

Abstract
Eighteen analogues of the marine cytotoxic linear peptide tasiamide were designed, synthesized and screened for their inhibitory activities against the growth of human nasopharyngeal carcinoma (KB) and human non-small cell lung tumor (A549) cell lines. The results indicated that minor modifications of the C-terminuswith aromatic groups were tolerated, with the IC50 values between 1.29 and 12.88 μM against these two cancer cell lines. Truncation, minor modifications at the N-terminus or elimination of the N-methyl groups in N-Me-d-Gln and/or N-Me-d-Phe residues resulted in inactive analogues.
Keywords: tasiamide, analogues, synthesis, cytotoxicity, marine peptide
1. Introduction
Marine cyanobacteria are a well-known prolific source of novel bioactive compounds [1,2,3], most of which are linear or cyclic (depsi)peptides with distinctive structures. Tasiamide (Figure 1) is a linear peptide isolated from the marine cyanobacterium Symploca sp. in 2002, which showed moderate cytotoxicity against human nasopharyngeal carcinoma (KB) and human colon carcinoma (LoVo) cell lines with IC50 values of 0.48 and 3.47 μg/mL, respectively [4]. The first total synthesis of tasiamide was reported by our group [5] in 2008. The result indicated that the previously assumed N-Me-l-glutamine residue should be N-Me-d-glutamine by comparing the physical data of the synthetic and natural products (1H-NMR, 13C-NMR and the optical rotation). As a part of our ongoing efforts to find bioactive marine peptides [6,7,8], herein we report the design, synthesis and biological evaluation of some analogues of this natural product.
Figure 1.
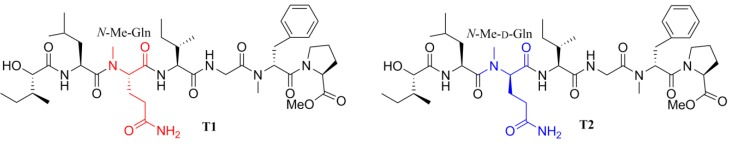
Previously assumed (T1) and revised (T2) structures of tasiamide.
2. Results and Discussion
2.1. Chemistry
Tasiamide is an acyclic peptide composed of six amino acid residues and an α-hydroxy acid (2-hydroxy-3-methylpentanoic acid (Hmp)). In order to determine the minimal active fragment of this natural product, we decided to design and prepare some truncated analogues (Figure 2). Truncating the Hmp-Leu residues or the Hmp-Leu-N-Me-d-Gln residues from the N-terminus of tasiamide gave analogues S1 and S2, respectively. Truncating the N-Me-d-Phe-Pro residues or Gly-N-Me-d-Phe-Pro residues from the C-terminus resulted in analogues S3 and S4, respectively.
Figure 2.

Structures of the designed truncated analogues of tasiamide.
The preparation of these four truncated analogues are shown in Scheme 1. Dipeptide 1 was prepared by coupling commercially available reagents H-Pro-OMe and Boc-N-Me-d-Phe-OH using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride/1-hydroxy-7-azabenzotriazole (EDC/HOAt) in dichloromethane (DCM). Removal of the Boc protecting group yielded 1a, which was used directly in the next cycle of coupling, giving the corresponding tripeptide 2 in good yield. Next, the N-Fmoc of 2 was removed under a mild condition by diethylamine (DEA), which was coupled with N,N-Me2-Ile-OH, leading to the desired analogue S2. On the other hand, the free amine 2a was coupled with Boc-Ile-OH, giving tetrapeptide 3 in a 73% yield. After revealing the amino group of compound 3, N,N-Me2-d-Gln(Trt)-OH was coupled with 3a to give the fully protected pentapeptide 4 in a yield of 95%. Removal of the Trt group under an acidic condition gave the desired truncated analogue S1 in a 97% yield.
Scheme 1.

Reagents and conditions: (a) 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride/1-hydroxy-7-azabenzotriazole (EDC/HOAt), NaHCO3, dichloromethane (DCM), 12 h, 97%; (b) 4 mol/L HCl/EtOAc; (c) Fmoc-Gly-OH, EDC/HOAt, NaHCO3, 12 h, 77%; (d) diethylamine (DEA), CH3CN; (e) N,N-Me2-Ile-OH, EDC/HOAt, diisopropylethylamine (DIEA), DCM, 24%; (f) Boc-Ile-OH, EDC/HOAt, NaHCO3, 73%; (g) N,N-Me2-d-Gln(Trt)-OH, EDC/HOAt, 95%; (h) trifluoroacetic acid (TFA), DCM, 97%.
As shown in Scheme 2, dipeptides 5 and 6 were conveniently synthesized from commercially available amino acids. Deprotections of 5 with DEA and 6 through Pd-mediated hydrogenation liberated the amine and carboxylic acid, respectively, and was subsequently coupled to afford S3. Repeating similar procedures, compound S3a with carboxylic acid was coupled with H-Gly-OBn to yield target S4.
Scheme 2.

Reagents and conditions: (a) EDC/HOAt, N-methylmorpholine (NMM), DCM, 12h, 92% for 5, 91% for 6, 85% for S3, 60% for S4; (b) DEA/CH3CN; (c) Pd-C, H2, EtOAc.
In order to determine the effect of C-terminus modifications, several full-length analogues were designed, with Pro residue being replaced by an amino group having different aromatic groups (C1–C9, Figure 3). Starting from Boc-N-Me-d-Phe-OH, the desired compounds were obtained by following the standard solution phase peptide synthesis procedure (Scheme 3).
Figure 3.

Structures of analogues C1–C9.
Scheme 3.

Reagents and conditions: (a) R2H, EDC/HOAt, NaHCO3, 57%~95%; (b) HCl/EtOAc; (c) Fmoc-Gly-OH, EDC/HOAt, NaHCO3, 52%~99%; (d) DEA/CH3CN; (e) H2, Pd-C, EtOAc; (f) EDC/HOAt. AA = amino acid residue.
In order to investigate the role of the Hmp residue on the N-terminus, two analogues were designed. The α-hydroxyl or sec-butyl group was truncated to afford N1 or N2, respectively (Scheme 4). With the pentapeptide fragment 10d in hand, these two compounds were prepared easily in two steps.
Scheme 4.

Synthesis of analogues with modification on the N-terminus. Reagents and conditions: (a) DEA/CH3CN; (b) EDC/HOAt, pentanoic acid, 17%; (c) EDC/HOAt, hydroxyacetic acid, 26%.
Structurally, there are two N-methyl amino acids in the parent compound. Usually, N-methyl amino acids are believed to play important roles in the conformational alteration and increasing stability over proteolytic enzymes [9]. In order to investigate the role of the N-methyl amino acids, three analogues were designed (M1, M2 and M3). Starting from easily obtained peptide fragments 11, 12 and some commercially available reagents, these three compounds were obtained smoothly, as shown in Scheme 5.
Scheme 5.

Reagents and conditions: (a) 4 mol/L HCl/EtOAc; (b) EDC/HOAt, Fmoc-Me-d-Gln-OH or Boc-d-Gln-OH. PG = protecting group.
2.2. Biological Results and Discussion
All these eighteen tasiamide derivatives prepared above were evaluated for cytotoxicity against KB and A549 cancer cell lines using etoposide as the positive control.
As shown in Table 1, none of the truncated analogues (S1–S4) were effective against KB or A549 cell lines even at 50 μM. This result indicated that a certain length of the peptide might be necessary for cytotoxicity. Analogues with full length, but some minor modifications at the C-terminus and/or glutamine residue (C1–C8) showed moderate activities against cancer cell lines. According to the IC50 values of eight compounds (C1–C8), l-Gln residue-containing analogues showed slightly better activity than their d-Gln counterparts (C1 vs. C2, C3 vs. C4, C5 vs. C6, C7 vs. C8). On the other hand, if Gln was replaced by Ala (C9), the cytotoxicity reduced dramatically. However, further structural optimizations of whether simplification on the Hmp residue (N1, N2) or modification on the N-methylated amino acids (M1–M3) led to inactive analogues.
Table 1.
