

Abstract
Prions are infectious, self-propagating protein conformations. [PSI+], [RNQ+] and [URE3] are well characterized prions in Saccharomyces cerevisiae and represent the aggregated states of the translation termination factor Sup35, a functionally unknown protein Rnq1 and a regulator of nitrogen metabolism Ure2, respectively. Overproduction of Sup35 induces the de novo appearance of the [PSI+] prion in [RNQ+] or [URE3] strain, but not in non-prion strain. However, [RNQ+] and [URE3] prions themselves, as well as overexpression of a mutant Rnq1 protein, Rnq1Δ100 and Lsm4, hamper the maintenance of [PSI+]. These findings point to a bipolar activity of [RNQ+], [URE3], Rnq1Δ100 and Lsm4, and probably other yeast prion proteins as well, for the fate of [PSI+] prion. Possible mechanisms underlying the apparent bipolar activity of yeast prions will be discussed.
Key words: yeast prion, [PSI+], Pin+, [RNQ+], [RNQ1Δ100+], [URE3], Lsm4, New1
Prions are transmissible agents caused by the self-propagating conformational change of proteins.1 According to the “protein only” hypothesis,1 the prion protein (PrP) is the sole agent responsible for causing numerous infectious diseases including scrapie (sheep), bovine spongiform encephalopathy (BSE, cow) and chronic wasting (deer and elk) as well as kuru and Creutzfeld-Jacob disease (humans). In yeast Saccharomyces cerevisiae, prions have also been characterized as non-Mendelian inheritable elements, notably [PSI+], [URE3] and [RNQ+].2,3 Molecular and genetic studies of these yeast prions have greatly facilitated the elucidation of the molecular basis for prion conversion and propagation.
The yeast prion [PSI+]2,4 is the amyloid-like structure of the eRF3 polypeptide release factor, Sup35, that is essential for terminating protein synthesis at stop codons5,6 (reviewed in ref. 7). [PSI+] cells are marked by an altered catalytic protein conformation of Sup35 whereby the Sup35 protein is converted from a soluble, active state to an aggregated inactive state. When Sup35 is in the [PSI+] state, ribosomes exhibit an increased rate of stop codon readthrough, causing a non-Mendelian trait easily detected by nonsense suppression.8–10
The de novo appearance of [PSI+] is induced by overexpression of the Sup35NM domain (NM domain is also called prion domain). This appearance can be enhanced by Pin+ protein (designated for [PSI+] inducibility). The best studied Pin+ protein is [RNQ+] prion. [RNQ+] is the prion form of Rnq1 protein of unknown function.3 [rnq−], the non-prion form of Rnq1, does not have the Pin+ activity. Liebman and coworkers have identified many Pin+ proteins by genetic screen in S. cerevisiae.11 Those factors contain glutamine/asparagine (Q/N) rich domain, and some of these Pin+ proteins form prions similarly to [RNQ+] prion. These include the transcription regulator Ure2 that forms [URE3] prion,2 chromatin remodeling factor Swi1 that forms [SWI+] prion,12 global transcriptional co-repressor Cyc8 that forms [OCT+] prion,13 a Lsm-family protein, Lsm4, that likely forms a prion,14 and a chimera protein of N-terminal domain of New1 and C-terminal domain of Sup35 that forms [NU+] prion.15,16 These Pin+ proteins markedly stimulate the de novo appearance of [PSI+] upon overproduction of Sup35NM, but are neither required for nor inhibitory to the maintenance of [PSI+] prion.17
In the past few years, we have conducted genome-wide screens for [PSI+]- and [URE3]-eliminating factors or mutants18–20 (Oishi K and Nakamura Y, unpublished). These screens provided us with known or unknown host factors or their mutant forms. What surprised us was that many of them were potential Pin+ proteins, suggesting that Pin+ proteins might be inhibitory to the maintenance of the [PSI+] prion when overproduced19 (Oishi K and Nakamura Y, unpublished). We will overview these apparently contradictory observations and discuss possible mechanisms underlying the bipolar activity of yeast prions.
