google Scholar]
- 69.Sczepanski JT, Jacobs AC, Greenberg MM. J Am Chem Soc. 2008;130:9646–9647. doi: 10.1021/ja8030642. [DOI] [PubMed] [Google Scholar]
- 70.Sczepanski JT, Heimstra CN, Greenberg MM. Bioorg Med Chem. 2011;19:5788–5793. doi: 10.1016/j.bmc.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holton S, Runquist O. J Org Chem. 1961;26:5193–5195. [Google Scholar]
- 72.Dirksen A, Dawson PE. Bioconjugate Chem. 2008;19:2543–2548. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thygesen MB, Munch H, Sauer J, Cló E, Jorgensen MR, Hindsgaul O, Jensen KJ. J Org Chem. 2010;75:1752–1755. doi: 10.1021/jo902425v. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y, Wang Y. Anal Chem. 2003;75:6306–6313. doi: 10.1021/ac034683n. [DOI] [PubMed] [Google Scholar]
- 75.Cao H, Hearst JE, Corash L, Wang Y. Anal Chem. 2008;80:2932–2938. doi: 10.1021/ac7023969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lai C, Cao H, Hearst JE, Corash L, Luo H, Wang Y. Anal Chem. 2008;80:8790–8798. doi: 10.1021/ac801520m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanderson PN, Sweatman BC, Farrant RD, Lindon JC. Carbohydate Res. 1996;284:51–60. [Google Scholar]
- 78.Regulus P, Duroux B, Bayle PA, Favier A, Cadet J, Ravanat JL. Proc Nat Acad Sci USA. 2007;104:14032–14037. doi: 10.1073/pnas.0706044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kallen RG, Jencks WP. J Biol Chem. 1966;241:5851–5863. [PubMed] [Google Scholar]
- 80.Bomann MD, Guch IC, DiMare M. J Org Chem. 1995;60:5995–5996. [Google Scholar]
- 81.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Xa, Song D, Lippard SJ. J Org Chem. 2008;73:734–737. doi: 10.1021/jo702394v. [DOI] [PubMed] [Google Scholar]
- 83.Dallmann A, Dehmel L, Peters T, Mügge C, Griesinger C, Tuma J, Ernsting NP. Angew Chem Int Ed Eng. 2010;49:5989–5992. doi: 10.1002/anie.201001312. [DOI] [PubMed] [Google Scholar]
- 84.Belowich ME, Stoddart JF. Chem Rev. 2012;41:2003–2024. doi: 10.1039/c2cs15305j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.