In vitro inhibitory activities of the tasiamide analogues (IC50 (µM) a).
| Compounds | KB b | A549 b | Compounds | KB b | A549 b |
|---|---|---|---|---|---|
| Etoposide c | 0.85 ± 0.08 | 0.99 ± 0.09 | C5 | 2.45 ± 0.41 | 5.24 ± 1.12 |
| T2 | 0.58 ± 0.07 | >50 | C6 | 8.52 ± 1.41 | 12.88 ± 1.32 |
| S1 | >50 | >50 | C7 | 3.80 ± 0.46 | 3.67 ± 0.66 |
| S2 | >50 | >50 | C8 | 3.64 ± 0.55 | >50 |
| S3 | >50 | >50 | C9 | >50 | >50 |
| S4 | >50 | >50 | N1 | >50 | >50 |
| C1 | 3.21 ± 0.6 | 4.17 ± 0.51 | N2 | >50 | >50 |
| C2 | 8.26 ± 1.22 | >50 | M1 | >50 | >50 |
| C3 | 2.08 ± 0.33 | 2.24 ± 0.44 | M2 | >50 | >50 |
| C4 | 1.29 ± 0.31 | 8.48 ± 1.39 | M3 | >50 | >50 |
a The concentration of compound that inhibits 50% (IC50, µM) of the growth of human tumor cell line after 72 h of drug exposure. IC50 values are taken as the means ± standard deviation from three independent experiments; b KB, human nasopharyngeal carcinoma cell line; A549, human non-small cell lung tumor cell line; c Used as a positive control.
3. Experimental Section
3.1. Chemistry
3.1.1. Materials and Methods
Solvents were processed by conventional methods. Thin layer chromatography (TLC) was performed on pre-coated Merck silica gel 60 F254 plates. Flash column chromatography was performed on silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd, Qingdao, China). Optical rotations were determined with a JASCO P-1020 polarimeter. NMR spectra were recorded on a Jeol JNM-ECP 600 MHz spectrometer (Jeol Ltd., Tokyo, Japan) with Me4Si as the internal standard, and chemical shifts were recorded in δ value. Mass spectra were obtained on a Q-TOF GIOBAL mass spectrometer (Waters, Wilford, MA, USA).
3.1.2. Synthesis of Boc-Ile-Gly-N-Me-d-Phe-Pro-OMe (3)
DEA (5 mL) was added to a solution of tripeptide 2 (428.4 mg, 0.75 mmol) in CH3CN (5 mL). The solution was concentrated in vacuo after stirring at room temperature (rt) for 2 h. Subsequently, the residue was then redissolved in CH3CN (5 mL), concentrated in vacuo again and dried under vacuum for 2 h. This free amine was dissolved in dry tetrahydrofuran (THF) (10 mL) and cooled with an ice-water bath for 10 min. Then, Boc-Ile-OH (208.0 mg, 0.9 mmol), EDC (173.0 mg, 0.9 mmol), HOAt (123.0 mg, 0.9 mmol) and NaHCO3 (130.0 mg, 1.5 mmol) were added, respectively. The mixture was stirred at 0 °C for 2 h and then rt overnight. The solvent was removed, and the residue was diluted with EtOAc (200 mL), washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4, then concentrated in vacuo. The crude product was purified by flash chromatography providing Tetrapeptide (3) (306.8 mg, 73%): Rf (AcOEt/hexane 1:1) 0.41; 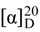 = 19.1 (c 0.05, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.31–7.08 (m, 5H), 6.82 (brs, 1H), 5.58 (t, J = 7.0 Hz, 1H), 5.03–4.99 (m, 1H), 4.42 (dd, J = 8.3, 5.5 Hz, 1H), 4.07–4.00 (m, 1H), 3.92–3.84 (m, 1H), 3.72 (s, 3H), 3.39–3.35 (m, 2H), 3.28 (dd, J = 13.7, 8.3 Hz, 1H), 2.99 (s, 3H), 2.84 (dd, J = 13.7, 7.1 Hz, 1H), 2.16–2.10 (m, 1H), 1.93–1.87 (m, 4H), 1.44–1.42 (m, 10H), 1.10–0.96 (m, 1H), 0.93–0.84 (m, 6H); ESI-MS m/z: 583.3 [M + Na]+; HRESIMS calcd. for C29H44N4O7Na [M + Na]+ 583.3108, found 583.3120.
= 19.1 (c 0.05, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.31–7.08 (m, 5H), 6.82 (brs, 1H), 5.58 (t, J = 7.0 Hz, 1H), 5.03–4.99 (m, 1H), 4.42 (dd, J = 8.3, 5.5 Hz, 1H), 4.07–4.00 (m, 1H), 3.92–3.84 (m, 1H), 3.72 (s, 3H), 3.39–3.35 (m, 2H), 3.28 (dd, J = 13.7, 8.3 Hz, 1H), 2.99 (s, 3H), 2.84 (dd, J = 13.7, 7.1 Hz, 1H), 2.16–2.10 (m, 1H), 1.93–1.87 (m, 4H), 1.44–1.42 (m, 10H), 1.10–0.96 (m, 1H), 0.93–0.84 (m, 6H); ESI-MS m/z: 583.3 [M + Na]+; HRESIMS calcd. for C29H44N4O7Na [M + Na]+ 583.3108, found 583.3120.
3.1.3. Synthesis of N,N-Me2-d-Gln-Ile-Gly-N-Me-d-Phe-Pro-OMe (S1)
A solution of compound 3 (91 mg, 0.162 mmol) in HCl/EtOAc (4 mol/L, 2 mL) was concentrated in vacuo after stirring for 45 min at rt. The residue was redissolved in EtOAc (5 mL) and concentrated in vacuo again. The resulting yellow solid was dried under vacuum for 2 h and then dissolved in dry THF (10 mL). After being cooled with an ice-water bath for 10 min, N,N-Me2-d-Gln(Trt)-OH (81.1 mg, 0.195 mmol), EDC (46.6 mg, 0.243 mmol), HOAt (33.1 mg, 0.243 mmol) and NaHCO3 (27.2 mg, 0.324 mmol) were added, respectively. The mixture was stirred at this temperature for 2 h and then at rt for another 12 h. The solvent was removed, and the residue was dissolved in EtOAc (100 mL) and washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired pentapeptide 4 as a pale-yellow solid (133 mg, 95%).
125 mg (0.146 mmol) of compound 4 was dissolved in 5 mL of DCM and treated with TFA (5 mL) for 2 h, then concentrated in vacuo. The residue was redissolved in 5 mL of DCM and concentrated in vacuo again. The residue was purified by flash column chromatography to give the desired S1 as a white solid (85 mg, 97%): Rf (CHCl3/MeOH 1:1) 0.30; = 28.1 (c 0.14, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 9.30–9.10 (m, 1H), 8.79–8.71 (m, 1H), 8.00 (t, J = 7.8 Hz, 1H), 7.36–7.30 (m, 1H), 7.22–7.12 (m, 5H), 5.42 (t, J = 7.3 Hz, 1H), 4.51–4.49 (m, 1H), 4.35–4.31 (m, 2H), 4.10–3.99 (m, 1H), 3.82–3.79 (m, 1H), 3.70 (s, 3H), 3.26–3.24 (m, 1H), 3.23–3.19 (m, 1H), 3.00–2.95 (m, 4H), 2.85 (brs, 6H), 2.76–2.74 (m, 1H), 2.65–2.59 (m, 2H), 2.45–2.30 (m, 1H), 2.14–2.03 (m, 2H), 1.88–1.85 (m, 2H), 1.79–1.77 (m, 1H), 1.73–1.69 (m, 1H), 1.42–1.39 (m, 1H), 1.20–1.18 (m, 1H), 0.88–0.76 (m, 6H); 13C-NMR (CDCl3, 150 MHz) δ 172.6, 167.8, 136.9, 129.4, 128.5, 126.9, 59.5, 59.2, 59.1, 56.7, 52.4, 46.8, 41.2, 36.4, 35.1, 30.2, 29.8, 28.9, 25.2, 25.1, 24.9, 18.5, 15.6, 11.1; HRESIMS calcd. for C31H48N6O7Na [M + Na]+ 639.3482, found 639.3472.