A Bipolar Activity of [RNQ+] Prion and its Prion Variants
Sondheimer and Lindquist searched for prion candidates using yeast protein database as unusual high concentration of the polar residues glutamine and asparagine,3 which is the known landmark for the prion-determining domains of [PSI+] and [URE3].9,21,22 They found that one such novel protein, Rnq1 (named so for being rich in asparagine [N] and glutamine [Q]), forms a prion, designated [RNQ+]. The Rnq1 protein is composed of the non-Q/N rich N-terminal (i.e., nonprion) domain (1–152 amino acids [a.a.]) and the Q/N-rich C-terminal (i.e., prion) domain (153–405 a.a).3 It has been reported that a null rnq1 mutation produces the doubling of spores in the asci, suggesting a regulatory role of Rnq1 in preventing an additional mitotic division during ascus formation.23 However, the phenotype of the null rnq1 mutation could be due to concurrent abolition of the adjacent BIK1 promoter.24 In any case, this phenotype or the function of Rnq1 remains to be investigated firmly. Shortly after the identification of Rnq1 and [RNQ+] prion, Weissman and Liebman groups independently reported that the [RNQ+] prion has the Pin+ activity,11,16 showing a positive role of [RNQ+] for [PSI+] prion.
Different strains of the PrP prion diseases have distinct characteristics such as incubation times prior to neurodegeneration, variable distribution of PrPSc in brain tissue and distinct PrPSc proteolysis patterns.25,26 It is also thought that the conformation of the pathologic isoform of PrP accounts for different prion strains.27 Yeast prions have also prion strains (or variants). [PSI+] variants show distinct readthrough phenotype on YPD: weak [PSI+] can read through nonsense mutations less efficiently than strong [PSI+].28 In the subsequent studies of [RNQ+] prion, Liebman and coworkers have defined several prion variants with distinct Pin+ activities (i.e., low, medium, high and very high) and distinct fluorescent patterns of Rnq1-GFP fusion protein [i.e., single dot (s.d.) and multiple dot (m.d.)].29,30 They found that, among these prion variants, s.d. [RNQ+] variants drastically destabilize weak [PSI+] variant when analyzed by the cytoduction experiment. This might be the first report of a negative influence of [RNQ+] on [PSI+].
In our screen for factors inhibitory to the maintenance of [PSI+], we found that overproduction of Rnq1Δ100 that deletes the N-terminally 100 amino acids (a.a.) of Rnq1 inhibits the maintenance of [PSI+] in [RNQ+] cells, but not in [rnq−] cells,19 and yet stimulates the de novo appearance of [PSI+].31 Whereas Rnq1Δ100 forms SDS-unstable co-aggregates with Rnq1 in [rnq−], Rnq1Δ100 forms SDS-stable co-aggregates with Rnq1 in [RNQ+], probably through the interaction between the C-terminal prion domains of Rnq1 and Rnq1Δ100. Importantly, Rnq1Δ100 per se is capable of forming a prion, when Rnq1 is replaced by Rnq1Δ100 in [RNQ+] strain by a plasmid shuffle strategy.31 The resulting prion was designated [RNQ1Δ100+]. [RNQ1Δ100+] has a similar Pin+ activity as it stimulates the de novo induction of [PSI+], and inherits the inhibitory activity to hamper the maintenance of [PSI+] though less efficiently than [RNQ+] made of Rnq1-Rnq1Δ100 co-aggregates.31 Interestingly, [RNQ1Δ100+] prion was eliminated by de novo [PSI+] induction, demonstrating the selfish activity to eliminate a heterologous prion in S. cerevisiae.31
Finally, we obtained the missense mutations in N-terminal nonprion domain of Rnq1 that impaired the stable maintenance of [RNQ+].32 Those mutant Rnq1s have the Pin+ activity in their prion states.32 When these mutant Rnq1s are overexpressed, [PSI+] was inhibited in [RNQ+] but not in [rnq−].33 Furthermore, prions of Rnq1Δ100 and the Rnq1 missense mutant proteins are unstable under their native expression levels but are stable when overexpressed.31,32 Therefore, Rnq1Δ100 and the missense mutant proteins share the phenotype. These findings strongly suggest that these N-terminal nonprion-domain mutants exhibit the same activity as Rnq1Δ100.
