

Abstract
Studying phage codon adaptation is important not only for understanding the process of translation elongation, but also for reengineering phages for medical and industrial purposes. To evaluate the effect of mutation and selection on phage codon usage, we developed an index to measure selection imposed by host translation machinery, based on the difference in codon usage between all host genes and highly expressed host genes. We developed linear and nonlinear models to estimate the C→T mutation bias in different phage lineages and to evaluate the relative effect of mutation and host selection on phage codon usage. C→T-biased mutations occur more frequently in single-stranded DNA (ssDNA) phages than in double-stranded DNA (dsDNA) phages and affect not only synonymous codon usage, but also nonsynonymous substitutions at second codon positions, especially in ssDNA phages. The host translation machinery affects codon adaptation in both dsDNA and ssDNA phages, with a stronger effect on dsDNA phages than on ssDNA phages. Strand asymmetry with the associated local variation in mutation bias can significantly interfere with codon adaptation in both dsDNA and ssDNA phages.
Keywords: bacteriophage, codon adaptation, mutation bias, tRNA-mediated selection, strand asymmetry, Escherichia coli
CODON adaptation has been well documented in bacterial and fungal genomes (Gouy and Gautier 1982; Ikemura 1981, 1992; Xia 1998) as well as in mitochondrial genomes in vertebrates (Xia 2005; Xia et al. 2007) and fungi (Carullo and Xia 2008; Xia 2008). Optimizing codon usage according to the codon usage of highly expressed host genes has been shown to increase the production of viral proteins (Haas et al. 1996; Ngumbela et al. 2008) or transgenic genes (Hernan et al. 1992; Kleber-Janke and Becker 2000; Koresawa et al. 2000). Studies on codon–anticodon adaptation have progressed in theoretical elaboration (Bulmer 1987, 1991; Xia 1998, 2008; Higgs and Ran 2008; Jia and Higgs 2008; Palidwor et al. 2010) as well as in critical tests of alternative theoretical predictions (Xia 1996, 2005; Carullo and Xia 2008; van Weringh et al. 2011).
Codon–anticodon adaptation has been documented in bacteriophage (referred to as phage hereafter), partly because several phage species have been used to treat human infections (Sau et al. 2005; Ranjan et al. 2007; Sau 2007; Abedon et al. 2011) or remove infectious biofilms (Azeredo and Sutherland 2008) and need to be reengineered to improve translation efficiency. While phage codon adaptation is shaped mainly by mutation and transfer (t)RNA-mediated selection, previous studies (Grosjean et al. 1978; Gouy 1987; Kunisawa et al. 1998; Sahu et al. 2005; Carbone 2008; Lucks et al. 2008) have focused mainly on the tRNA-mediated selection on codon usage and have not assessed quantitatively the joint effect of mutation and selection on codon usage bias of phages.
Here we aim to elucidate how biased mutation and selection mediated by host translation machinery will alter the trajectory of codon adaptation in phage protein-coding genes. Many DNA phages, especially single-stranded DNA (ssDNA) phages, experience strong C→T mutation bias mediated by spontaneous or enzymatic deamination (Duncan and Miller 1980; Lindahl 1993; Xia and Yuen 2005). In particular, the spontaneous deamination rate is ∼100 times higher in ssDNA than in double-stranded DNA (dsDNA) based on experimental evidence (Frederico et al. 1990), which may explain why some ssDNA viruses, including ssDNA phages, evolve much faster than dsDNA viruses, with their evolutionary rate comparable to that of RNA viruses (Umemura et al. 2002; Shackelton et al. 2005; Xia and Yuen 2005; Shackelton and Holmes 2006; Duffy and Holmes 2008, 2009). Oxidative deamination leading to high C→U/T transitional mutation rates has been reported in ssDNA phage M13 (Kreutzer and Essigmann 1998).
The Effect of C→T Mutation Bias
When selection is absent, if the C→T mutation bias experienced by a phage genome is strong enough to overcome stochastic fluctuation of codon frequencies of viral protein-coding genes, then all Y-ending codon families and subfamilies (where Y stands for pyrimidine) in viral protein-coding genes will tend to have the same proportion of U-ending codons; i.e.,
| (1) |
where NU.i and NC.i are the numbers of codons ending with U or C, respectively, in codon family i, and BC→T is a constant representing C→T mutation bias (being 0.5 when there is no C→T mutation bias). When BC→T increases, PU for all codon families will tend to increase synchronously if not checked by selection. Equation 1 represents a purely mutation-only model of codon usage bias in the viral Y-ending codon families.
When the effect of selection on viral codon usage is negligible, BC→T can be approximated simply as the average of PU.i values for all Y-ending codon families in the viral protein-coding genes; i.e.,
| (2) |
where NY is the number of codon families with Y-ending codons. For simplicity, we refer to both Y-ending codon families (e.g., Asn codon family AAY) and Y-ending codon subfamilies (e.g., GGY codons in the Gly codon family) as Y-ending codon families.
Some ssDNA phages have high values; e.g., at the third codon position of Chlamydia phage Chp1 (Microviridae, NC_001741) is 0.9518, with U-ending codons being invariably the most frequent in all Y-ending or N-ending codon families. Some dsDNA phages can also have high , e.g., being 0.9014 at the third codon position of Clostridium phage phi3626 (Siphoviridae, NC_003524). However, codon usage bias is almost always the result of both mutation bias and selection.
The Effect of tRNA-Mediated Selection and Its Characterization
A bacterial host may have many tRNAs to read the U-ending codons and few to read the C-ending codons in certain codon families. In such a codon family, a U-ending codon is expected to be decoded efficiently (U-friendly); i.e., tRNA-mediated selection will favor U-ending codons. Similarly, we refer to a codon in which C-ending codons can be decoded more efficiently than U-ending codons as U-hostile. A strong C→T mutation bias would accelerate/enhance codon adaptation in a U-friendly codon family, but would go against codon adaptation in U-hostile codon families. Thus, the degree of U friendliness in the host is expected to be a major determinant of phage codon evolution.
How do we measure U friendliness (i.e., selection in favor of U-ending codons)? We develop a simple index, numerically illustrated in Figure 1, based on the comparison of codon frequency (CF) between highly expressed host genes (HEGs) and all other genes (non-HEGs), designated by CFHEG and CFnon-HEG, respectively. Take, for example, the Ala (A) and Phe (F) codon families, where the Y-ending codons are translated by tRNA with a wobble G. In the Ala codon family, GCC is more frequent than GCU when all coding sequences (CDSs) are included. This alone may suggest that the host translation machinery favors C-ending codons. However, with only HEGs from Escherichia coli, GCC is much less frequent than GCU, suggesting that U-ending codons are more efficiently translated than C-ending codons in the Ala codon family. The mechanistic explanation for this is that GCC can be decoded only by tRNAAla/GGC, whereas GCU can be decoded by both tRNAAla/GGC and tRNAAla/UGC, where the wobble U in the anticodon is modified to cmo5U, which pairs efficiently with U at the third codon position (Mitra et al. 1977, 1979; Nasvall et al. 2007). Similarly, the Phe codon family has more UUU codons than UUC codons when all CDSs are included, but fewer UUU than UUC codons when only E. coli HEGs are included. This is also expected because these codons are decoded by tRNAPhe/GAA, which prefers UUC over UUU. The observation that UUC is preferred by HEGs suggests that the Phe codon family is not U-friendly (it is C friendly). These illustrations lead us to adopt the association coefficient φ as a proxy for U friendliness. The Ala codon family is U-friendly and has a positive φ-value, whereas the Phe codon family is U-hostile and has a negative φ-value (Figure 1). φ takes values between −1 and 1 and is equivalent to the Pearson correlation coefficient for continuous variables. Because φi measures the selection (preference of the host machinery) in favor of the U-ending codons, it is expected to be positively correlated with PU.i.
Figure 1.