3.1.4. Synthesis of N,N-Me2-Ile-Gly-N-Me-d-Phe-Pro-OMe (S2)
DEA (5 mL) was added to a solution of tripeptide 2 (62.5 mg, 0.11 mmol) in CH3CN (5 mL). After the solution was stirred at rt for 2 h, the solvent was concentrated in vacuo. The residue was redissolved in CH3CN (5 mL), concentrated in vacuo and dried under vacuum for 2 h. This free amine was dissolved in dry DCM (10 mL) and cooled with an ice-water bath for 10 min. Then, N, N-Me2-Ile-OH (35.0 mg, 0.22 mmol), EDC (31.6 mg, 0.17 mmol), HOAt (22.5 mg, 0.17 mmol), and DIEA (38.4 μL, 0.122 mmol) were added, respectively. The mixture was stirred at this temperature for 2 h and then at rt overnight. The reaction mixture was diluted with 80 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compound S2 as a pale-yellow solid (13.0 mg, 24%): Rf (CHCl3/MeOH 10:1) 0.72; = 26.5 (c 0.13, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.32–7.19 (m, 5H), 6.80–6.78 (m, 1H), 5.50 (t, J = 7.8 Hz, 1H), 4.50–4.44 (m, 1H), 4.41 (m, 1H), 4.10 (d, J = 4.1 Hz, 1H), 3.90 (d, J = 3.2 Hz, 1H), 3.70 (s, 3H), 3.37 (t, J = 4.6 Hz, 2H), 3.28 (dd, J = 13.7, 8.2 Hz, 1H), 3.00 (s, 3H), 2.85 (dd, J = 13.7, 7.3 Hz, 1H), 2.26–2.00 (brs, 6H), 1.97–1.81 (m, 4H), 1.72–1.70 (m, 1H), 1.55–1.42 (m, 2H), 0.99–0.81 (m, 6H); ESI-MS m/z: 511.2 [M + Na]+; HRESIMS calcd. for C26H40N4O5Na [M + Na]+ 511.2896, found 511.2903.
3.1.5. Synthesis of HO-Hmp-Leu-N-Me-d-Gln-Ile-OBn (S3)
Hydrogenation of the benzyl ester, 6 (69.4 mg, 0.21 mmol), was carried out in EtOAc (10 mL) in the presence of a catalytic amount of Pd–C (10%) under hydrogen for 4 h. The Pd–C was removed by filtration, and the filtrate was concentrated in vacuo to yield the corresponding carboxylic acid, 6a, which was used for the next step without further purification.
DEA (5 mL) was added to a solution of dipeptide 5 (111.3 mg, 0.19 mmol) in CH3CN (5 mL). After the solution was stirred at rt for 2 h, the solvent was concentrated in vacuo. The residue was redissolved in CH3CN (5 mL), concentrated in vacuo again, then dried under vacuum for 2 h. This free amine (5a) was dissolved in DCM (10 mL) and cooled with an ice-water bath. EDC (54.1 mg 0.28 mmol), HOAt (38.4 mg, 0.28 mmol) and NaHCO3 (23.7 mg, 0.28 mmol) were added, respectively. The mixture was stirred at this temperature for 2 h and then at rt overnight. The reaction mixture was diluted with 100 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compound S3 as a white solid (94.4 mg, 85%): Rf (CHCl3/MeOH 15:1) 0.30; = −48.5 (c 0.29, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.35–7.28 (m, 5H), 7.20–7.18 (m, 1H), 7.12–7.09 (m, 1H), 6.94 (brs, 1H), 6.64 (brs, 1H), 5.20 (d, J = 12.1 Hz, 1H), 5.10 (d, J = 12.1 Hz, 1H), 4.76–4.73 (m, 1H), 4.50–4.47 (m, 1H), 3.95–3.91 (m, 1H), 3.11–2.82 (m, 4H), 2.30–2.12 (m, 3H), 2.05–1.83 (m, 3H), 1.71–1.65 (m, 3H), 1.44–1.11 (m, 4H), 0.98–0.73 (m, 18H); 13C-NMR (CDCl3, 150 MHz) δ 175.9, 175.2, 174.5, 171.9, 170.4, 135.4, 128.7, 128.6, 128.5, 76.3, 67.2, 56.8, 54.6, 48.1, 40.7, 40.1, 38.6, 37.4, 32.0, 30.7, 29.8, 25.0, 23.7, 23.4, 21.3, 15.7, 14.2, 11.9, 11.6; HRESIMS calcd. for C31H50N4O7Na [M + Na]+ 613.3577, found 613.3589.
3.1.6. Synthesis of HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-OBn (S4)
Hydrogenation of S3 (23.6 mg, 0.04 mmol) was carried out in EtOAc-EtOH (1:4, 10 mL) in the presence of a catalytic amount of Pd–C (10%) under hydrogen for 4 h. The Pd–C was removed by filtration, and the filtrate was concentrated in vacuo to yield the corresponding carboxylic acid S3a, which was used for next step without further purification.
H-Gly-OBn tosylate (16.4 mg, 0.05 mmol) and the carboxylic acid obtained above were dissolved in dry THF and cooled with an ice-water bath for 15 min. EDC (9.4 mg, 0.05 mmol), HOAt (6.7 mg, 0.05 mmol) and NMM (5.4 μL, 0.05 mmol) were added, respectively. The mixture was stirred at this temperature for 2 h and then at rt overnight. The reaction mixture was diluted with 50 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compound S4 as a white solid (15.6 mg, 60%): Rf (AcOEt/hexane 1:1) 0.25; = −37 (c 0.07, MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.37–7.32 (m, 5H), 7.25 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 6.25 (brs, 1H), 5.98 (brs, 1H), 5.15 (d, J = 13.3 Hz, 2H), 5.15 (t, J = 7.7 Hz, 1H), 4.84–4.78 (m, 1H), 4.53 (brs, 1H), 4.32 (t, J = 7.7 Hz, 1H), 4.07 (m, 1H), 3.94–3.93 (m, 1H), 3.92 (d, J = 4.7 Hz, 1H), 3.08 (s, 3H), 2.34–2.27 (m, 1H), 2.26–2.20 (m, 1H), 2.19–2.14 (m, 1H), 2.01–1.95 (m, 1H), 1.87–1.82 (m, 1H), 1.80–1.77 (m, 1H), 1.68–1.60 (m, 2H), 1.58–1.52 (m, 1H), 1.51–1.45 (m, 1H), 1.44–1.37 (m, 1H), 1.24–1.19 (m, 1H), 1.12–1.06 (m, 1H), 0.97 (d, J = 6.6 Hz, 3H), 0.94 (d, J = 7.0 Hz, 3H), 0.90–0.83 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ: 174.7, 174.6, 172.0, 169.9, 169.5, 135.0, 128.6, 128.5, 128.4, 76.3, 67.2, 58.2, 56.4, 47.7, 41.3, 40.6, 38.4, 36.8, 32.3, 31.3, 25.0, 24.9, 23.7, 23.2, 23.1, 21.9, 15.6, 15.4, 11.8, 11.2; ESI-MS m/z: 670.4 [M + Na]+; HRESIMS calcd. for C33H53N5O8Na [M + Na]+ 670.3792, found 670.3808.
3.1.7. General Procedure for the Preparation of Compounds 7a–d
To a solution of Boc-N-Me-d-Phe-OH (1.0 mmol) and amine (1.1 mmol of aniline, phenylmethanamine, tetrahydroisoquinoline or tetrahydroisoquinoline carboxylate) in DCM-DMF (1:1, 10 mL) at 0 °C were added EDC (1.2 mmol), HOAt (1.2 mmol) and NaHCO3 (1.2 mmol), respectively. The mixture was stirred at this temperature for 2 h and then at rt overnight. The reaction mixture was diluted with 100 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3 and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compounds.
Boc-N-Me-d-Phe-NH-Ph (7a): Yield 65%; Rf (AcOEt/hexane 1:1) 0.80; 1H-NMR (CDCl3, 600 MHz) δ 8.31 (brs, 1H), 7.60–7.09 (m, 10H), 5.00 (brm, 1H), 3.54–3.37 (m, 1H), 3.08–3.01 (m, 1H), 2.81 (s, 3H), 1.43(s, 9H, Boc); ESI-MS m/z: 377.2 [M + Na]+; HRESIMS calcd. for C21H26N2O3Na [M + Na]+ 377.1841, found 377.1853.
Boc-N-Me-d-Phe-NH-Bn (7b): Yield 57%; Rf (AcOEt/hexane 1:1) 0.81; 1H-NMR (CDCl3, 600 MHz) δ 7.34–7.15 (m, 10H), 6.50 (brs, 1H), 4.92 (t, J = 7.8 Hz, 1H), 4.51 (dd, J = 14.6, 6.4 Hz, 1H), 4.43 (dd, J = 14.6, 5.0 Hz, 1H), 3.51–3.36 (m, 1H), 3.00–2.89 (m, 1H), 2.76 (s, 3H), 1.35 (s, 9H); ESI-MS m/z: 391.3 [M + Na]+; HRESIMS calcd. for C22H28N2O3Na [M + Na]+ 391.1998, found 391.2006.