A Bipolar Activity of [URE3] Prion
The [URE3] prion is the amyloid like structure of the nitrogen-catabolite-repression transcriptional regulator Ure2 that inhibits Gln3 transcription factor under rich nitrogen condition. Similar to other prion proteins, Ure2 overexpression and [URE3] prion shows the Pin+ activity.11 On the other hand, Schwimmer and Masison showed the antagonistic interaction between [PSI+] and [URE3] in the presence of [RNQ+].34 They made a [PSI+] [URE3] double-prion strain and examined the mitotic stability of each prion by monitoring the [PSI+] state by colony color (white for [PSI+] and red for [psi−] on YPD plate by ade2-1 nonsense suppression), and the [URE3] state by colony size (smaller for [URE3] and larger for [ure-o], nonprion state). The original [PSI+][URE3] colonies were slightly pink, showing a reduced ability of [PSI+] to mediate nonsense suppression. The [PSI+] [URE3] cells also grew more slowly than isogenic [ure-o] cells. When these cells were grown nonselectively, there was a noticeable frequency of appearance of faster-growing white colonies that contained [PSI+] but lost [URE3]. Thus, the loss of [URE3] from cells with both prions restored the normal [PSI+] phenotype and wildtype growth, indicating that [URE3] impaired [PSI+]-mediated nonsense suppression and was inhibitory to growth.
A [psi−][URE3] variant was obtained by selectively eliminating [PSI+] from the [PSI+][URE3] strain by transient overexpression of Hsp104. The resulting [psi−][URE3] cells grew even more slowly than the [PSI+][URE3] cells, indicating that the growth-inhibitory effect of [URE3] was diminished by the presence of [PSI+]. Together, these results showed that both prions could propagate in the same cell, but the presence of [PSI+] impaired [URE3] propagation and [URE3] modestly impaired [PSI+] propagation and inhibited growth independently of [PSI+].34
A mechanism of the antagonistic interaction between [PSI+] and [URE3] was proposed as the cellular response model.34,35 Expressions of Hsp70 (Ssa protein family) and Hsp104 chaperones were elevated in the presence of prion, and more elevated in the co-presence of two prions.34,36 The elevated Hsp104 expression causes the elimination of [PSI+],37 and the elevated Ssa expression cause elimination of [URE3].34 Therefore the bipolar activity of [URE3] might be the cross-seeding and the cellular response as the positive and negative interactions with [PSI+], respectively. However, to our knowledge there is no data that Ure2 fibrils accelerate Sup35 fibril formation in vitro.
A Bipolar Activity of Lsm4
Highly conserved from archaebacteria to humans, Lsm4 is a member of the Lsm protein family that is involved in multiple mRNA-related processes.38 In the nucleus, Lsm4 and other members form a ring-shaped heteroheptameric complex Lsm2–8 to stabilize U6 snRNA, thus playing essential roles in mRNA processing.39 In the cytoplasm, a similar heteroheptameric complex Lsm1–7 participates in P-bodies, an intracellular supercomplex of mRNA decay factors.40 These complex formations are dependent on the characteristic “Sm motifs,” which Lsm4 possesses in its N-terminal region.41 Intriguingly, the C-terminal region of Lsm4 bears a conspicuous, amyloid-prone Q/N-rich stretch, which has been reported to play a role in the localization of P-bodies.42
Liebman and coworkers reported that Lsm4 is one of the Pin+ proteins whose overproduction stimulates the de novo induction of [PSI+].11 This suggested Lsm4 could form prions because Pin+ factors are considered to be prions. Furthermore, Lindquist and coworkers have conducted a bioinformatic proteome-wide survey for prionogenic proteins in S. cerevisiae.14 They found an amino acid bias in aggregation-prone candidates and discovered that 19 of these could form prions including Lsm4.14 We found that overproduction of Lsm4 inhibits the maintenance of [PSI+] (Oishi K and Nakamura Y, unpublished). Interestingly, [URE3] prion was more prone to being eliminated by overproduced Lsm4 (and Rnq1Δ100 as well) compared with [PSI+] (Oishi K and Nakamura Y, unpublished observation). This probably reflects the number of prion seeds of [URE3] and [PSI+]: the [URE3] seeds were estimated to be ∼20/cell,43 while those of [PSI+] were estimated to be 60/cell by the same procedure or 500–1,000/cell by a newer procedure.44,45 Therefore, one might speculate that high curability of [URE3] is due to the fewer number of prion seeds compared with [PSI+] and the seed replication inhibited by Lsm4.