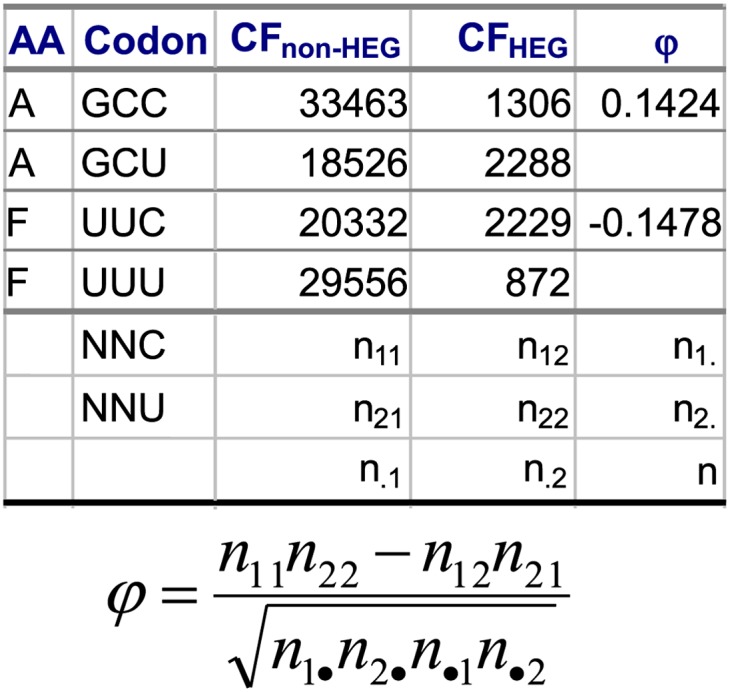
Rationale of using the φ-coefficient as a proxy for U friendliness, based on the codon frequencies (CF) between highly expressed genes (HEGs) and non-HEGs from E. coli. φ can take values within the range between −1 and 1.
Should we develop an index of selection based only on the highly expressed genes? The following scenario suggests that we should not. Suppose the codon frequencies of NNC and NNU from HEGs are 80 and 90, respectively, but those for all CDSs are 200 and 600, respectively. A proper interpretation of this scenario is that extremely high T-biased mutation leads to the dominance of NNU codons. However, the host translation machinery prefers C-ending codons and this selection acts against the T-biased mutation so that codon usage in HEGs is not as U-biased as that in all CDSs. If we have codon usage of only HEGs, we may conclude that the host translation machinery prefers U-ending codons.
A Simple Model of the Joint Effect of Mutation and Selection
The development of the φ-coefficient as a measure of selection for each Y-ending codon family allows us to extend the mutation-only model in Equation 1 to include the effect of selection on PU.i; i.e.,
| (3) |
Because PU.i can be readily computed from viral protein-coding genes, and φi can be derived from codon frequencies of host HEGs and non-HEGs (Figure 1), we can use Equation 3 to quantify the relative importance of mutation (BC→T) and selection (φ) on the codon usage bias of Y-ending codon families. If φi-values differ little among Y-ending codon families in a host, then PU.i will largely depend on BC→T, and we will observe little variation in PU.i values among different codon families. In contrast, with increasing intensity of selection (a large b) or increasing variation in preference of U-ending codons by E. coli (i.e., large variation in φ), PU.i will become more dependent on bφi. Similarly, if BC→T becomes very large (i.e., very strong mutation bias), then bφi naturally would become relatively small and we would conclude that the mutation bias is the dominant factor in shaping codon usage in Y-ending codon families.
One may also argue that PU.i cannot be >1 or <0, so it will asymptotically approach 1 with increasing φi and approach 0 with decreasing φi. This implies a sigmoidal relationship between PU and φ. For this reason, we have also fitted the following sigmoid function,
| (4) |
where parameters C and D are constants. The maximum and minimum values for PU, according to Equation 4, are 1 and 0, respectively. When φ = 0 or D = 0, the expected PU is 1/(1 + C), which is equivalent to BC→T in Equation 3. In most cases, BC→T and 1/(1 + C) are nearly identical and we will use BC→T to refer to both as an index of C→T mutation bias. D measures the benefit of codon adaptation for the phage. If D = 0 (i.e., codon adaptation is not important for the phage), then which codon is favored by the host machinery (measured by φ) is irrelevant to phage codon usage. If D is very large, then even a codon that is weakly favored by the host will be strongly favored by the phage.
Note that for a given viral species, BC→T is constant and affects uniformly the codon usage bias of all Y-ending codon families. In contrast, φi is specific to individual codon families. BC→T is estimated by the intercept of the linear regression model and selection intensity b is the slope. Also note that the correlation coefficient between PU and φ is also a measure of the effect of selection on codon usage bias (a measure of adaptation) in the Y-ending codon families in phages. We interpret adaptation broadly. For example, suppose that a phage species has evolved good codon adaptation to host species A. If the phage subsequently invaded host species B, and if the codon preference in host species B is exactly the same as that in host species A, then we will state that the phage exhibits good codon adaptation to host species B, although it is preadaptation that is applicable here.
We use Equation 3 to characterize mutation bias and selection intensity based on existing genomic data from dsDNA and ssDNA phages and their hosts. We detected the effect of φ in most dsDNA and ssDNA phage species. However, increasing C→T mutation bias significantly reduced the effect of selection in ssDNA phages and shifted the phage codon usage away from the optimum. Some E. coli phages such as phage PRD1 whose close relatives are all parasitizing gram-positive bacteria may have recently invaded E. coli and have codon usage highly different from that of E. coli HEGs. Strand asymmetry with the associated local variation in mutation bias (U-biased in one-half of the genome and C-biased in the other half) can significantly interfere with codon adaptation in both dsDNA and ssDNA phages. Much of the variation in codon adaptation among dsDNA phages can be attributed to lineage effects, with some phage lineages having uniformly strong codon adaptation and some other lineages having uniformly weak codon adaptation.
Materials and Methods
Genomic data and processing
The genome sequences of 469 dsDNA phages, 41 ssDNA phages, and their corresponding bacterial hosts were downloaded from GenBank, of which 71 have E. coli specified as their host in the “/HOST” tag in the “FEATURES” table, including 60 dsDNA phages and 11 ssDNA phages. The CDSs and codon usage data, as well as three codon positions, were extracted using DAMBE (Xia 2013b). All phage genomes were searched for encoded tRNAs by using the tRNAscan-SE Search Server (Schattner et al. 2005). The local TC skew plot, with the TC skew computed as (NT − NC)/(NT + NC), where Ni is the number of nucleotides i along a moving window, was generated from DAMBE (Xia 2013b). All statistical analyses were done with SAS (SAS Institute 1994), with the linear regression fitted by the GLM procedure and the sigmoid function by the NLIN procedure.
E. coli has 29 strains with RefSeq genomic sequences, but the /HOST tag in a viral genome gives only species name (i.e., E. coli), with no strain-specific information. For this reason, all 29 RefSeq genomic sequences were downloaded and E. coli codon usage is computed as the average of all CDSs from these 29 genomes. The codon usage of highly expressed E. coli genes was compiled in the Eeco_h.cut file distributed with EMBOSS (Rice et al. 2000). It is almost perfectly correlated with our own compilation of codon usage from all E. coli ribosomal proteins (which are necessarily highly expressed because of the high density of ribosomes in the cell). There is little variation in codon usage in highly expressed genes among different E. coli genomes.
Indexes of codon usage bias
While we mainly focus on modeling mutation and selection on PU in Equations 3 and 4, two indexes of codon usage bias were used to aid in the interpretation of the results: the codon adaptation index (CAI) (Sharp and Li 1987) with the improved implementation (Xia 2007) and the effective number of codons (Nc) (Wright 1990) with the improved implementation (Sun et al. 2013). All these indexes were computed using DAMBE (Xia 2013b). For computing the phage CAI, the host highly expressed genes are used as the reference set of genes. Only CDSs with at least 33 codons (99 nt) are included in computing the indexes of codon usage bias to alleviate stochastic noise in computing these indexes with few codons.
Phylogenetic analysis
Coancestry of phage species is difficult to establish. Although some dsDNA phage genomes are annotated to contain a DNA polymerase gene, the gene sequences from different phage lineages are often not homologous and cannot be aligned. We build phage “phylogenetic” trees by using a composition vector approach called CVTree (Xu and Hao 2009) that does not require aligned sequences but implicitly assumes the sharing of ancestral peptides as phylogenetic signals. The method uses amino acid sequences and is conceptually based on the sharing of ancient peptides that give individual evolutionary lineages their uniqueness. Computationally, the method is built upon the similarities in the sharing of words of length k (a1a2 … ak) after subtracting its random expectation based on the frequencies of a1a2 … ak−1, a2a3 … ak, and a2a3 … ak−1. The CVTree method has been implemented in the most recent version of DAMBE (Xia 2013b). We used a k value of 5, which has been recommended for viral genomes (Xu and Hao 2009). The data for reconstructing phylogenetic trees with the CVTree method are .faa files downloaded from GenBank, with each .faa file containing all annotated amino acid sequences for each phage species.
Results and Discussion
Codon preference by the E. coli translation machinery: φ
The φ-values for E. coli Y-ending codons are generally small, ranging from −0.1512 to 0.1424 (Table 1). Among the 16 Y-ending codon families and subfamilies, 7 are U-friendly (with φ > 0, with the mean = 0.0861) and 9 are U-hostile (with φ < 0, with the mean = −0.1025). Thus, C-ending codons overall should be slightly favored over U-ending codons to achieve the codon usage pattern of highly expressed host genes, which is consistent with the proportion of U-ending codons in Y-ending codon families in E. coli highly expressed genes (PU.E.coli = 0.4421). Increased C→T mutation bias will improve codon adaptation for the 7 U-friendly codon families, but will lead to deterioration in the 9 U-hostile codon families (Table 1).