Boc-N-Me-d-Phe-NH-MTC (7c, MTC = methyl (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylate): Yield 95%; Rf (AcOEt/hexane 1:1) 0.78; 1H-NMR (CDCl3, 600 MHz) δ 7.29–7.12 (m, 9H), 5.45 (dd, J = 5.0, 4.5 Hz, 1H), 5.39 (dd, J = 7.8, 7.3 Hz, 1H), 4.52–4.49 (m, 1H), 4.35–4.32 (m, 1H), 3.65 (s, 3H), 3.60–3.18 (m, 2H), 3.15–3.02 (m, 2H), 2.96 (s, 3H), 1.38 (s, 9H); ESI-MS m/z: 475.2 [M + Na]+; HRESIMS calcd. for C26H32N2O5Na [M + Na]+ 475.2209, found 475.2231.
Boc-N-Me-d-Phe-NH-TIQ (7d, TIQ = 1,2,3,4-tetrahydroisoquinoline): Yield 63%; Rf (AcOEt/hexane 1:1) 0.68; 1H-NMR (CDCl3, 600 MHz) δ 7.30–7.08 (m, 9H), 5.38–5.36 (m, 1H), 5.05–4.49 (m, 2H), 4.11–3.19 (m, 2H), 3.22–2.96 (m, 2H), 2.85–2.81 (m, 1H), 2.76–2.65 (m, 1H), 2.79 (s, 3H), 1.35 (s, 9H); ESI-MS m/z: 417.2 [M + Na]+; HRESIMS calcd. for C24H30N2O3Na [M + Na]+ 417.2154, found 417.2161.
3.1.8. General Procedure for the Preparation of Compounds 8a–d
Compound 7a (or 7b–d) (0.13 mmol) was treated with HCl/EtOAc (4 mol/L, 2 mL) for 45 min then concentrated in vacuo. The residue was redissolved in EtOAc (5 mL) and concentrated in vacuo again. The resulting solid was dried under vacuum for 2 h and then dissolved in 4 mL of dry DCM-DMF (3:1). After being cooled with an ice-water bath for 10 min, Fmoc-Gly-OH (0.13 mmol), EDC (0.16 mmol), HOAt (0.16 mmol) and NaHCO3 (0.16 mmol) were added, respectively. The mixture was stirred at this temperature for 2 h and then at rt for another 12 h. The mixture was diluted with 80 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3, water and brine. The organic layer was dried over Na2SO4 and then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compound 8a (or 8b–d).
Fmoc-Gly-N-Me-d-Phe-NH-Ph (8a): Yield 64%; Rf (AcOEt/hexane 1:1) 0.41; 1H-NMR (CDCl3, 600 MHz) δ 7.76 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 5.9 Hz, 2H), 7.47 (d, J = 7.8 Hz, 1H), 7.40 (t, J = 7.3 Hz, 2H), 7.31 (t-like, J = 7.3, 6.4 Hz, 2H), 7.29–7.22 (m, 10H), 5.67 (brm, 1H), 5.41 (t, J = 7.8 Hz, 1H), 4.38 (d, J = 7.3 Hz, 2H), 4.21 (t-like, J = 7.3, 6.8 Hz, 1H), 4.04 (dd, J = 17.0, 4.6 Hz, 1H), 3.89 (dd, J = 17.0, 4.6 Hz, 1H), 3.42 (dd, J = 14.2, 7.3 Hz, 1H), 3.06 (dd, J = 14.2, 8.3 Hz, 1H) , 3.00 (s, 3H); ESI-MS m/z: 556.2 [M + Na]+; HRESIMS calcd. for C33H31N3O4Na [M + Na]+ 556.2212, found 556.2231.
Fmoc-Gly-N-Me-d-Phe-NH-Bn (8b): Yield 99%; Rf (AcOEt/hexane 1:1) 0.43; 1H-NMR (CDCl3, 600 MHz) δ 7.76 (d, J = 7.8 Hz, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.3 Hz, 2H), 7.31 (t, J = 7.3 Hz, 2H), 7.30–7.09 (m, 10H), 6.31 (br, 1H), 5.63 (br, 1H), 5.31 (t-like, J = 8.2, 7.8 Hz, 1H), 4.44–4.42 (m, 1H), 4.36 (d, J = 6.8 Hz, 2H), 4.31–4.29 (m, 1H), 4.21 (t, J = 7.3 Hz, 1H), 3.99 (dd, J = 16.9, 4.6 Hz, 1H), 3.84 (dd, J = 17.4, 4.6 Hz, 1H), 3.37 (dd, J = 13.7, 7.8 Hz, 1H), 3.00 (dd, J = 14.2, 8.3 Hz, 1H) , 2.94 (s, 3H); ESI-MS m/z: 570.2 [M + Na]+; HRESIMS calcd. for C34H33N3O4Na [M + Na]+ 570.2369, found 570.2381.
Fmoc-Gly-N-Me-d-Phe-NH-MTC (8c): Yield 53%; Rf (AcOEt/hexane 1:1) 0.38; 1H-NMR (CDCl3, 600 MHz) δ 7.78 (d, J = 7.8 Hz, 2H), 7.62 (m, 2H), 7.41 (t-like, J = 7.3, 7.8 Hz, 2H), 7.33 (t, J = 7.3 Hz, 2H), 7.26–7.06 (m, 9H), 5.69–5.67 (m, 1H), 4.60 (d, J = 15.6 Hz, 1H), 4.39–4.37 (m, 2H), 4.24–4.21 (m, 1H), 4.12 (dd, J = 14.6, 7.3 Hz, 1H), 4.11–4.08 (m, 1H), 3.88–3.85 (m, 1H), 3.79–3.77 (m, 1H), 3.65 (s, 3H), 3.30–3.26 (m, 1H), 3.11 (dd, J = 15.5, 5.0 Hz, 1H), 3.09 (dd, J = 16.5, 6.4 Hz, 1H), 3.00 (s, 3H); 2.98–2.94 (m, 1H); ESI-MS m/z: 632.3 [M + H]+, 654.3 [M + Na]+; HRESIMS calcd. for C38H37N3O6Na [M + Na]+ 654.2580, found 654.2598.
Fmoc-Gly-N-Me-d-Phe-NH-TIQ (8d): Yield 97%; Rf (AcOEt/hexane 1:1) 0.34; 1H-NMR (CDCl3, 600 MHz) δ 7.77 (dd, J = 7.3, 3.2 Hz, 2H), 7.61 (t, J = 6.4 Hz, 2H), 7.41 (m, 2H), 7.32 (dd, J = 13.0, 7.0 Hz, 2H), 7.24–7.05 (m, 9H), 6.93 (d, J = 7.8 Hz, 1H), 5.65 (m, 1H), 4.76 (d, J = 16.5 Hz, 1H), 4.38–4.35 (m, 2H), 4.22 (dd, J = 12.8, 6.8 Hz, 1H), 4.12 (dd, J = 14.2, 6.8 Hz, 1H), 4.03–3.98 (m, 1H), 3.90–3.86 (dd, J = 16.9, 4.1 Hz, 1H), 3.69–3.64 (m, 1H), 3.64–3.54 (m, 1H), 3.32–3.24 (m, 1H), 3.01 (s, 3H), 2.98–2.88 (m, 1H), 2.86–2.84 (m, 1H), 2.79–2.55 (m, 1H); ESI-MS m/z: 574.3 [M + H]+, 596.3 [M + Na]+; HRESIMS calcd. for C36H35N3O4Na [M + Na]+ 596.2525, found 596.2547.
3.1.9. General Procedure for the Preparation of Compounds C1–C9
Following the similar procedure described previously [5]:
HO-Hmp-Leu-N-Me-Gln-Ile-Gly-N-Me-d-Phe-NH-Ph (C1): Yield 75%; Rf (CHCl3/MeOH 20:1) 0.21; = −17.0 (c 0.01 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.83 (d, J = 7.7 Hz, 1H), 7.68 (d, J = 7.7 Hz, 1H), 7.51–7.06 (m, 11H), 5.34 (t, J = 7.7 Hz, 1H), 5.07 (dd, J = 10.4, 3.3 Hz, 1H), 4.80 (brs, 1H), 4.69–4.66 (m, 1H), 4.42 (dd, J = 8.1, 3.7 Hz, 1H), 4.12–4.07 (m, 1H), 3.93 (dd, J = 13.2, 4.0 Hz, 1H), 3.88 (d, J = 3.3 Hz, 1H), 3.43 (dd, J = 13.9, 8.0 Hz, 1H), 3.15 (s, 3H), 2.99 (s, 3H), 2.99–2.93 (m, 1H), 2.33–2.15 (m, 3H), 2.10–1.93 (m, 3H), 1.83–1.67 (m, 3H), 1.46–1.39 (m, 1H), 1.26–1.09 (m, 3H), 0.96–0.79 (m, 18H); 13C-NMR (CDCl3, 150 MHz) δ 177.5, 173.2, 171.3, 170.4, 169.6, 167.3, 138.5, 137.4, 129.1, 129.0, 128.8, 128.7, 126.9, 124.1, 120.2, 119.7, 63.8, 57.9, 54.1, 49.7, 41.4, 38.6, 38.5, 38.0, 36.7, 36.4, 34.4, 31.9, 31.1, 30.4, 30.3, 30.2, 30.0, 29.9, 29.7, 29.4, 28.8, 27.1, 25.1, 24.5, 24.1, 23.9, 23.8, 23.7, 23.5, 23.4, 22.7, 20.9, 20.5, 15.7, 15.6, 15.4, 15.3, 14.1, 12.2, 12.0, 11.9, 11.5; ESI-MS m/z: 794.4 [M + H]+, 816.4 [M + Na]+; HRESIMS calcd. for C42H63N7O8Na [M + Na]+ 816.4636, found 816.4668.
HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-Ph (C2): Yield 67%; Rf (CHCl3/MeOH 20:1) 0.23; = −27.5 (c 0.02 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.30–7.06 (m, 13H), 6.15 (brs, 1H), 5.89 (brs, 1H), 5.28–5.22 (m, 1H), 5.05 (t, J = 9.1 Hz, 1H), 4.95–4.90 (m, 1H), 4.82 (d, J = 4.8 Hz, 1H), 4.31–4.29 (m, 1H), 4.04–4.00 (m, 1H), 3.91 (d, J = 3.7 Hz, 1H), 3.78–3.73 (m, 1H), 3.37 (dd, J = 17.4, 6.9 Hz, 1H), 3.08–2.92 (m, 4H), 2.82 (s, 3H), 2.39 (brs, 1H), 2.33–2.28 (m, 1H), 2.23–2.14 (m, 1H), 1.99–1.97 (m, 1H), 1.84–1.82 (m, 2H), 1.64–1.57 (m, 3H), 1.45–1.41 (m, 2H), 1.24–1.17 (m, 1H), 1.13–1.04 (m, 1H), 0.99–0.93 (m, 9H), 0.89–0.84 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ 174.4, 174.3, 174.0, 171.4, 170.3, 169.7, 168.2, 136.5, 128.7, 128.6, 127.0, 76.5, 58.8, 57.8, 47.2, 41.3, 41.1, 40.6, 38.5, 38.4, 38.3, 37.3, 34.7, 32.2, 31.6, 31.1, 29.7, 25.0, 24.8, 23.7, 23.1, 23.0, 22.9, 22.1, 21.7, 15.6, 14.2, 11.8, 11.3; ESI-MS m/z: 794.4 [M + H]+; HRESIMS calcd. for C42H63N7O8Na [M + Na]+ 816.4636, found 816.4670.
HO-Hmp-Leu-N-Me-Gln-Ile-Gly-N-Me-d-Phe-NH-Bn (C3): Yield 63%; Rf (CHCl3/MeOH 20:1) 0.28; = −19.7 (c 0.01 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.99 (brs, 1H), 7.73 (brs, 1H), 7.54 (d, J = 8.5 Hz, 1H),7.27–7.16 (m, 5H), 7.11 (d, J = 7.0 Hz, 2H), 5.31 (t, J = 7.7 Hz, 1H), 5.13 (dd, J = 10.3, 4.4 Hz, 1H), 4.76–4.71 (m, 1H), 4.45 (dd, J = 14.6, 6.6 Hz, 1H), 4.31 (dd, J = 7.7, 5.5 Hz, 1H), 4.25 (dd, J = 9.0, 5.4 Hz, 1H), 3.94–3.80 (m, 3H), 3.47 (s, 3H), 3.38 (dd, J = 14.3, 8.0 Hz, 1H), 2.96 (s, 3H), 2.96–2.86 (m, 1H), 2.33–1.94 (m, 4H), 1.88–1.92 (m, 1H), 1.77–1.70 (m, 2H), 1.45–1.29 (m, 3H), 1.21–1.15 (m, 2H), 1.08–1.01 (m, 1H), 0.98–0.80 (m, 18H); 13C-NMR (CDCl3, 150 MHz) δ 176.8, 176.0, 175.7, 174.7, 171.3, 170.7, 170.3, 169.2, 138.4, 138.2, 137.5, 136.8, 129.2, 129.0, 128.9, 128.6, 128.5, 127.7, 127.5, 127.3, 127.2, 126.8, 76.6, 76.5, 60.4, 58.1, 58.0, 50.8, 49.0, 48.2, 48.1, 43.6, 43.3, 41.3, 39.5, 38.5, 38.4, 36.6, 34.5, 34.4, 31.3, 30.5, 25.0, 24.9, 24.8, 24.6, 24.0, 23.9, 23.5, 23.4, 21.6, 21.1, 20.8, 15.7, 15.5, 15.4, 14.2, 11.9, 11.8, 11.4; ESI-MS m/z: 808.4 [M + H]+; HRESIMS calcd. for C43H65N7O8Na [M + Na]+ 830.4792, found 830.4814.
HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-Bn (C4): Yield 77%; Rf (CHCl3/MeOH 20:1) 0.22; = −7.3 (c 0.05 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.27–7.06 (m, 12H), 5.95 (brs, 1H), 5.69 (brs, 1H), 5.31 (t, J = 8.0 Hz, 1H), 4.96 (t, J = 7.3 Hz, 1H), 4.94–4.91 (m, 1H), 4.43 (dd, J = 14.9, 6.0 Hz, 1H), 4.37 (dd, J = 13.7, 6.1 Hz, 1H), 4.26 (dd, J = 14.8, 5.3 Hz, 1H), 4.03 (dd, J = 17.6, 4.7 Hz, 1H), 3.89 (d, J = 4.2 Hz, 1H), 3.78 (d, J = 17.2 Hz, 1H), 3.33 (dd, J = 14.1, 8.3 Hz, 1H), 3.00 (s, 3H), 2.99–2.92 (m, 1H), 2.96 (s, 3H), 2.25–2.19 (m, 1H), 2.18–2.07 (m, 2H), 1.97–1.91 (m, 1H), 1.86–1.84 (m, 1H), 1.80–1.76 (m, 1H), 1.64–1.58 (m, 1H), 1.58–1.55 (m, 1H), 1.47–1.39 (m, 3H), 1.21–1.14 (m, 1H), 1.12–1.04 (m, 1H), 0.95–0.93 (m, 6H), 0.92 (d, J = 7.0 Hz, 3H), 0.89–0.84 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ 175.1, 174.7, 174.5, 174.3, 174.1, 171.6, 171.5, 169.9, 169.4, 168.8, 168.3, 168.2, 138.3, 138.0, 137.1, 136.7, 129.1, 128.9, 128.5, 127.6, 127.5, 127.3, 127.2, 127.1, 126.8, 77.1, 62.0, 58.0, 57.9, 57.7, 56.7, 53.4, 47.4, 47.2, 43.5, 43.3, 41.3, 41.2, 40.4, 38.5, 38.4, 36.9, 36.1, 35.1, 34.8, 32.0, 31.9, 31.3, 30.1, 30.4, 29.7, 29.4, 24.9, 24.7, 24.2, 23.8, 23.2, 23.1, 22.7, 22.0, 21.7, 15.7, 15.5, 15.2, 14.1, 11.8, 11.7, 11.3, 11.0; ESI-MS m/z: 808.5 [M + H]+, 830.5 [M + Na]+; HRESIMS calcd. for C43H65N7O8Na [M + Na]+ 830.4792, found 830.4828.