A Bipolar Activity of New1
Recently, it has been reported that New1 might have an inhibitory effect on [PSI+].46 New1 has been proposed to form a prion because overproduction of its N-terminal fusion to the Sup35C domain resulted in the appearance of a [PSI+]-like prion, [NU+].15 The N-terminal domain of New1 carries a prion-domain signature including a Q/N-rich tract, a QQG GYQ SYN motif similar to oligopeptide repeat (Q/P)GG YQQ YN of Sup35, and octa-repeats of asparagine/tyrosine/asparagine (NYN) tripeptide.47 The C-terminal domain of New1 is homologous to the eukaryotic translation elongation factor eEF3 and has two putative ATP-binding sites. Yoshida and coworkers showed that New1 breaks Sup35NM amyloid fibrils in the presence of ATP in vitro.46 Overproduction of New1 in [PSI+] cells altered dot-like Sup35NM-GFP foci to dispersed fluorescence with very small dots or string-like aggregates. However, [PSI+] was not cured under these conditions.46 We also confirmed that overproduction of New1 does not eliminate [PSI+] prion (Oishi K and Nakamura Y, unpublished). However, overproduction of New1 results in loss of [URE3] prion (Oishi K and Nakamura Y, unpublished), probably reflecting different susceptibility of each prion to inhibitors as described above.
Some Pin+ proteins that have been reported by Derkatch form prions [SWI+] and [OCT+].11–13 We found that overproduction of Swi1 inhibits the maintenance of [URE3] prion (Oishi K and Nakamura Y, unpublished). It remains to be investigated whether Cyc8 overproduction affects [PSI+] and/or [URE3].
Mechanistic Insight into the [PSI+] Amyloids Hampered by Overproduced Pin+ Proteins
A key to answer the question of why [PSI+] is eliminated by overproduced Pin+ proteins is probably to investigate the fate of [PSI+] amyloids. One idea is to assume that [PSI+] amyloids are disassembled into Sup35 monomer or oligomers that no longer retain the prion-seeding activity as seen with overproduced Hsp104.10,48 In yeast, the molecular chaperone Hsp104 has been shown to play a crucial role in the fragmentation process37,49,50 and is required for the propagation of yeast prions because it breaks up amyloid filaments to generate prion seeds for efficient prion transmission.10,50,51 It is also well established that Hsp104 overproduction causes loss of [PSI+] prion phenotypes in yeast,37 most likely because abundant Hsp104 exceedingly breaks the prion aggregates, resulting in insufficient population of functional prion seeds. Another possible mechanism by which overproduced Pin+ proteins inhibit [PSI+] propagation is their direct interaction with the [PSI+] growing tip, which leads to the inability of further extension of amyloids (called capping model).35
To investigate these plausible scenarios, we examined the dynamics of [PSI+] aggregates by semi-denaturing detergent-agarose gel electrophoresis (SDD-AGE) and fluorescence correlation spectroscopy (FCS) using strains with GFP integrated in the endogenous SUP35 ORF.33,52 In the SDD-AGE analysis, to our surprise, the average size of detergent-resistant Sup35-GFP ([PSI+]) aggregates increased after overproduction of Rnq1Δ100. The amount of the enlarged aggregates tended to decrease upon longer incubation, and the amount of monomers increased, in accordance with the [PSI+] elimination phenotype.33
Another method, FCS, is a technique to determine the diffusion coefficients of fluorescence molecules by calculating the autocorrelation function in a microscopic detection volume under 10−15 L (1 femtoliter) defined by a tightly focused laser beam and pinhole, providing us an estimation of the size of aggregates.53 Consistent with the SDD-AGE result, the FCS data indicated that the average size of Sup35-GFP ([PSI+]) aggregates was increased at 24 h after Rnq1Δ100 induction, and then the amount of aggregates was dramatically reduced and instead the amount of monomer was increased at 48 h after Rnq1Δ100 induction.33