Table 1. Codon frequencies (CF) for Y-ending codons in E. coli, compiled for highly expressed genes (HEG) and all other genes (non-HEG), together with the gene copy number of tRNA in the genome (strain K12) whose anticodon matches the codon, and φ as a measure of codon preference of the host translation machinery (a large φ corresponds to greater preference of U-ending codons over C-ending codons).
| AA | Codon | CFnon-HEG | CFHEG | tRNA | φ |
|---|---|---|---|---|---|
| A | GCC | 33,463 | 1,306 | 2 | 0.1424 |
| A | GCT | 18,526 | 2,288 | ||
| C | TGC | 8,397 | 475 | 1 | −0.0362 |
| C | TGT | 6,802 | 270 | ||
| D | GAC | 23,226 | 2,786 | 3 | −0.0993 |
| D | GAT | 41,472 | 2,345 | ||
| F | TTC | 20,332 | 2,229 | 2 | −0.1478 |
| F | TTT | 29,556 | 872 | ||
| G | GGC | 37,418 | 2,987 | 4 | 0.0566 |
| G | GGT | 30,154 | 3,583 | ||
| H | CAC | 12,144 | 1,160 | 1 | −0.1331 |
| H | CAT | 17,170 | 477 | ||
| I | ATC | 30,787 | 3,488 | 3 | −0.1232 |
| I | ATT | 39,788 | 1,640 | ||
| L | CTC | 14,591 | 541 | 1 | −0.0353 |
| L | CTT | 14,679 | 357 | ||
| N | AAC | 26,674 | 2,832 | 4 | −0.1512 |
| N | AAT | 23,652 | 539 | ||
| P | CCC | 7,443 | 38 | 1 | 0.1032 |
| P | CCT | 9,235 | 343 | ||
| R | CGC | 28,473 | 1,530 | 0.1011 | |
| R | CGT | 25,528 | 2,995 | 3a | |
| S | AGC | 20,868 | 1,015 | 1 | −0.0842 |
| S | AGT | 11,802 | 168 | ||
| S | TCC | 10,649 | 1,110 | 2 | 0.0327 |
| S | TCT | 10,217 | 1,320 | ||
| T | ACC | 29,335 | 2,533 | 2 | 0.0408 |
| T | ACT | 10,950 | 1,286 | ||
| V | GTC | 19,972 | 824 | 2 | 0.1262 |
| V | GTT | 22,297 | 2,669 | ||
| Y | TAC | 15,094 | 1,569 | 3 | −0.1122 |
| Y | TAT | 21,207 | 865 |
P-values from a chi-square test of 2 × 2 contingency tables, with the null hypothesis that φ = 0, are all <0.0001.
The anticodon has a wobble A modified to inosine.
Effect of mutation and selection on codon usage of E. coli ssDNA phages
If selection in favor of U-ending codons by the host translation machinery (φ) is efficient, then we expect PU to increase with φ. This expectation is consistent with data from E. coli Enterobacteria phage G4 (NC_001420) showing PU increasing roughly linearly with φ (Figure 2). Fitting the linear model in Equation 3 results in BC→T = 0.5707, and b = 0.6609, with the relationship being statistically significant (P = 0.0159). Fitting the sigmoid function in Equation 4 yields C = 0.7475, D = 2.7241, and 1/(1 + C) = 0.5722, which is equivalent to BC→T in the linear model, i.e., both being the expected PU value when Dφ in Equation 4 is zero (i.e., no selection on phage codon usage in Y-ending codon families). The predicted values from the linear model in Equation 3 and the nonlinear model in Equation 4 are identical to the first two digits after the decimal point, indicating sufficiency of the linear model.
Figure 2.

Relationship between PU (the proportion of U-ending codons in Y-ending codon families) and φ (selection in favor of U-ending codons), based on codon usage data from E. coli Enterobacteria phage G4 (NC_001420). Also shown is the linear fit to the data. Applying the sigmoid function in Equation 4 generated effectively the same predicted values.
The estimated BC→T from applying the regression model in Equation 3 to all 11 ssDNA Enterobacteria phages parasitizing E. coli varies from 0.5443 to 0.7419 (Table 2). These would be the PU values when selection mediated by the host translation machinery is absent. With the slopes in Table 2, the effect of selection on viral PU values is small relative to BC→T. We thus expect the estimated BC→T values to be close to the empirical values defined in Equation 2, which is true (Figure 3).
Table 2. Results of fitting the linear regression model in Equation 3 to codon usage in ssDNA Enterobacteria phages parasitizing E. coli, with viral genome accession number (ACCN), viral genome length (L), number of viral genes (Ng), the estimated intercept (BC→T) and slope (b), the Pearson correlation between PU and φ for each phage species, and the statistical significance (two-tailed P) of the relationship.
| Phage | ACCN | L | Ng | BC→T | b | R | P |
|---|---|---|---|---|---|---|---|
| Phage alpha3 | NC_001330 | 6087 | 10 | 0.7022 | 0.3790 | 0.3936 | 0.1315 |
| Phage G4 | NC_001420 | 5577 | 11 | 0.5707 | 0.6609 | 0.5912 | 0.0159 |
| Phage ID18 | NC_007856 | 5486 | 11 | 0.5876 | 0.6551 | 0.5105 | 0.0433 |
| Phage ID2 | NC_007817 | 5486 | 11 | 0.5443 | 0.5820 | 0.5055 | 0.0458 |
| Phage phiX174 | NC_001422 | 5386 | 11 | 0.6965 | 0.2840 | 0.2969 | 0.2641 |
| Phage St-1 | NC_012868 | 6094 | 11 | 0.6881 | 0.2895 | 0.3048 | 0.2511 |
| Phage WA13 | NC_007821 | 6068 | 10 | 0.7122 | 0.2190 | 0.2354 | 0.3801 |
| Phage I2-2 | NC_001332 | 6744 | 9 | 0.6987 | 0.3301 | 0.2649 | 0.3214 |
| Phage If1 | NC_001954 | 8454 | 10 | 0.6551 | −0.0763 | −0.0819 | 0.7629 |
| Phage Ike | NC_002014 | 6883 | 10 | 0.7419 | 0.1614 | 0.1097 | 0.6858 |
| Phage M13 | NC_003287 | 6407 | 10 | 0.7390 | 0.1717 | 0.1899 | 0.4812 |
Figure 3.

The average defined in Equation 2 is similar to BC→T estimated from fitting the linear model in Equation 3, based on 11 ssDNA Enterobacteria phages parasitizing E. coli (Table 2).
The standard error associated with the BC→T values is on the order of 0.02 (not shown), so that BC→T values in Table 2 are all significantly greater than the observed (= 0.4421) in E. coli HEGs. If we assume that the codon usage of E. coli HEGs represents the optimum achievable given the counterbalance between mutation and selection, then the large BC→T values in Table 2 suggest that C→T-biased mutation in ssDNA has shifted the codon usage of ssDNA phages away from the optimum.
BC→T has a strong effect on the effective number of codons (Nc), as expected. Nc is at its maximum when BC→T ∼ 0.5, but decreases sharply as BC→T increases, leading to U-ending codons dominating over C-ending codons. However, BC→T has little effect on CAI, partly because E. coli translation machinery favors U-ending codons in about half of the Y-ending codon families and C-ending codons in the other half (Table 1). A large BC→T will increase the frequency of U-ending codons in both the U-friendly and U-hostile codon families. The positive effect in the U-friendly codon families is offset by the negative effect on U-hostile codon families.
A well-adapted codon usage in a phage species in E. coli should have PU positively and highly correlated with φ, i.e., large PU in U-friendly codon families (with large φ-values) and small PU in U-hostile codon families (with small φ-values). However, a strong C→T mutation bias (a large BC→T) will lead to high PU in all Y-ending codon families, resulting in reduced correlation between PU and φ. We therefore expect the correlation between PU and φ (R) to decrease with increasing BC→T. This expectation is consistent with the empirical data (Figure 4), although there is one outlying point (Enterobacteria phage If1; NC_001954) for which we offer an explanation later.
Figure 4.

The correlation (R) between PU and φ decreases with increasing BC→T. The outline point is E. coli Enterobacteria phage If1 (NC_001954). The negative association is statistically significant (P = 0.0292 with the outlying point included). The four red circles form a monophyletic taxon and the rest form another monophyletic taxon (Figure 5).