HO-Hmp-Leu-N-Me-Gln-Ile-Gly-N-Me-d-Phe-NH-MTC (C5): Yield 60%; Rf (CHCl3/MeOH 20:1) 0.29; = −9.9 (c 0.05 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.36 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 7.7 Hz, 1H), 7.28–6.99 (m, 9H), 6.96 (t, J = 4.4 Hz, 1H), 6.73 (brs, 1H), 6.18 (brs, 1H), 5.78 (t, J = 7.5 Hz, 1H), 5.21 (t, J = 5.5 Hz, 1H), 5.13 (t, J = 7.7 Hz, 1H), 4.85–4.81 (m, 1H), 4.53 (d, J = 15.8 Hz, 1H), 4.43 (d, J = 15.4 Hz, 1H), 4.29 (t, J = 7.3 Hz, 1H), 4.17 (dd, J = 17.4, 5.0 Hz, 1H), 3.98 (brs, 1H), 3.84 (dd, J = 17.6, 3.7 Hz, 1H), 3.64 (s, 3H), 3.27 (dd, J = 13.7, 7.5 Hz, 1H), 3.21–3.15 (m, 1H), 3.14 (s, 3H), 3.11–3.04 (m, 1H), 3.01 (s, 3H), 2.93 (dd, J = 13.7, 7.5 Hz, 1H), 2.36–2.32 (m, 1H), 2.23–2.18 (m, 2H), 2.07–2.02 (m, 1H), 1.96–1.93 (m, 1H), 1.86–1.82 (m, 1H), 1.73–1.68 (m, 1H), 1.66–1.61 (m, 1H), 1.47–1.38 (m, 3H), 1.25–1.20 (m, 1H), 1.16–1.09 (m, 1H), 0.99–0.94 (m, 9H), 0.91–0.85 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ 175.0, 174.1, 171.1, 170.5, 169.2, 168.1, 136.7, 132.2, 132.1, 129.3, 128.5, 128.1, 127.5, 127.1, 126.9, 126.1, 76.2, 58.0, 55.4, 55.2, 52.6, 52.4, 47.9, 45.1, 41.2, 40.5, 38.5, 36.6, 35.2, 31.5, 30.7, 30.5, 29.7, 24.9, 24.7, 23.9, 23.6, 23.4, 21.3, 15.7, 15.6, 11.9, 11.3; ESI-MS m/z: 892.4 [M + H]+, 914.4 [M + Na]+; HRESIMS calcd. for C47H69N7O10Na [M + Na]+ 914.5004, found 914.5018.
HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-MTC (C6): Yield 67%; Rf (CHCl3/MeOH 20:1); = −29.1 (c 0.01 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.29–6.97 (m, 12H), 5.81 (t, J = 7.7 Hz, 2H), 5.57 (brs, 1H), 5.20 (dd, J = 6.2, 4.8 Hz, 1H), 5.07 (t, J = 7.5 Hz, 1H), 4.98 (dd, J = 15.4, 6.9 Hz, 1H), 4.53 (d, J = 15.4 Hz, 1H), 4.45 (d, J = 15.4 Hz, 1H), 4.33 (dd, J = 9.2, 6.6 Hz, 1H), 4.10 (dd, J = 17.2, 4.7 Hz, 1H), 3.93 (dd, J = 4.2 Hz, 1H), 3.80 (dd, J = 17.4, 3.5 Hz, 1H), 3.64 (s, 3H), 3.28 (dd, J = 13.9, 7.7 Hz, 1H), 3.17 (dd, J = 15.8, 6.4 Hz, 1H), 3.06 (dd, J = 15.8, 6.2 Hz, 1H), 3.02 (s, 3H), 3.00 (s, 3H), 2.92 (dd, J = 13.7, 7.5 Hz, 1H), 2.33 (td, J = 13.9, 7.4 Hz, 1H), 2.28–2.22 (m, 1H), 2.15–2.12 (m, 1H), 2.05–1.97 (m, 1H), 1.95 (brs, 1H), 1.88–1.80 (m, 1H), 1.65–1.60 (m, 2H), 1.51–1.39 (m, 3H), 1.23–1.19 (m, 1H), 1.17–1.10 (m, 1H), 0.97–0.95 (m, 9H), 0.93–0.86 (m, 9H); ESI-MS m/z: 892.4 [M + H]+; HRESIMS calcd. For C47H69N7O10Na [M + Na]+ 914.5004, found 914.5032.
HO-Hmp-Leu-N-Me-Gln-Ile-Gly-N-Me-d-Phe-NH-TIQ (C7): Yield 60%; Rf (CHCl3/MeOH 20:1) 0.28; = −3.3 (c 0.02 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.57 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.24–7.05 (m, 9H), 7.00 (t, J = 4.2 Hz, 1H), 6.80 (brs, 1H), 6.53 (brs, 1H), 5.76 (t, J = 7.5 Hz, 1H), 5.17 (dd, J = 9.8, 5.9 Hz, 1H), 4.81–4.76 (m, 1H), 4.72 (d, J = 16.5 Hz, 1H), 4.58 (d, J = 16.5 Hz, 1H), 4.26 (t, J = 7.3 Hz, 1H), 4.06 (dd, J = 17.6, 4.7 Hz, 1H), 4.00 (dt, J = 12.8, 5.9 Hz, 1H), 3.96 (d, J = 2.9 Hz, 1H), 3.88 (dd, J = 17.6, 3.7 Hz, 1H), 3.72 (t, J = 3.8 Hz, 1H), 3.64 (dd, J = 7.3, 4.7 Hz, 1H), 3.57 (dd, J = 8.4, 4.7 Hz, 1H), 3.51 (dd, J = 7.7, 4.7 Hz, 1H), 3.30 (dd, J = 13.6, 8.0 Hz, 1H), 3.15 (s, 3H), 2.94–2.88 (m, 1H), 2.86–2.82 (m, 1H), 2.79 (s, 3H), 2.79–2.76 (m, 1H), 2.26–2.19 (m, 1H), 2.18–2.13 (m, 2H), 2.08–2.01 (m, 1H), 1.93–1.88 (m, 1H), 1.84–1.81 (m, 1H), 1.75–1.70 (m, 1H), 1.69–1.64 (m, 1H), 1.45–1.38 (m, 3H), 1.25–1.19 (m, 1H), 1.12–1.07 (m, 1H), 0.98–0.93 (m, 9H), 0.83–0.91 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ 175.5, 175.4, 174.2, 171.2, 171.0, 170.6, 168.6, 168.2, 167.9, 136.7, 134.2, 134.0, 132.8, 132.7, 129.2, 128.6, 128.5, 128.4, 127.1, 126.9, 126.6, 126.5, 126.4, 76.4, 57.9, 54.8, 54.7, 54.0, 48.0, 47.1, 44.7, 43.1, 41.1, 41.3, 41.2, 40.1, 40.0, 38.5, 36.6, 36.5, 35.7, .35.4, 31.4, 30.6, 30.0, 29.7, 29.5, 29.3, 24.9, 24.7, 24.0, 23.7, 23.4, 21.2, 15.6, 15.5, 11.8, 11.3; ESI-MS m/z: 834.3 [M + H]+, 856.3 [M + Na]+; HRESIMS calcd. For C45H67N7O8Na [M + Na]+ 856.4949, found 856.4991.
HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-TIQ (C8): Yield 59%; Rf (CHCl3/MeOH 20:1) 0.22; = −15.1 (c 0.04 MeOH); 1H-NMR (CDCl3, 600 MHz) δ 7.24–7.04 (m, 11H), 5.95 (brs, 1H), 5.76 (dd, J = 15.4, 7.7 Hz, 1H), 5.70 (brs, 1H), 5.07 (t, J = 7.5 Hz, 1H), 4.95 (dd, J = 14.5, 7.1 Hz, 1H), 4.71 (d, J = 17.2 Hz, 1H), 4.62 (d, J = 16.8 Hz, 1H), 4.58 (s, 1H), 4.30 (t, J = 7.0 Hz, 1H), 4.04 (dd, J = 17.4, 4.9 Hz, 1H), 4.01–3.96 (m, 1H), 3.92 (dd, J = 4.0 Hz, 1H), 3.80 (dd, J = 17.6, 3.7 Hz, 1H), 3.59–3.54 (m, 1H), 3.29 (dd, J = 13.7, 8.6 Hz, 1H), 3.03 (s, 3H), 3.00 (s, 3H), 2.97–2.89 (m, 1H), 2.87–2.81 (m, 1H), 2.79–2.74 (m, 1H), 2.32 (td, J = 13.9, 7.0 Hz, 1H), 2.26-2.20 (m, 1H), 2.19–2.14 (m, 1H), 1.99 (td, J = 14.3, 7.7 Hz, 1H), 1.84–1.80 (m, 2H), 1.63–1.57 (m, 3H), 1.48–1.41 (m, 2H), 1.26–1.19 (m, 1H), 1.17–1.09 (m, 1H), 0.98–0.94 (m, 9H), 0.91–0.85 (m, 9H); 13C-NMR (CDCl3, 150 MHz) δ 174.5, 174.3, 174.1, 171.5, 171.3, 169.7, 168.7, 168.3, 167.7, 167.4, 136.6, 134.3, 134.0, 132.8, 132.7, 129.1, 128.9, 128.6, 128.5, 128.4, 127.1, 126.9, 126.6, 126.5, 126.3, 125.6, 76.5, 57.8, 56.3, 54.6, 53.9, 47.9, 44.7, 43.1, 41.3, 41.2, 41.1, 41.0, 38.3, 37.1, 35.8, 35.5, 32.2, 31.0, 29.9, 29.7, 29.5, 29.3, 28.1, 24.9, 24.8, 23.8, 23.1, 23.0, 22.1, 15.6, 11.8, 11.3; ESI-MS m/z: 834.3 [M + H]+, 856.3 [M + Na]+; HRESIMS calcd. for C45H67N7O8Na [M + Na]+ 856.4949, found 856.4977.