The dynamics of Sup35-GFP in single living cells was further investigated. Single cell analysis showed slower diffusion in mother cell but fast diffusion in daughter cell upon Rnq1Δ100 overproduction, characteristic of [PSI+] and [psi−] states, respectively.33 Strikingly, the mother cell had freely diffusing Sup35 aggregates with high fluorescent intensity over average intensity, which had much larger diffusional component than [PSI+] cells than those in the absence of Rnq1Δ100 overproduction. The number of aggregates in the mother cell was fewer than that in the control cells with similar average fluorescent intensity. In contrast, the daughter cell only had stationary fluctuation of fluorescent intensity with fast diffusion similar to that of [psi−] cells.33 Therefore, it is most likely that the size of the diffusing and enlarged aggregates observed in the mother cell upon Rnq1Δ100 overproduction exceeds the physical size limitation of the aggregate for transmission from mother to daughter cells, leading to a loss of [PSI+].54,55
Surprisingly, such enlargement of Sup35-GFP aggregate and reduction of the number of [PSI+] seeds are not limited to Rnq1Δ100. The series of the N-terminal nonprion domain mutations described above, which give the same phenotypic change as Rnq1Δ100, caused similar aggregate aberrations in the course of the prion curing.32,33 Furthermore, the enlargement of [PSI+] aggregates was also observed upon overproduction of Lsm4 (Oishi K and Nakamura Y, unpublished). These data suggest that the prion aggregate enlargement is a crucial on-pathway event in the Pin+ protein-driven prion loss, disabling efficient transmission of prion seeds from mother to daughter cells. Besides Pin+ proteins, our genome-wide screen for [PSI+]-eliminating factors pointed to a G-protein γ subunit mimic, Gpg1.20 Although functionally uncharacterized, Gpg1 also caused a size enlargement of [PSI+] aggregates upon overproduction (Kurahashi H and Nakamura Y, unpublished).
Why Overproduced Pin+ Proteins Lead to a Size Enlargement of Prion Aggregates?
It is widely accepted that the enhanced de novo appearance of [PSI+] by Pin+ proteins is mediated by direct interactions between a preexisting Pin+ prion and Sup35, in which a heterologous Pin+ protein is used as a template for the conversion of Sup35 into its prion form.11,16 This model, designated “seeding model,” predicts that Pin+ prion aggregates provide a “friendly” nidus on which the first seeds of a heterologous prion can form. In fact, Rnq1 and New1 proteins showed cross-seeding activity to Sup35 in the in vitro fibril assembly,46,56 and Sup35 amyloid extension at Rnq1 amyloid was visualized in transmission electron microscopy.57 The seeding model predicts that the interaction occurs at the growing tip of each prion aggregates.35 Given this tip-to-tip interaction happens between overproduced Pin+ proteins and [PSI+] aggregates, the size enlargement of prion aggregates might be explained, at least in part, by assuming that abundant Pin+ proteins accelerate the growing speed of [PSI+]-Pin+ heterologous aggregates (Fig. 1). In fact, Rnq1 and Rnq1Δ100 were partially co-localized with Sup35 in [RNQ+],19,56 and Gpg1 was also co-localized with Sup35 aggregates.20 However, the growing tip interaction model does not readily account for the reduced number of enlarged aggregates in the mother cell33 as well as the bipolar action of Pin+ proteins.
Figure 1.
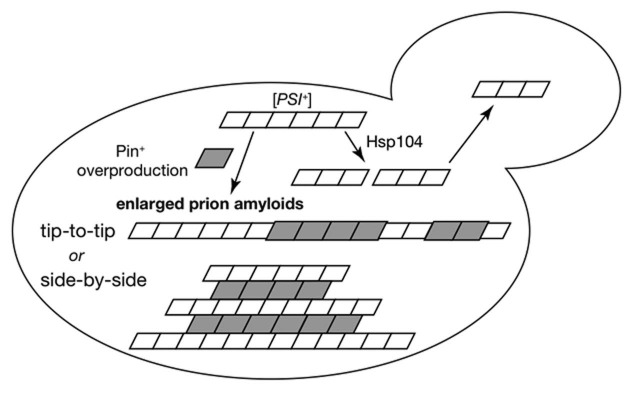
A possible mechanism for the inhibition of the maintenance of [PSI+] prion by overproduced Pin+ proteins. Under normal situations, Hsp104 breaks up [PSI+] amyloid filaments to generate prion seeds for efficient prion transmission. Under Pin+ overproduction conditions, Pin+ proteins accelerates a size enlargement of [PSI+] amyloid fibrils through tip-to-tip or side-by-side interactions, giving rise to overgrown [PSI+] amyloids that are incapable of reaching daughter cell.