The four ssDNA phage species (phage I2-2, IF1, Ike, and M13) with low correlation between PU and φ (red diamonds in Figure 4) have codon usages significantly correlated with each other, which suggests that they might be phylogenetically related. The tree built with the CVTree algorithm (Xu and Hao 2009) implemented in DAMBE (Xia 2013b) does cluster these four species, all belonging to Inoviridae, into a monophyletic taxon (Figure 5). Other viral proteomic trees (Rohwer and Edwards 2002; Edwards and Rohwer 2005) also group these four E. coli ssDNA phages into the same clade. The other seven ssDNA phages, all belonging to Microviridae, are also clustered into a monophyletic taxon (Figure 5).
Figure 5.

Phylogenetic tree of ssDNA phages reconstructed by using the CVTree method (Xu and Hao 2009) implemented in Xia (2013b). The four phage species colored in blue belong to Inoviridae whereas the other seven phages colored in red belong to Microviridae. The Operational Taxonomic Units (OTUs) are formed by a combination of host (the first letter of the genus name and the first four letters of the host species name), phage species name, GenBank accession number, R (correlation between PU and φ), estimated BC→T, and number of tRNA genes (N_tRNA) in the phage genome.
Because of the phylogenetic structure (Figure 5), one may argue that the 11 points are not statistically independent and question the validity of using conventional regression to test the significance of the negative association between R and BC→T. For example, the ancestor of the seven phages in Microviridae (colored red in Figure 5) may have codon usage similar to that in E. coli, which was then inherited by all seven descendant phage lineages. Similarly, the ancestor of the four phage species in Inoviridae (colored blue in Figure 5) may have codon usage different from that of E. coli, which was then inherited by its four descendant phage lineages. Thus, we would, in an extreme case, have only two data points. To overcome this problem, we used the subtree for the 11 species in Figure 5 and performed independent-contrasts analysis (Felsenstein 1985, 2004, pp. 435–443) implemented in DAMBE (Xia 2013a, pp. 24–29; Xia 2013b) and found the negative association still significant (P < 0.05).
We may conclude from the results above that the overall effect of selection in ssDNA phage is statistically significant as the mean of the 11 b values is significantly >0 (mean b = 0.3324, SE = 0.0685, t = 4.8546, degrees of freedom (d.f.) = 10, P = 0.0007). However, when the false discovery rate method (Benjamini and Hochberg 1995; Benjamini and Yekutieli 2001) is used to control for type I error rate involving multiple comparisons, none of the 11 individual P-values is statistically significant at the 0.05 significance level. The false discovery rate method has been numerically illustrated (Xia 2012b) and implemented in DAMBE (Xia 2013b).
Effect of mutation and selection, as well as evolutionary history, on codon usage of E. coli dsDNA phages
Some dsDNA phages show strong response to selection by the host translation machinery (φ), e.g., NC_010324 phage Phieco32, with PU strongly dependent on φ (Figure 6). The estimated BC→T spans a wide range (Table 3), but on average is significantly smaller than that of ssDNA phages (t-test, t = 2.1379, d.f. = 69, two-tailed P = 0.0361), suggesting a weaker effect of C→T mutation on codon usage in dsDNA phages than in ssDNA phages. The b values also vary substantially (Table 3). For example, phage BP-4795 (NC_004813), phage cdtI (NC_009514), and 10 other dsDNA phages have negative slopes between PU and φ (Table 3), contrary to what we would have predicted based on codon adaptation. While it is easy to understand why phages should exhibit codon adaptation (with a positive slope b) to the host, it is puzzling why some dsDNA phages do not evolve codon usage similar to that of the host.
Figure 6.

Relationship between PU (the proportion of U-ending codons in Y-ending codon families) and φ (selection in favor of U-ending codons), based on codon usage data from E. coli Enterobacteria phage Phieco32 (NC_010324).
Table 3. Results of fitting the linear regression model in Equation 3 to codon usage in dsDNA E. coli phages, with viral genome accession number (ACCN), the estimated intercept (BC→T) and slope (b), the Pearson correlation between PU and φ for each phage species, and the statistical significance (two-tailed P) of the relationship.
| Phage | ACCN | BC→T | b | R | Pa |
|---|---|---|---|---|---|
| Phage 13a | NC_011045 | 0.5690 | 1.3782 | 0.8381 | 0.00005b |
| Phage 285P | NC_015249 | 0.5769 | 1.4737 | 0.8751 | 0.00001b |
| Phage 933W | NC_000924 | 0.5543 | 0.0089 | 0.0129 | 0.96205 |
| Phage BP-4795 | NC_004813 | 0.5210 | −0.2133 | −0.2768 | 0.29931 |
| Phage CC31 | NC_014662 | 0.7277 | 1.0780 | 0.8437 | 0.00004b |
| Phage cdtI | NC_009514 | 0.5632 | −0.2310 | −0.2948 | 0.26777 |
| Phage EcoDS1 | NC_011042 | 0.5477 | 1.4925 | 0.8077 | 0.00015b |
| Phage EPS7 | NC_010583 | 0.7368 | 0.8949 | 0.9011 | 0.00000b |
| Phage HK022 | NC_002166 | 0.4858 | 0.1603 | 0.1659 | 0.53920 |
| Phage HK97 | NC_002167 | 0.4951 | 0.2864 | 0.2699 | 0.31203 |
| Phage IME08 | NC_014260 | 0.7156 | 0.6011 | 0.6583 | 0.00557c |
| Phage JK06 | NC_007291 | 0.6157 | 0.7818 | 0.5302 | 0.03462 |
| Phage JS10 | NC_012741 | 0.7206 | 0.6104 | 0.6538 | 0.00601c |
| Phage JS98 | NC_010105 | 0.7200 | 0.6300 | 0.6684 | 0.00464c |
| Phage JSE | NC_012740 | 0.6931 | 0.6136 | 0.5988 | 0.01425c |
| Phage K1-5 | NC_008152 | 0.6612 | 0.9844 | 0.8118 | 0.00013b |
| Phage K1E | NC_007637 | 0.6605 | 0.9377 | 0.8209 | 0.00010b |
| Phage K1F | NC_007456 | 0.6605 | 0.9377 | 0.8209 | 0.00010b |
| Phage lambda | NC_001416 | 0.4971 | −0.1962 | −0.1908 | 0.47894 |
| Phage Min27 | NC_010237 | 0.5504 | 0.0858 | 0.1302 | 0.63088 |
| Phage Mu | NC_000929 | 0.4980 | −0.6273 | −0.5245 | 0.03700 |
| Phage N15 | NC_001901 | 0.4834 | 0.0208 | 0.0231 | 0.93224 |
| Phage N4 | NC_008720 | 0.7556 | 0.9110 | 0.8554 | 0.00002b |
| Phage P1 | NC_005856 | 0.5475 | 0.0698 | 0.0885 | 0.74454 |
| Phage P2 | NC_001895 | 0.4768 | −0.4703 | −0.4765 | 0.06202 |
| Phage P4 | NC_001609 | 0.5065 | −0.6610 | −0.5315 | 0.03412 |
| Phage Phi1 | NC_009821 | 0.6905 | 0.5815 | 0.5728 | 0.02039c |
| Phage Phieco32 | NC_010324 | 0.7277 | 1.1889 | 0.9436 | 0.00000b |
| Phage phiEcoM-GJ1 | NC_010106 | 0.6421 | 1.0372 | 0.8690 | 0.00001b |
| Phage phiP27 | NC_003356 | 0.5451 | −0.3341 | −0.3569 | 0.17473 |
| Phage PRD1 | NC_001421 | 0.5324 | −0.5638 | −0.2643 | 0.32262 |
| Phage RB16 | NC_014467 | 0.6257 | 1.0301 | 0.7718 | 0.00046b |
| Phage RB49 | NC_005066 | 0.6919 | 0.5836 | 0.5790 | 0.01878c |
| Phage RB69 | NC_004928 | 0.7549 | 0.6396 | 0.7414 | 0.00101b |
| Phage RTP | NC_007603 | 0.6092 | 0.8781 | 0.5433 | 0.02964 |
| Phage SfV | NC_003444 | 0.5316 | −0.1209 | −0.1431 | 0.59705 |
| Phage SPC35 | NC_015269 | 0.7544 | 0.6200 | 0.8677 | 0.00001b |
| Phage SSL-2009a | NC_012223 | 0.3735 | 0.2555 | 0.3003 | 0.25843 |
| Phage T1 | NC_005833 | 0.5786 | 0.9154 | 0.5723 | 0.02053c |
| Phage T3 | NC_003298 | 0.5336 | 1.3661 | 0.8663 | 0.00001b |
| Phage T4 | NC_000866 | 0.7967 | 0.3770 | 0.5538 | 0.02603 |
| Phage T5 | NC_005859 | 0.7386 | 0.5950 | 0.8399 | 0.00005b |
| Phage T7 | NC_001604 | 0.5641 | 1.3971 | 0.8461 | 0.00004b |
| Phage TLS | NC_009540 | 0.6377 | 0.5840 | 0.3261 | 0.21766 |
| Phage vB_EcoM-VR7 | NC_014792 | 0.6945 | 0.4866 | 0.6418 | 0.00736c |
| Phage VT2-Sakai | NC_000902 | 0.5434 | 0.0377 | 0.0517 | 0.84906 |
| Phage WV8 | NC_012749 | 0.7372 | 1.0841 | 0.9077 | 0.00000b |
| Phage bV_EcoS_AKFV3 | NC_017969 | 0.7444 | 0.5894 | 0.8094 | 0.00015b |
| Phage D108 | NC_013594 | 0.4994 | −0.5957 | −0.4791 | 0.06045 |
| Phage HK639 | NC_016158 | 0.4491 | 0.2538 | 0.2437 | 0.36316 |
| Phage HK75 | NC_016160 | 0.4839 | 0.3029 | 0.3018 | 0.25602 |
| Phage phiV10 | NC_007804 | 0.5705 | 0.4618 | 0.4478 | 0.08199 |
| Phage rv5 | NC_011041 | 0.6296 | 0.8548 | 0.6931 | 0.00291b |
| Phage vB_EcoM_CBA12 | NC_016570 | 0.5988 | 0.3685 | 0.4449 | 0.08425 |
| Stx1-converting bac | NC_004913 | 0.5512 | 0.0121 | 0.0165 | 0.95162 |
| Phage BA14 | NC_011040 | 0.5691 | 1.4057 | 0.8765 | 0.00001b |
| Stx2-converting phage II | NC_004914 | 0.5479 | 0.0495 | 0.0706 | 0.79503 |
| Stx2-converting phage 1717 | NC_011357 | 0.5011 | −0.1374 | −0.1724 | 0.52309 |
| Stx2-converting phage 86 | NC_008464 | 0.5681 | 0.0285 | 0.0417 | 0.87813 |
| Stx2-converting phage I | NC_003525 | 0.5132 | −0.0566 | −0.0986 | 0.71646 |
Significant at the 0.05 level when experimentwise error rate is controlled by the false discovery rate method.