HO-Hmp-Leu-N-Me-Ala-Ile-Gly-N-Me-d-Phe-NH-Bn (C9): Yield 63%; Rf (CHCl3/MeOH 20:1) 0.42; = −20.2 (c 0.1 MeOH); 1H-NMR (dimethyl sulfoxide-d6 (DMSO-d6), 600 MHz) δ 8.69 (t, J = 5.9 Hz, 1H), 7.91 (br, 1H), 7.67 (br, 1H), 7.50 (br, 1H), 7.30–7.10 (m, 10H), 5.20–5.18 (m, 1H), 4.98–4.96 (m, 1H), 4.80–4.77 (m, 1H), 4.30–4.27 (m, 2H), 4.20–4.16 (m, 1H), 4.00–3.96 (m, 1H), 3.70–3.67 (m, 2H), 3.25–3.18 (m, 1H), 2.92–2.88 (m, 1H), 2.90 (s, 3H), 2.87 (s, 3H), 1.70–1.66 (m, 2H), 1.57–1.55 (m, 1H), 1.45–1.42 (m, 1H), 1.36–1.31 (m, 2H), 1.27–1.23 (m, 1H), 1.20–1.18 (m, 3H), 1.14–0.97 (m, 2H), 0.90–0.74 (m, 18H); 13C-NMR (DMSO-d6, 150 MHz) δ 173.7, 173.6, 171.9, 171.1, 170.8, 169.3, 138.1, 136.8, 129.0, 128.7, 128.6, 127.6, 127.4, 126.9, 76.4, 58.6, 58.1, 57.6, 47.5, 43.4, 41.6, 41.5, 38.9, 37.2, 37.0, 34.8, 29.8, 24.9, 24.7, 23.6, 23.5, 22.0, 21.5, 15.7, 15.6, 11.9, 11.5; HRESIMS calcd. for C41H62N6O7Na [M + Na]+ 773.4578, found 773.4577.
3.1.10. General Procedure for the Preparation of Compounds N1–N2
Prepared by following similar method used for C1–C9, but using n-pentanoic acid or 2-hydroxyacetic acid to block the N-terminus.
n-Pentanoyl-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-Bn (N1): Yield 17%; Rf (CHCl3/MeOH 10:1) 0.61; = −21.8 (c 0.10, CHCl3); 1H-NMR (DMSO-d6, 600 MHz) δ 8.63 (br, 1H), 8.10 (br, 1H), 7.95 (br, 1H), 7.80 (br, 1H), 7.28–7.17 (m, 11H), 6.75–6.72 (m, 1H), 5.22–5.18 (m, 1H), 4.90–4.85 (m, 1H), 4.29–4.17 (m, 3H), 4.04–4.00 (m, 1H), 3.70–3.66 (m, 1H), 3.50–3.47 (m, 1H), 3.20–3.17 (m, 1H), 2.93–2.71 (m, 6H), 2.58–2.55 (m, 1H), 2.31–1.88 (m, 6H), 1.77–1.73 (m, 2H), 1.50–1.23 (m, 8H), 0.93–0.76 (m, 15H); 13C-NMR (DMSO-d6, 150 MHz) δ 174.4, 173.1, 172.9, 170.1, 169.4, 169.2, 169.0, 139.7, 138.4, 129.3, 128.7, 128.6, 127.7, 127.5, 127.1, 58.7, 47.9, 42.9, 42.8, 41.4, 41.0, 35.4, 34.6, 31.9, 31.4, 29.6, 29.3, 28.0, 27.9, 24.8, 23.5, 22.3, 22.1, 21.9, 15.8, 14.5, 14.2, 11.5; HRESIMS calcd. for C42H64N7O7 [M + H]+ 778.4867, found 778.4860.
2-Hydroxyacetyl-Leu-N-Me-d-Gln-Ile-Gly-N-Me-d-Phe-NH-Bn (N2): Yield 26%; Rf (CHCl3/MeOH 10:1) 0.62; = −9.3 (c 0.11, CHCl3); 1H-NMR (CDCl3, 600 MHz) δ 7.64 (br, 1H), 7.45 (br, 2H), 7.36 (br, 1H), 7.16–7.03 (m, 10H), 6.92–6.88 (m, 2H), 5.16–5.12 (m, 1H), 4.80–4.76 (m, 2H), 4.49–4.37 (m, 2H), 4.21–4.17 (m, 1H), 4.08–3.90 (m, 2H), 3.76–3.58 (m, 2H), 3.30–3.26 (m, 1H), 2.97–2.95 (m, 1H), 2.88 (s, 3H), 2.81 (s, 3H), 2.25–2.10 (m, 3H), 1.66–1.34 (m, 3H), 1.29–1.03 (m, 4H), 0.90–0.63 (m, 12H); HRESIMS calcd. for C39H58N7O8 [M + H]+ 752.4347, found 752.4344.
3.1.11. General Procedure for the Preparation of Compounds 13a–c
Compound 11 or 12 (0.2 mmol) was treated with HCl/EtOAc (4 mol/L, 2 mL) for 1 h then concentrated in vacuo. The residue was redissolved in EtOAc (10 mL) and concentrated in vacuo again. The resulting solid was dried under vacuum for 2 h and then dissolved in 8 mL of dry DCM-DMF (3:1). After being cooled with an ice-water bath for 10 min, Fmoc-N-Me-d-Gln-OH (0.2 mmol) or Boc-d-Gln-OH (0.2 mmol), EDC (0.24 mmol), HOAt (0.24 mmol) and NaHCO3 (0.3 mmol) were added consecutively. The mixture was stirred at this temperature for 2 h and then at rt for another 12 h. The mixture was diluted with 80 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3, water and brine. The organic layer was dried over Na2SO4, then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compounds 13a–c. These three compounds were used directly without further structural characterizations.
3.1.12. General Procedure for the Preparation of Compounds M1–M3
Compounds 13a~c (0.1 mmol) were treated with HCl/EtOAc (4 mol/L, 2 mL) (13a, 13c) or DEA/DCM (13b) for 1 h, then concentrated in vacuo. The residue was redissolved in EtOAc (10 mL) and concentrated again. The resulting solid was dried under vacuum for 2 h and then dissolved in 8 mL of dry DCM-DMF (3:1). After being cooled with an ice-water bath for 10 min, Hmp-Leu-OH (0.1 mmol), EDC (0.12 mmol), HOAt (0.12 mmol) and NaHCO3 (0.2 mmol) were added consecutively. The mixture was stirred at this temperature for 2 h and then at rt for another 12 h. The mixture was diluted with 80 mL of EtOAc and washed with 10% citric acid, 5% NaHCO3, water and brine. The organic layer was dried over Na2SO4, then concentrated in vacuo. The residue was purified by flash column chromatography to give the desired compounds M1–M3.
HO-Hmp-Leu-d-Gln-Ile-Gly-N-Me-d-Phe-NH-Bn (M1): Yield 75%; Rf (CHCl3/MeOH 10:1) 0.53; = −3.7 (c 0.11, MeOH); 1H-NMR (DMSO-d6, 600 MHz) δ 8.70 (t, J = 5.9 Hz, 1H), 8.22 (br, 1H), 7.93 (t, J = 5.0 Hz, 1H), 7.71 (d, J = 9.4 Hz, 1H), 7.60 (d, J = 8.7 Hz, 1H), 7.30–7.14 (m, 11H), 6.75–6.73 (m, 1H), 5.25–5.21 (m, 1H), 4.45–4.43 (m, 1H), 4.40–4.17 (m, 4H), 4.01–3.97 (m, 1H), 3.77–3.72 (m, 1H), 3.46–3.40 (m, 1H), 3.24 (dd, J = 14.6, 5.9 Hz, 1H), 2.95–2.90 (m, 1H), 2.87 (s, 3H), 2.12–2.09 (m, 2H), 1.84–1.80 (m, 1H), 1.70–1.64 (m, 3H), 1.55–1.51 (m, 1H), 1.49–1.33 (m, 3H), 1.25–1.21 (m, 1H), 1.16–1.01 (m, 3H), 0.88–0.76 (m, 18H); 13C-NMR (DMSO-d6, 150 MHz) δ 174.4, 173.4, 172.5, 171.4, 170.2, 169.5, 169.0, 139.9, 138.5, 129.7, 128.9, 128.8, 128.7, 127.8, 127.5, 127.2, 126.8, 75.6, 58.7, 57.2, 52.6, 50.8, 42.7, 41.4, 38.7, 37.5, 34.7, 32.0, 31.5, 28.1, 24.7, 24.6, 23.8, 23.6, 22.2, 16.1, 15.8, 12.3, 11.7; HRESIMS calcd. For C42H64N7O8 [M + H]+ 794.4816, found 794.4801.