An alternative model is to assume a side-by-side interaction between Q/N-rich domains of overproduced Pin+ proteins and preexisting [PSI+] or [URE3] prion, which results in a size enlargement of prion amyloids (Fig. 1). This model might explain the bipolar activity of Pin+ proteins on [PSI+] and [URE3] prions on the common protein-protein interaction basis. In the case of de novo appearance of [PSI+], Sup35 forms soluble oligomeric intermediates in vitro before achieving the amyloid state at 4°C.58 Lansbury's nucleation-dependent polymerization (NP) model states that the oligomeric precursor formation is reversible and kinetically unfavorable, but once amyloid nucleus is achieved it starts to elongate in a favorable manner (reviewed in ref. 59). At 37°C, amyloid formation occurs at a certain frequency in vitro without the oligomeric precursor formation.58 In either case, we speculate that the in vivo amyloid nucleation event should be even rarer than the in vitro implication because nascent minimal nuclei are likely to be disassembled by molecular chaperone Hsp104 or external force.
Our model predicts that the side-by-side binding of Pin+ proteins accelerates the [PSI+] elongation and thus stabilizes its nucleation. This is consistent with the finding by Lindquist and coworkers that the de novo prion induction takes place on the prion amyloid bundles on IPOD, a cellular compartment for irreversibly aggregated protein deposition.60 The biophysical mechanism by which Pin+ proteins influence on [PSI+] elongation remains to be studied. One possibility is that the activation energy of amyloid elongation, and possibly of nucleation, may be lowered when its diffusion coefficient is reduced via a second amyloid accompaniment.
The overgrowth of [PSI+] might be caused not only by acceleration of amyloid growth but also by disaggregation inefficiency of the amyloids composed of heterologous proteins by chaperones in either model (tip-to-tip or side-by-side). Because the disaggregation of amyloids is catalyzed cooperatively by Hsp104, Ssa proteins and Hsp40s,61 the protein heterogeneity in an amyloid may lead to the failure in efficient recognition and cleavage by the molecular chaperones. In the case of prion loss, it is assumed that overproduced Pin+ proteins facilitate overgrowth of [PSI+] aggregates by the side-by-side interaction of abundant Pin+ proteins and [PSI+] aggregates. Serio and coworkers recently demonstrated that there exists an upper limit of amyloid size for prion transmission to daughter cells.55 This implies that the overgrown [PSI+] amyloid after Pin+ protein overproduction may be incapable of reaching daughter cell, which ends up with the failure in adequate prion inheritance. In fact, several studies have observed that overproduction of prion proteins in yeast abolishes their own prion propagation,55,62,63 accompanied by the amyloid enlargement.55,63 The side-by-side interaction model that we propose here nicely accounts for the observation that the efficiency of Pin+ activities of Rnq1, its mutants, Lsm4 and some other Pin+ proteins well correlates with the degree of [PSI+] or [URE3] elimination activity (Oishi K and Nakamura Y, unpublished).
Acknowledgments
This work was supported in part by grants from The Ministry of Education, Sports, Culture, Science and Technology of Japan (MEXT).
Abbreviation
- S. cerevisiae
Saccharomyces cerevisiae
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 3.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/S1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 4.Cox BS. Ψ, a cytoplasmic suppressor of super-suppressor in yeast. Heredity. 1965;20:505–521. doi: 10.1038/hdy.1965.65. [DOI] [Google Scholar]
- 5.Stansfield I, Jones KM, Kushnirov VV, Dagkesamanskaya AR, Poznyakovski AI, Paushkin SV, et al. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995;14:4365–4373. doi: 10.1002/j.1460-2075.1995.tb00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, et al. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14:4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrenberg M, Hauryliuk V, Crist CG, Nakamura Y. Translation termination, prion [PSI+] and ribosome recycling. In: Mathews MB, Sonenberg N, Hershey JWB, editors. Translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press; 2007. [Google Scholar]