Significant with the more conservative approach of Benjamini and Yekutieli (2001).
Significant with the approach of Benjamini and Hochberg (1995).
We noted that all phages that have negative correlation between PU and φ share similar codon usages. For example, PU values from phage BP-4795 (NC_004813) and those from phage cdtI (NC_009514) have a correlation coefficient of 0.9349. The observation that they have similar codon usages that are different from that of their host increases the plausibility that they may have adapted to a common host that has codon usage different from that of E. coli and that they have invaded E. coli recently and have not yet had enough time to evolve codon adaptation. The shared correlation among the 12 phages with negative slopes (Table 3), summarized in the first principal component (PC1), accounts for 81% of the total variation. All 12 phages with a negative slope have PU positively correlated with PC1 (Figure 7). Here we offer two explanations for the lack of codon adaptation in these 12 phages with empirical substantiation.
Figure 7.

Phage species that do not exhibit codon adaptation to E. coli (negative correlation between PU and φ) nevertheless exhibit codon usage similar to each other in their Y-ending codon families, with the shared correlation summarized in the first principal component (PC1) that accounts for 81% of total variation. The phage genomes are identified by their GenBank accession number and species name.
Phylogenetic inertia:
If a phage has only recently invaded E. coli, it would have little time to evolve codon adaptation to E. coli translation machinery. This explanation may be applicable to phage PRD1, which has the fourth most negative slope (b = −0.5638, Table 3). Phage PRD1 belongs to the peculiar Tectiviridae family with members parasitizing both gram-negative and gram-positive bacteria. Phage PRD1 is the only species in the family known to parasitize a variety of gram-negative bacteria, including Salmonella, Pseudomonas, Escherichia, Proteus, Vibrio, Acinetobacter, and Serratia species (Bamford et al. 1995; Grahn et al. 2006). This wide host range might lead one to think that the poor codon adaptation of phage PRD1 to E. coli is because the phage is not E. coli specific. However, other lines of evidence suggest that this is not true. First, the gram-negative hosts that phage PRD1 parasitizes have similar codon usage and adaptation to any one of them or to the average of all of them and will not lead to a negative b (Table 3). Second, other members of the phage family, i.e., phages PR3, PR4, PR5, L17, and PR772, parasitize gram-positive bacteria. Phage PRD1 is extremely similar to its sister lineages parasitizing gram-positive bacteria. For example, there is only one amino acid difference in the coat protein between PRDl and PR4 (Bamford et al. 1995). It is thus quite likely that the ancestor of phage PRD1 parasitizes gram-positive bacteria. The lineage leading to phage PRD1 may have switched to gram-negative bacterial hosts only recently and still has its codon usage similar to that of its ancestral gram-positive bacterial host. In support of this, among 87 bacterial genomes covering major groups of bacterial species, the host species with codon usage most similar to that of phage PRD1 are strains in the gram-positive Geobacillus (NC_014206, NC_012793, NC_014650, NC_014915, and NC_013411).
Phylogenetic reconstruction with the CVTree method (Xu and Hao 2009) suggests that evolutionary history may have contributed to the differences in codon adaptation in E. coli dsDNA phages because codon usage captured by the intercept (BC→T) and the correlation (R) is similar among related phage species. Descendants of node A (Figure 8) belong to phage family Podoviridae, and all have high R and a narrow range of BC→T values. None of them encode tRNA genes in their genomes, whereas tRNA genes are present in many other E. coli dsDNA phage lineages. Another podovirus, phage Phi eco32 (NC_010324), has the highest correlation between PU and φ (r = 0.9436). It is likely that the ancestor of podoviruses evolved good codon adaptation to an E. coli-like bacterial species, which was then inherited by its descendants. Because of the good codon adaptation, there is no need for the phages to carry their own tRNA genes, and all E. coli podoviruses studied do not encode tRNA genes in their genomes, except for phage Phi eco32 (NC_010324), which carried one putative tRNAArg of uncertain function. The sequence is incorrectly annotated because the tRNAArg sequence cannot be folded into a proper 7-nt anticodon loop for Arg. It also has an extraordinarily long branch when aligned and clustered with any of the E. coli tRNA genes, suggesting that it is unlikely to be used by E. coli translation machinery even if it is transcribed.
Figure 8.

Phylogenetic tree of dsDNA phages reconstructed by using the CVTree method with k (peptide length) = 5. The OTUs are formed by a combination of host (the first letter of the genus name and the first four letters of the host species name), phage species name, GenBank accession number, R (correlation between PU and φ), estimated BC→T, and number of tRNA genes (tRNA) in the phage genome.
In contrast to podoviruses, a number of myoviruses have phage-encoded tRNA genes. For example, Enterobacteria phage WV8 (NC_012749) and Erwinia phage phiEa21-4 (NC_011811) have 19 and 23 tRNA genes, respectively (excluding one tRNA pseudogene in phage WV8). Enterobacteria phage WV8 has excellent codon adaptation, with R between PU and φ being 0.9077. One may wonder why Enterobacteria phage WV8, with excellent codon adaptation at least for the Y-ending codon families and subfamilies, should still keep its set of tRNA genes. One possibility is that it is a generalist with more than one host. Previous studies have already suggested an association of host diversity and the number of tRNA genes carried on phage genomes (Sau et al. 2007; Enav et al. 2012). Another possibility is that the Enterobacteria phage WV8 is already in the process of losing its tRNA genes because it has fewer tRNA genes than its sister lineage Erwinia phage phiEa21-4, which has 23 tRNA genes. Furthermore, similar to the annotated tRNAArg “gene” in phage Phi eco32, annotated tRNA genes in phage WV8 are also quite different from their E. coli counterparts and may not be used by E. coli translation machinery. They may be either nonfunctional or functional in non-E. coli hosts.
While phage WV8 exhibits high codon adaptation to E. coli, other myoviruses such as those under node C have poor codon adaptation with the correlation either close to zero or negative (Figure 8, Table 3). Strong heterogeneity in codon adaptation among myoviruses is also visible among species under cluster F (Figure 8), with one myovirus having negative R and the rest have positive R. Another cluster of species with poor codon adaptation to E. coli (having R close to 0 or negative) are those under node B, made mainly of lambdoid phages. While the term “lambdoid” is never intended as a taxonomic term, the clustering of these species together suggests phylogenetic affiliations.