HO-Hmp-Leu-N-Me-d-Gln-Ile-Gly-d-Phe-NH-Bn (M2): Yield 35%; Rf (CHCl3/MeOH 10:1) 0.50; = −34.7 (c 0.11, MeOH); 1H-NMR (DMSO-d6, 600 MHz) δ 8.43 (t, J = 5.8 Hz, 1H), 8.27 (t, J = 6.1 Hz, 1H), 8.13 (br, 1H), 7.68 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.28–7.18 (m, 10H), 7.13–7.09 (m, 2H), 4.94–4.90 (m, 1H), 4.77–4.72 (m, 1H), 4.51–4.44 (m, 1H), 4.31–4.27 (m, 1H), 4.21–4.18 (m, 1H), 4.11–4.07 (m, 1H), 3.77–3.71 (m, 2H), 3.62–3.59 (m, 1H), 3.05–3.01 (m, 1H), 2.94 (s, 3H), 2.83–2.79 (m, 1H), 1.82–1.77 (m, 2H), 1.71–1.62 (m, 5H), 1.38–1.23 (m, 3H), 1.18–1.08 (m, 2H), 1.05–0.98 (m, 1H), 0.93–0.76 (m, 18H); 13C-NMR (DMSO-d6, 150 MHz) δ 175.1, 174.1, 173.4, 171.9, 171.4, 170.6, 169.1, 139.6, 138.3, 129.7, 128.8, 128.7, 127.6, 127.2, 126.9, 73.0, 57.7, 56.0, 54.9, 47.3, 43.2, 42.6, 42.5, 41.7, 38.7, 38.2, 37.0, 32.1, 29.6, 24.8, 24.4, 23.8, 23.7, 21.9, 16.0, 15.8, 12.2, 11.5; HRESIMS calcd. for C42H64N7O8 [M + H]+ 794.4816, found 794.4782.
HO-Hmp-Leu-d-Gln-Ile-Gly-d-Phe-NH-Bn (M3): Yield 32%; Rf (CHCl3/MeOH 10:1) 0.51; = 46.0 (c 0.1, MeOH); 1H-NMR (DMSO-d6, 600 MHz) δ 8.4 (t, J = 5.9 Hz, 1H), 8.28 (t, J = 5.5 Hz, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.15 (br, 1H), 7.85 (d, J = 6.6 Hz, 1H), 7.58 (d, J = 8.7 Hz, 1H), 7.28–7.17 (m, 10H), 7.13 (d, J = 7.3 Hz, 2H), 4.55–4.50 (m, 1H), 4.48–4.42 (m, 1H), 4.34–4.18 (m, 3H), 4.11–4.02 (m, 1H), 3.79–3.72 (m, 2H), 3.60–3.55 (m, 1H), 3.03–2.99 (m, 1H), 2.85–2.79 (m, 1H), 2.15–2.04 (m, 2H), 1.86–1.83 (m, 1H), 1.73–1.66 (m, 3H), 1.53–1.49 (m, 1H), 1.48–1.41 (m, 3H), 1.34–1.32 (m, 1H), 1.14–1.02 (m, 2H), 0.86–0.75 (m, 18H); 13C-NMR (DMSO-d6, 150 MHz) δ 174.5, 173.4, 172.6, 172.2, 171.9, 171.4, 169.2, 139.6, 138.4, 129.7, 128.8, 127.5, 127.2, 126.9, 75.5, 57.7, 55.4, 55.1, 52.7, 50.7, 42.6, 42.2, 38.7, 38.1, 37.0, 32.1, 27.9, 24.8, 24.7, 23.8, 23.6, 22.1, 16.0, 15.7, 12.3, 11.6; HRESIMS calcd. for C41H62N7O8 [M + H]+ 780.4660, found 780.4648.
3.2. Cytotoxicity Assays
All of the prepared analogues (S1–S4, C1–C9, N1–N2, M1–M3) were tested for their in vitro anticancer activities against KB and A549 cell lines. Briefly, cells were seeded into 96-well plates and cultured overnight, treated with tested samples for 72 h. Then, the cells were fixed with 10% trichloroacetic acid (100 µL) and stained with sulforhodamine B (Sigma, St. Louis, MO, USA). The sulforhodamine B (100 µL) in the cells was dissolved in 10 mmol/L Tris-HCl (150 µL) and measured at 515 nm using a multiwell spectrophotometer (VERSAmax, Molecular Devices, Sunnyvale, CA, USA). The inhibition rate on cell proliferation was calculated for each well as (A515control cells − A515treated cells)/A515control cells × 100% (A515: optical density value at 515 nm). The average IC50 values were determined by the Logit method from at least three independent tests.
4. Conclusions
In conclusion, eighteen analogues of tasiamide were designed and synthesized to investigate the structure-activity relationship (SAR) of this marine linear peptide. The result indicated that a certain length of the peptide chain might be essential for cytotoxicity. Modifications on the C-terminus with aromatic groups were tolerated. On the other hand, simplifications on the Hmp residue or modifications on the N-methylated amino acids led to inactive analogues. Further structural optimization and SAR research are ongoing in our laboratory and will be reported in the future.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81001392, 81172975), the Fundamental Research Funds for the Central Universities (10FX070), the National High Technology Research and Development Program of China (863 Program, no. 2013AA092903) and the Project to Enhance the Research Ability of Young Teachers (Fudan University). We thank Josette Wilkes and Chengli Zong (The University of Georgia, Athens, GA, USA) for reading the manuscript and offering valuable advice and suggestions.
Author Contributions
Conceived and designed the experiments: W.Z.; Y.L. Performed the experiments: T.S.; Z.M.; W.Z. Analyzed the data: W.Z.; Wrote the paper: W.Z.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Gademann K., Portmann C. Secondary metabolites from cyanobacteria: Complex structures and powerful bioactivities. Curr. Org. Chem. 2008;12:326–341. doi: 10.2174/138527208783743750. [DOI] [Google Scholar]
- 2.Tan L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 2007;68:954–979. doi: 10.1016/j.phytochem.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Burjar A.M., Banaigs B., Abuo-Mansour E., Burgess J.G., Wright P.C. Marine cyanobacteria—A prolific source of natural products. Tetrahedron. 2001;57:9347–9377. doi: 10.1016/S0040-4020(01)00931-0. [DOI] [Google Scholar]
- 4.Williams P.G., Yoshida W.Y., Moore R.E., Paul V.J. Tasiamide, a cytotoxic peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2002;65:1336–1339. doi: 10.1021/np020184q. [DOI] [PubMed] [Google Scholar]
- 5.Ma Z.-H., Song N., Li C.-X., Zhang W., Wang P., Li Y.-X. Total synthesis and stereochemical reassignment of tasiamide. J. Pept. Sci. 2008;14:1139–1147. doi: 10.1002/psc.1051. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Zhang W., Li L., Salvador L.A., Chen T., Chen W., Felsenstein K.M., Ladd T.B., Price A.R., Golde T.E., et al. Cyanobacterial peptides as a prototype for the design of potent β-secretase inhibitors and the development of selective chemical probes for other proteases. J. Med. Chem. 2012;55:10749–10765. doi: 10.1021/jm301630s. [DOI] [PubMed] [Google Scholar]
- 7.Yang S., Zhang W., Ding N., Lo J., Liu Y., Clare-Salzler M.J., Luesch H., Li Y. Total synthesis of grassystatin A, a probe for cathepsin E function. Bioorg. Med. Chem. 2012;20:4774–4780. doi: 10.1016/j.bmc.2012.05.077. [DOI] [PubMed] [Google Scholar]
- 8.Sun T., Zhang W., Zong C., Wang P., Li Y. Total synthesis and stereochemical reassignment of tasiamide B. J. Pept. Sci. 2010;16:364–374. doi: 10.1002/psc.1254. [DOI] [PubMed] [Google Scholar]
- 9.Aurelio L., Brownlee R.T.C., Hughes A.B. Synthetic preparation of N-methyl-α-amino acids. Chem. Rev. 2004;104:5823–5846. doi: 10.1021/cr030024z. [DOI] [PubMed] [Google Scholar]