- 8.Liebman SW, Sherman F. Extrachromosomal Ψ+ determinant suppresses nonsense mutations in yeast. J Bacteriol. 1979;139:1068–1071. doi: 10.1128/jb.139.3.1068-1071.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 10.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 11.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN+] Cell. 2001;106:171–182. doi: 10.1016/S0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 12.Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet. 2008;40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat Cell Biol. 2009;11:344–349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277–288. doi: 10.1016/S0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 16.Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion. Cell. 2001;106:183–194. doi: 10.1016/S0092-8674(01)00440-8. [DOI] [PubMed] [Google Scholar]
- 17.Derkatch IL, Bradley ME, Masse SV, Zadorsky SP, Polozkov GV, Inge-Vechtomov SG, et al. Dependence and independence of [PSI+] and [PIN+]: a two-prion system in yeast? EMBO J. 2000;19:1942–1952. doi: 10.1093/emboj/19.9.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurahashi H, Nakamura Y. Channel mutations in Hsp104 hexamer distinctively affect thermotolerance and prion-specific propagation. Mol Microbiol. 2007;63:1669–1683. doi: 10.1111/j.1365-2958.2007.05629.x. [DOI] [PubMed] [Google Scholar]
- 19.Kurahashi H, Ishiwata M, Shibata S, Nakamura Y. A regulatory role of the Rnq1 nonprion domain for prion propagation and polyglutamine aggregates. Mol Cell Biol. 2008;28:3313–3323. doi: 10.1128/MCB.01900-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishiwata M, Kurahashi H, Nakamura Y. A G-protein γ subunit mimic is a general antagonist of prion propagation in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2009;106:791–796. doi: 10.1073/pnas.0808383106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 23.Orlowska-Matuszewska G, Wawrzycka D. A novel phenotype of eight spores asci in deletants of the prion-like Rnq1p in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2006;340:190–193. doi: 10.1016/j.bbrc.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Strawn LA, True HL. Deletion of RNQ1 gene reveals novel functional relationship between divergently transcribed Bik1p/CLIP-170 and Sfi1p in spindle pole body separation. Curr Genet. 2006;50:347–366. doi: 10.1007/s00294-006-0098-6. [DOI] [PubMed] [Google Scholar]
- 25.Bruce ME, McBride PA, Farquhar CF. Precise targeting of the pathology of the sialoglycoprotein, PrP and vacuolar degeneration in mouse scrapie. Neurosci Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-X. [DOI] [PubMed] [Google Scholar]
- 26.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 28.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci USA. 2002;99:16392–16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley ME, Liebman SW. Destabilizing interactions among [PSI+] and [PIN+] yeast prion variants. Genetics. 2003;165:1675–1685. doi: 10.1093/genetics/165.4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurahashi H, Shibata S, Ishiwata M, Nakamura Y. Selfish prion of Rnq1 mutant in yeast. Genes Cells. 2009;14:659–668. doi: 10.1111/j.1365-2443.2009.01297.x. [DOI] [PubMed] [Google Scholar]
- 32.Shibata S, Kurahashi H, Nakamura Y. Localization of prion-destabilizing mutations in the N-terminal non-prion domain of Rnq1 in Saccharomyces cerevisiae. Prion. 2009;3:250–258. doi: 10.4161/pri.3.4.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurahashi H, Pack CG, Shibata S, Oishi K, Sako Y, Nakamura Y. [PSI+] aggregate enlargement in rnq1 nonprion domain mutants, leading to a loss of prion in yeast. Genes Cells. 2011;16:576–589. doi: 10.1111/j.1365-2443.2011.01511.x. [DOI] [PubMed] [Google Scholar]
- 34.Schwimmer C, Masison DC. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol Cell Biol. 2002;22:3590–3598. doi: 10.1128/MCB.22.11.3590-8.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derkatch IL, Liebman SW. Prion-Prion Interactions. Prion. 2007;1:161–169. doi: 10.4161/pri.1.3.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung G, Jones G, Wegrzyn RD, Masison DC. A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress. Genetics. 2000;156:559–570. doi: 10.1093/genetics/156.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]
- 38.Tharun S. Roles of eukaryotic Lsm proteins in the regulation of mRNA function. Int Rev Cell Mol Biol. 2009;272:149–189. doi: 10.1016/S1937-6448(08)01604-3. [DOI] [PubMed] [Google Scholar]
- 39.Mayes AE, Verdone L, Legrain P, Beggs JD. Characterization of Sm-like proteins in yeast and their association with U6 snRNA. EMBO J. 1999;18:4321–4331. doi: 10.1093/emboj/18.15.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tharun S, He W, Mayes AE, Lennertz P, Beggs JD, Parker R. Yeast Sm-like proteins function in mRNA decapping and decay. Nature. 2000;404:515–518. doi: 10.1038/35006676. [DOI] [PubMed] [Google Scholar]