The lambdoid phages must have evolved in E. coli and E. coli-like hosts for a long time, and it would be weak to invoke phylogenetic inertia as an explanation for the lack of concordance of their codon usage with that of E. coli. This forces us to reexamine the assumptions of our model in Equations 3 and 4 in search of an alternative explanation for phages with poor codon adaptation to E. coli. In the two models specified in Equations 3 and 4, we have assumed a uniform mutation bias that will affect all genes and all Y-ending codon families. However, because of the asymmetric replication of the two DNA strands with associated asymmetric mutation bias, local mutation bias is often not accounted for by PU.i. Differences in mutation bias between the two DNA strands has been documented in organisms ranging from viruses and organelles to prokaryotic and eukaryotic genomes (Marin and Xia 2008; Xia 2012a,c).
Strand asymmetry, local mutation bias, and phage codon adaptation:
We found that E. coli dsDNA phages with negative slopes exhibit stronger local strand asymmetry than those phages with positive slopes. For example, among the 60 dsDNA E. coli phages, the three phages (P4, NC_001609; Mu, NC_000929; and D108, NC_013594) with the most negative b values (−0.6610, −0.6273, and −0.5957, respectively, Table 3) exhibit strong local TC skew, defined as (NT − NC)/(NT + NC), relative to the three phage species (phage BA14, NC_011040; phage 285P, NC_015249; and phage EcoDS1, NC_011042) with the largest b values (Figure 9).These results suggest that codon adaptation may be difficult to achieve when one part of the genome experiences strong T-biased mutation and the other part strong C-biased mutation. Such local mutation bias is obscured if we consider only global codon frequencies over all phage genes.
Figure 9.

TC skew, defined as (NT − NC)/(NT + NC), for three phages with the most negative slopes and three phages with the most positive slopes, plotted over sliding windows (window width = 2000 nt and step size = 200). Phage P4 is much shorter than the others and requires coinfection by phage P2 to complete its lytic cycle. The key shows the phage name with the GenBank accession number and the slope from regression of PU on φ.
How strong local strand asymmetry affects codon adaptation is not immediately obvious, so we offer an illustration here. Take, for example, the phage P4 genome (NC_001609) with 14 genes. We first need to recognize that C→T mutations often lead to not only increased U at the third codon position, but also increased U at the first and second codon positions (Figure 10). A similar response of nonsynonymous mutation rate to directional mutation pressure has also been documented in several other studies (Sueoka 1961; Lobry 2004; Urbina et al. 2006). The sites before genomic position 4500 (five genes) are relatively T rich and those after (nine genes) are relatively C rich (which is obvious from Figure 9). T-biased mutation reduces codons such as CCY (which is U-friendly as highly expressed E. coli genes strongly prefer CCU over CCC), so that CCY are found mostly at T-poor (and C-rich) regions and present mainly as CCC, which are not favored by E. coli translation machinery. In contrast, codons such as UUY (which is U-hostile because highly expressed E. coli genes strongly prefer UUC over UUU) are found mostly in T-rich regions and present mainly in the unfavored UUU form. Thus, the T-rich region features many unfavored UUU codons and C-rich regions feature many unfavored CCC codons, leading to poor codon adaptation.
Figure 10.

TC skew at the first and second codon positions (SKEWTC12) in the 14 genes in phage P4 increases with TC skew at the third codon position (SKEWTC3).
The effect of strand asymmetry on codon adaptation observed in dsDNA phages is also visible in ssDNA phages. To show this quantitatively, we used the same window size and step size and computed the variance of the window-specific skew values as an index of strand asymmetry (ISA). Take the six dsDNA phage species in Figure 9, for example. The ISA value would be much greater for the three species with negative slopes than for the three species with positive slopes. The R value is highly significantly and negatively correlated with ISA for ssDNA phages (P = 0.0008, Figure 11). The same negative relationship between R and ISA holds for dsDNA phages with P < 0.0001. Thus, mutation bias along different parts of the phage genome in opposite directions can significantly reduce the efficiency of selection on codon usage of both ssDNA and dsDNA phages. To properly assess the effect of mutation bias and selection by the host translation machinery, it is important to apply Equations 3 and 4 to phage genomic segments with relatively homogenous mutation bias.
Figure 11.

The effect of selection on Y-ending codons, measured by the correlation (R) between PU and φ, decreases with the degree of strand asymmetry, measured by the index of strand asymmetry (ISA, which is the variance of the window-specific TC skew values).
The relationship between R and ISA in Figure 11 offers an explanation for the outlying point in Figure 4, where Enterobacteria phage If1 (NC_001954) has an R value much smaller than expected from the general trend. This phage has the strongest strand asymmetry (i.e., the largest ISA value) among ssDNA phages and in this new light is expected to be associated with a low R value (Figure 11). In short, the mutation effect (on the x-axis) for Enterobacteria phage If1 is underestimated in Figure 4, which does not take local strand asymmetry into consideration. When local strand asymmetry is accounted for (Figure 11), the point is shifted rightward along the x-axis to its proper location.
We may conclude that selection on Y-ending codons, represented by φ, detectable in dsDNA phages as the mean of the b values (Table 3) is significantly >0 (mean b = 0.4622, SE = 0.07436, t = 6.2156, d.f. = 59, P < 0.0001). When the false discovery method is used to control for type I error rate, about half of the b values are statistically significant at the 0.05 level (Table 3).
Other factors that may contribute to phage codon usage
One factor that may contribute to phage codon usage bias is phage-encoded tRNA genes. Note that E. coli dsDNA phages other than those in clusters A, B, and C (Figure 8) are scattered in clusters that frequently have phage lineages with phage-encoded tRNA genes. The presence of phage-encoded tRNA genes can alter the host tRNA pool, so that φ may no longer reflect the selection on codon usage. For example, if the host tRNA for an NNY codon favors U-ending codons, but phage-encoded tRNA favors C-ending codons, then φ would not be a good predictor of phage codon usage. It is noteworthy that phage species in cluster A that do not have phage-encoded tRNA genes all have uniformly high R values, suggesting that selection by the host translation machinery may be more effective on phage codon usage in phages with no tRNA encoded in the phage genome. Alteration of the host tRNA pool through selective local tRNA enrichment in favor of viral gene translation has been documented in several viral species, including HIV-1 (van Weringh et al. 2011) and vaccinia and influenza A (Pavon-Eternod et al. 2013).
While almost all Y-ending codons are translated by tRNAs with a wobble G (except for Ile and Arg codon families where Y-ending codons are decoded by tRNAs with a wobble A chemically modified to inosine), different tRNAs with a wobble G appear to have different codon preferences, with some favoring C-ending codons, some favoring U-ending codons, and some with no detectable preference. At present, such a preference is not well understood and cannot be properly measured. This is in contrast to R-ending codon families that are typically translated by two types of tRNAs, one with a wobble U consistently preferring A-ending codons and the other with a wobble C consistently preferring G-ending codons. To properly assess the effect of phage-encoded tRNAs on phage codon usage, one needs minimally to assess whether the tRNA is actually functional and measure the synonymous codon preference of phage tRNAs. Currently we have no means of doing this bioinformatically.
Although our focus is on the joint effect of mutation and host tRNA-mediated selection on phage codon usage, we are aware of other factors that have been suggested to affect codon usage. Some bacterial hosts live in a high-temperature environment and have relatively high genomic GC. Their phages are also expected to have high GC to maintain genome stability at high temperature (Xia and Yuen 2005). Different host species may have the 4 nt in quite different concentrations (e.g., nucleotide C is typically rare and A typically abundant) and cytoplasmic parasites or symbionts such as virus or organelles should avoid using rare nucleotides in building their genome and RNA molecules (Xia 1996; Xia and Palidwor 2005; Marin and Xia 2008). Such avoidance would also be reflected in codon usage bias. However, these effects are likely additive and not expected to confound the relationships we aim to study here.
Dinucleotide frequencies are often used to explore the presence of site-dependent mutation patterns. Some bacterial species such as Mycoplasma pulmonis carry CpG-specific methyltransferase and exhibit strong CpG deficiency (Xia 2003) that could lead to context-dependent codon usage; e.g., C-ending codons are particularly rare if the next codon is GNN (where N stands for any nucleotide). However, neither E. coli nor any of its phages carries CpG-specific methyltransferase genes. The ratio PCpG/(PCPG) is close to 1 for both E. coli and its phages.