- 41.Kambach C, Walke S, Young R, Avis JM, de la Fortelle E, Raker VA, et al. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell. 1999;96:375–387. doi: 10.1016/S0092-8674(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 42.Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007;179:437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ripaud L, Maillet L, Cullin C. The mechanisms of [URE3] prion elimination demonstrate that large aggregates of Ure2p are dead-end products. EMBO J. 2003;22:5251–5259. doi: 10.1093/emboj/cdg488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eaglestone SS, Ruddock LW, Cox BS, Tuite MF. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2000;97:240–244. doi: 10.1073/pnas.97.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrne LJ, Cole DJ, Cox BS, Ridout MS, Morgan BJ, Tuite MF. The number and transmission of [PSI+] prion seeds (Propagons) in the yeast Saccharomyces cerevisiae. PLoS ONE. 2009;4:4670. doi: 10.1371/journal.pone.0004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue Y, Kawai-Noma S, Koike-Takeshita A, Taguchi H, Yoshida M. Yeast prion protein New1 can break Sup35 amyloid fibrils into fragments in an ATP-dependent manner. Genes Cells. 2011;16:545–556. doi: 10.1111/j.1365-2443.2011.01510.x. [DOI] [PubMed] [Google Scholar]
- 47.Osherovich LZ, Cox BS, Tuite MF, Weissman JS. Dissection and design of yeast prions. PLoS Biol. 2004;2:86. doi: 10.1371/journal.pbio.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kushnirov VV, Ter-Avanesyan MD. Structure and replication of yeast prions. Cell. 1998;94:13–16. doi: 10.1016/S0092-8674(00)81216-7. [DOI] [PubMed] [Google Scholar]
- 49.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 50.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 51.Ness F, Ferreira P, Cox BS, Tuite MF. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol. 2002;22:5593–5605. doi: 10.1128/MCB.22.15.5593-605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawai-Noma S, Pack CG, Tsuji T, Kinjo M, Taguchi H. Single mother-daughter pair analysis to clarify the diffusion properties of yeast prion Sup35 in guanidine-HCl-treated [PSI+] cells. Genes Cells. 2009;14:1045–1054. doi: 10.1111/j.1365-2443.2009.01333.x. [DOI] [PubMed] [Google Scholar]
- 53.Hess ST, Huang S, Heikal AA, Webb WW. Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry. 2002;41:697–705. doi: 10.1021/bi0118512. [DOI] [PubMed] [Google Scholar]
- 54.Taguchi H, Kawai-Noma S. Amyloid oligomers: diffuse oligomer-based transmission of yeast prions. FEBS J. 2010;277:1359–1368. doi: 10.1111/j.1742-4658.2010.07569.x. [DOI] [PubMed] [Google Scholar]
- 55.Derdowski A, Sindi SS, Klaips CL, DiSalvo S, Serio TR. A size threshold limits prion transmission and establishes phenotypic diversity. Science. 2010;330:680–683. doi: 10.1126/science.1197785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Derkatch IL, Uptain SM, Outeiro TF, Krishnan R, Lindquist SL, Liebman SW. Effects of Q/N-rich, polyQ and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc Natl Acad Sci USA. 2004;101:12934–12939. doi: 10.1073/pnas.0404968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vitrenko YA, Gracheva EO, Richmond JE, Liebman SW. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–1787. doi: 10.1074/jbc.M609269200. [DOI] [PubMed] [Google Scholar]
- 58.Ohhashi Y, Ito K, Toyama BH, Weissman JS, Tanaka M. Differences in prion strain conformations result from non-native interactions in a nucleus. Nat Chem Biol. 2010;6:225–230. doi: 10.1038/nchembio.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 60.Tyedmers J, Treusch S, Dong J, McCaffery JM, Bevis B, Lindquist S. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc Natl Acad Sci USA. 2010;107:8633–8638. doi: 10.1073/pnas.1003895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glover JR, Lindquist S. Hsp104, Hsp70 and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/S0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 62.Edskes HK, Gray VT, Wickner RB. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA. 1999;96:1498–1503. doi: 10.1073/pnas.96.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crapeau M, Marchal C, Cullin C, Maillet L. The cellular concentration of the yeast Ure2p prion protein affects its propagation as a prion. Mol Biol Cell. 2009;20:2286–2296. doi: 10.1091/mbc.E08-11-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]