Given nucleotide frequencies Pi (where i = A, C, G, or T), the observed dinucleotide frequencies Pij, assuming random association, are expected to be PiPj (where i, j = A, C, G, or T). The deviation of the observed frequency from the expected frequency may be expressed as Dij = (Pij – PiPj)/PiPj. A dinucleotide is in surplus if Dij > 0 and in deficiency if Dij < 0. The 11 ssDNA phages share similar dinucleotide frequencies, all with DAA being the highest and DAG the lowest. However, the surplus of AA dinucleotides is largely explainable by the usage of AAN codons, especially AAA codons, which is far more than expected. If we designate total number of codons in the 11 ssDNA phages as NT, then the expected number of AAA codons (designated EAAA) is 404.7 (= NT × PA3) whereas the observed AAA codon (designated OAAA) is 930. OAAG, OAAC, and OAAU codons are also greater than EAAG, EAAC and EAAU, although not as dramatic as AAA codons. In contrast, the deficiency of AG dinucleotides can be attributed to OAGA, OAGG, OAGC, and OAGT (= 104, 44, 163, and 179, respectively) all being smaller than EAGA, EAGG, EAGC, and EAGT (= 351.5, 305.3, 306.9, and 430.6, respectively).
One may ask why OAAN is far greater than EAAN whereas OAGN is far smaller than EAGN. One simple explanation invokes tRNA abundance (Xia 1998), which is well predicted by tRNA copy number (Percudani et al. 1997). In unicellular organisms such as E. coli, Salmonella typhimurium, and Saccharomyces cerevisiae, the frequency of an amino acid increases with the abundance of tRNA carrying the amino acid. As ssDNA phages do not carry their own tRNA genes and therefore depend entirely on the host tRNA pool to translate phage genes, we expect the frequency of amino acids to increase with the number of their cognate tRNA genes in the E. coli genome. This expectation is supported by a strong positive correlation between the frequency of an amino acid and the number of gene copies of the tRNA carrying the amino acid (r = 0.8426, P < 0.0001, Figure 12). This finding explains why there is a surplus of AA dinucleotides because there are six tRNA genes decoding AAA and AAG codons, leading to relative overuse of AAR codons. In contrast, there are only two tRNA genes for AGR codons (coding Arg1) and only one tRNA for the AGY codons (coding for Ser1), which explains the relatively rarity of AGR and AGY codons as well as the deficiency of AG dinucleotides mentioned above.
Figure 12.

Frequency of amino acids increases with the number of tRNA genes in E. coli K12 MG1655. Met tRNAs are not included because Met is translated by both initiator and elongator tRNAs while all other amino acids are by elongator tRNAs only.
We did not incorporate translation initiation into our model in this article. However, selection on codon adaptation is present only for mRNAs with efficient initiation; i.e., one should expect little selection for codon adaptation in genes whose mRNAs have poor translation initiation efficiency. The availability of ribosomal loading data and their analysis (Xia et al. 2011) may eventually lead to an index of translation initiation to facilitate more vigorous studies of codon adaptation.
We should mention that, although selection on codon usage appears more detectable in dsDNA phages than in ssDNA phages (Table 2 and Table 3), this does not mean that dsDNA phage mRNA will necessarily have better translation efficiency than ssDNA phage mRNA. A previous study suggested that dsDNA phage coat protein genes have higher CAI (Sharp and Li 1987), which has been known to be a good measure of translation efficiency (Comeron and Aguade 1996; Duret and Mouchiroud 1999; Coghlan and Wolfe 2000), than ssDNA phage genes. However, there is no coat gene that is homologous between dsDNA and ssDNA phages, so the comparison may be between apples and oranges. If all genes are included, then the difference is minimal (mean CAI = 0.4768 for dsDNA phages and 0.4743 for ssDNA phages, excluding the 22 dsDNA phages with phage-encoded tRNA genes) and not statistically significant. The mean CAI value for the 22 dsDNA phages with phage-encoded tRNA genes is even lower, but that is because the phage-encoded tRNAs may allow the phage to use codons frequently used in the phage but rare in the host (i.e., the phage tRNAs reduce the need for the phage to evolve codon usage similar to that of the host). Some ssDNA phages with increasing C→T mutation bias appear to increase the usage of codons in the “U-friendly” codon families, thereby achieving CAI values almost as large as those of dsDNA phages.
In summary, our results show that previous studies on phage codon adaptation are insufficient in at least two ways. First, codon frequencies from either all host CDSs or all highly expressed host genes are insufficient to capture the selection by host translation machinery. Second, it is crucially important to have explicit models to dissect the effect of mutation and selection. Our index (φ) is a proper measure of selection imposed by host translation machinery on phage codon usage, and our linear and nonlinear models allow us to estimate the C→T mutation bias (BC→T) and to evaluate the relative effect of the mutation bias and host translation machinery on phage codon usage. C→T mutations occur more frequently in ssDNA phages than in dsDNA phages and affect not only synonymous codon usage, but also nonsynonymous substitutions, especially in ssDNA phages. dsDNA phages exhibit better codon adaptation to host translation machinery than ssDNA phages, but much of the variation in codon usage may be attributed to phylogenetic inertia. Strand asymmetry strongly influences the efficiency of selection on codon adaptation and needs to be taken into account when studying codon adaptation.
Acknowledgments
We thank S. Aris-Brosou, N. Corradi, M. Gilchrist, A. Golshani, and J. Wang for discussion and comments. Two anonymous reviews and comments from J. Lawrence resulted in significant improvement of the manuscript. This study is supported by a Natural Science and Engineering Research Council of Canada discovery grant (to X.X.).
Footnotes
Communicating editor: J. Lawrence
Literature Cited
- Abedon S. T., Kuhl S. J., Blasdel B. G., Kutter E. M., 2011. Phage treatment of human infections. Bacteriophage 1: 66–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeredo J., Sutherland I. W., 2008. The use of phages for the removal of infectious biofilms. Curr. Pharm. Biotechnol. 9: 261–266. [DOI] [PubMed] [Google Scholar]
- Bamford D. H., Caldentey J., Bamford J. K., 1995. Bacteriophage PRD1: a broad host range DSDNA tectivirus with an internal membrane. Adv. Virus Res. 45: 281–319. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Benjamini Y., Yekutieli D., 2001. The control of the false discovery rate in multiple hypothesis testing under dependency. Ann. Stat. 29: 1165–1188. [Google Scholar]
- Bulmer M., 1987. Coevolution of codon usage and transfer RNA abundance. Nature 325: 728–730. [DOI] [PubMed] [Google Scholar]
- Bulmer M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A., 2008. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol. 66: 210–223. [DOI] [PubMed] [Google Scholar]
- Carullo M., Xia X., 2008. An extensive study of mutation and selection on the wobble nucleotide in tRNA anticodons in fungal mitochondrial genomes. J. Mol. Evol. 66: 484–493. [DOI] [PubMed] [Google Scholar]
- Coghlan A., Wolfe K. H., 2000. Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast 16: 1131–1145. [DOI] [PubMed] [Google Scholar]
- Comeron J. M., Aguade M., 1996. Synonymous substitutions in the Xdh gene of Drosophila: heterogeneous distribution along the coding region. Genetics 144: 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Holmes E. C., 2008. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 82: 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S., Holmes E. C., 2009. Validation of high rates of nucleotide substitution in geminiviruses: phylogenetic evidence from East African cassava mosaic viruses. J. Gen. Virol. 90: 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan B. K., Miller J. H., 1980. Mutagenic deamination of cytosine residues in DNA. Nature 287: 560–561. [DOI] [PubMed] [Google Scholar]
- Duret L., Mouchiroud D., 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 96: 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R. A., Rohwer F., 2005. Viral metagenomics. Nat. Rev. Microbiol. 3: 504–510. [DOI] [PubMed] [Google Scholar]
- Enav H., Beja O., Mandel-Gutfreund Y., 2012. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 6: 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J., 1985. Phylogenies and the comparative method. Am. Nat. 125: 1–15. [Google Scholar]
- Felsenstein J., 2004. Inferring Phylogenies. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Frederico L. A., Kunkel T. A., Shaw B. R., 1990. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry 29: 2532–2537. [DOI] [PubMed] [Google Scholar]
- Gouy M., 1987. Codon contexts in enterobacterial and coliphage genes. Mol. Biol. Evol. 4: 426–444. [DOI] [PubMed] [Google Scholar]
- Gouy M., Gautier C., 1982. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10: 7055–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahn A. M., Butcher S. J., Bamford J. K. H., Bamford D. H., 2006. PRD1: dissecting the genome, structure and entry, pp. 176–185 in The Bacteriophages, edited by Calendar R. Oxford University Press, Oxford. [Google Scholar]
- Grosjean H., Sankoff D., Jou W. M., Fiers W., Cedergren R. J., 1978. Bacteriophage MS2 RNA: a correlation between the stability of the codon: anticodon interaction and the choice of code words. J. Mol. Evol. 12: 113–119. [DOI] [PubMed] [Google Scholar]
- Haas J., Park E.-C., Seed B., 1996. Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr. Biol. 6: 315–324. [DOI] [PubMed] [Google Scholar]
- Hernan R. A., Hui H. L., Andracki M. E., Noble R. W., Sligar S. G., et al. , 1992. Human hemoglobin expression in Escherichia coli: importance of optimal codon usage. Biochemistry 31: 8619–8628. [DOI] [PubMed] [Google Scholar]
- Higgs P. G., Ran W., 2008. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol. Biol. Evol. 25: 2279–2291. [DOI] [PubMed] [Google Scholar]
- Ikemura T., 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 146: 1–21. [DOI] [PubMed] [Google Scholar]
- Ikemura T., 1992. Correlation between codon usage and tRNA content in microorganisms, pp. 87–111 in Transfer RNA in Protein Synthesis, edited by Hatfield D. L., Lee B. J., Pirtle R. M. CRC Press, Boca Raton, FL. [Google Scholar]
- Jia W., Higgs P. G., 2008. Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 25: 339–351. [DOI] [PubMed] [Google Scholar]
- Kleber-Janke T., Becker W. M., 2000. Use of modified BL21(DE3) Escherichia coli cells for high-level expression of recombinant peanut allergens affected by poor codon usage. Protein Expr. Purif. 19: 419–424. [DOI] [PubMed] [Google Scholar]
- Koresawa Y., Miyagawa S., Ikawa M., Matsunami K., Yamada M., et al. , 2000. Synthesis of a new Cre recombinase gene based on optimal codon usage for mammalian systems. J. Biochem. 127: 367–372. [DOI] [PubMed] [Google Scholar]
- Kreutzer D. A., Essigmann J. M., 1998. Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc. Natl. Acad. Sci. USA 95: 3578–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisawa T., Kanaya S., Kutter E., 1998. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 5: 319–326. [DOI] [PubMed] [Google Scholar]
- Lindahl T., 1993. Instability and decay of the primary structure of DNA. Nature 362: 709–715. [DOI] [PubMed] [Google Scholar]
- Lobry J. R., 2004. Life history traits and genome structure: aerobiosis and G+C content in bacteria. Lect. Notes Comput. Sci. 3039: 679–686. [Google Scholar]
- Lucks J. B., Nelson D. R., Kudla G. R., Plotkin J. B., 2008. Genome landscapes and bacteriophage codon usage. PLoS Comput. Biol. 4: e1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin A., Xia X., 2008. GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: new substitution models incorporating strand bias. J. Theor. Biol. 253: 508–513. [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Lustig F., Akesson B., Lagerkvist U., 1977. Codon-anticodon recognition in the valine codon family. J. Biol. Chem. 252: 471–478. [PubMed] [Google Scholar]
- Mitra S. K., Lustig F., Akesson B., Axberg T., Elias P., et al. , 1979. Relative efficiency of anticodons in reading the valine codons during protein synthesis in vitro. J. Biol. Chem. 254: 6397–6401. [PubMed] [Google Scholar]
- Nasvall S. J., Chen P., Bjork G. R., 2007. The wobble hypothesis revisited: uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 13: 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngumbela K. C., Ryan K. P., Sivamurthy R., Brockman M. A., Gandhi R. T., et al. , 2008. Quantitative effect of suboptimal codon usage on translational efficiency of mRNA encoding HIV-1 gag in intact T cells. PLoS ONE 3: e2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palidwor G. A., Perkins T. J., Xia X., 2010. A general model of codon bias due to GC mutational bias. PLoS ONE 5: e13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M., David A., Dittmar K., Berglund P., Pan T., et al. , 2013. Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res. 41: 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R., Pavesi A., Ottonello S., 1997. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J. Mol. Biol. 268: 322–330. [DOI] [PubMed] [Google Scholar]
- Ranjan A., Vidyarthi A. S., Poddar R., 2007. Evaluation of codon bias perspectives in phage therapy of Mycobacterium tuberculosis by multivariate analysis. In Silico Biol. 7: 423–431. [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A., 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16: 276–277. [DOI] [PubMed] [Google Scholar]
- Rohwer F., Edwards R., 2002. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 184: 4529–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu K., Gupta S. K., Sau S., Ghosh T. C., 2005. Comparative analysis of the base composition and codon usages in fourteen mycobacteriophage genomes. J. Biomol. Struct. Dyn. 23: 63–71. [DOI] [PubMed] [Google Scholar]
- SAS Institute, 1994 SAS/STAT User’s Guide, Vol. 2. GLM-VARCOMP, Cary, NC: SAS Institute Inc. [Google Scholar]
- Sau K., 2007. Studies on synonymous codon and amino acid usages in Aeromonas hydrophila phage Aeh1: architecture of protein-coding genes and therapeutic implications. J. Microbiol. Immunol. Infect. 40: 24–33. [PubMed] [Google Scholar]
- Sau K., Gupta S. K., Sau S., Ghosh T. C., 2005. Synonymous codon usage bias in 16 Staphylococcus aureus phages: implication in phage therapy. Virus Res. 113: 123–131. [DOI] [PubMed] [Google Scholar]
- Sau K., Gupta S. K., Sau S., Mandal S. C., Ghosh T. C., 2007. Studies on synonymous codon and amino acid usage biases in the broad-host range bacteriophage KVP40. J. Microbiol. 45: 58–63. [PubMed] [Google Scholar]
- Schattner P., Brooks A. N., Lowe T. M., 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33: W686–W689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A., Holmes E. C., 2006. Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J. Virol. 80: 3666–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. A., Parrish C. R., Truyen U., Holmes E. C., 2005. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 102: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P. M., Li W. H., 1987. The codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15: 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N., 1961. Correlation between base composition of deoxyribonucleic acid and amino acid composition of proteins. Proc. Natl. Acad. Sci. USA 47: 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. Y., Yang Q., and X. Xia, 2013. An improved implementation of effective number of codons (Nc). Mol. Biol. Evol. 30: 191–196. [DOI] [PubMed] [Google Scholar]
- Umemura T., Tanaka Y., Kiyosawa K., Alter H. J., Shih J. W., 2002. Observation of positive selection within hypervariable regions of a newly identified DNA virus (SEN virus)(1). FEBS Lett. 510: 171–174. [DOI] [PubMed] [Google Scholar]
- Urbina D., Tang B., Higgs P. G., 2006. The response of amino acid frequencies to directional mutation pressure in mitochondrial genome sequences is related to the physical properties of the amino acids and to the structure of the genetic code. J. Mol. Evol. 62: 340–361. [DOI] [PubMed] [Google Scholar]
- van Weringh A., Ragonnet-Cronin M., Pranckeviciene E., Pavon-Eternod M., Kleiman L., et al. , 2011. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 28: 1827–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F., 1990. The ‘effective number of codons’ used in a gene. Gene 87: 23–29. [DOI] [PubMed] [Google Scholar]
- Xia X., 1996. Maximizing transcription efficiency causes codon usage bias. Genetics 144: 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., 1998. How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics 149: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., 2003. DNA methylation and mycoplasma genomes. J. Mol. Evol. 57: S21–S28. [DOI] [PubMed] [Google Scholar]
- Xia X., 2005. Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene 345: 13–20. [DOI] [PubMed] [Google Scholar]
- Xia X., 2007. An improved implementation of codon adaptation index. Evol. Bioinform. 3: 53–58. [PMC free article] [PubMed] [Google Scholar]
- Xia X., 2008. The cost of wobble translation in fungal mitochondrial genomes: integration of two traditional hypotheses. BMC Evol. Biol. 8: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., 2012a DNA replication and strand asymmetry in prokaryotic and mitochondrial genomes. Curr. Genomics 13: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X., 2012b Position weight matrix, Gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica 2012: 917540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X., 2012c Rapid evolution of animal mitochondria, pp. 73–82 in Evolution in the Fast Lane: Rapidly Evolving Genes and Genetic Systems, edited by R. S. Singh, J. Xu, and R. J. Kulathinal. Oxford University Press, Oxford. [Google Scholar]
- Xia, X., 2013a Comparative Genomics. Springer Heidelberg. [Google Scholar]
- Xia X., 2013b DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30: 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Palidwor G., 2005. Genomic adaptation to acidic environment: evidence from Helicobacter pylori. Am. Nat. 166: 776–784. [DOI] [PubMed] [Google Scholar]
- Xia X., Yuen K. Y., 2005. Differential selection and mutation between dsDNA and ssDNA phages shape the evolution of their genomic AT percentage. BMC Genet. 6: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Huang H., Carullo M., Betran E., Moriyama E. N., 2007. Conflict between translation initiation and elongation in vertebrate mitochondrial genomes. PLoS ONE 2: e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., MacKay V., Yao X., Wu J., Miura F., et al. , 2011. Translation initiation: a regulatory role for poly(A) tracts in front of the AUG codon in Saccharomyces cerevisiae. Genetics 189: 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Hao B., 2009. CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Res. 37: W174–W178. [DOI] [PMC free article] [PubMed] [Google Scholar]