

Abstract
Little is known about either the basal or stimulated homeostatic mechanisms regulating nuclear tenure of Nf-e2-related factor 2 (Nrf2), a transcription factor that mediates expression of over 200 detoxification genes. Our data show that stress-induced nuclear Nrf2 accumulation is largely from de novo protein synthesis, rather than translocation from a pre-existing cytoplasmic pool. HepG2 cells were used to monitor nuclear Nrf2 24 hrs following treatment with the dithiol micronutrient (R)-α-lipoic acid (LA; 50 μM), or vehicle. LA caused a ≥2.5-fold increase in nuclear Nrf2 within 1 hr. However, pretreating cells with cycloheximide (50 μg/ml) inhibited LA-induced Nrf2 nuclear accumulation by 94%. Providing cells with the mTOR inhibitor, rapamycin, decreased basal Nrf2 levels by 84% after 4 hrs, but LA overcame this inhibition. LA-mediated de novo protein translation was confirmed using HepG2 cells transfected with a bicistronic construct containing an internal ribosome entry sequence (IRES) for Nrf2, with significant (P<0.05) increase in IRES use under LA treatment. These results suggest that a dithiol stimulus mediates Nrf2 nuclear tenure via cap-independent protein translation. Thus, translational control of Nrf2 synthesis, rather than reliance solely on pre-existing protein, may mediate the rapid burst of Nrf2 nuclear accumulation following stress stimuli.
Keywords: Nrf2, lipoic acid, mammalian target of rapamycin (mTOR), bicistronic, cap-independent translation, protein homeostasis, cellular stress
1. Introduction
Nf-e2-related factor 2 (Nrf2) is a transcription factor that regulates the expression of multiple Phase II detoxification and antioxidant genes, and is thus critical for cellular stress response [1]. While the Nrf2 gene is constitutively expressed and the protein contains a nuclear localization sequence [2], very little Nrf2 is typically found in the cell under quiescent conditions. The mechanism associated with Nrf2 nuclear accumulation under both quiescent and stress-induced conditions is complex and only currently being elucidated. It is thought that oxidation of or electrophilic addition to critical sulfhydryls on Keap1, a Kelch-like actin-binding protein that bridges Nrf2 to the Cul3 E3 ubiquitin ligase [3, 4], may prevent Nrf2 association or else causes its release from the Keap1 complex [5–7]. Either scenario increases Nrf2 half-life [8], which allows for enhanced nuclear accumulation and expression of stress response genes [9], including NAD(P)H quinone oxidoreductase-1 (nqo1), heme oxygenase-1 (ho1), and those associated with glutathione (GSH) synthesis (both the catalytic [gclc] and modulatory [gclm] subunits of glutamate-cysteine ligase, and glutathione synthetase). Indeed, Keap1 knockout mice demonstrate the importance of Keap1, as tissue from these animals exhibits constant and excessive nuclear Nrf2 even during times of quiescence, and the result is lethal [10].
In addition to Nrf2 regulation via Keap1 thiol redox control, previous studies revealed that protein kinase C (PKC)-induced Nrf2 phosphorylation at Ser40 causes its disassociation from Keap1 [11, 12], indicating that kinase-dependent stress signaling may regulate Nrf2 by discouraging Keap1-mediated degradation. However, it is not clear whether Nrf2 phosphorylation affects its nuclear accumulation [11]. Thus, Nrf2 activation may be subject to numerous redox and signal transduction-associated stimuli to elicit Nrf2 nuclear accumulation and induction of stress response genes.
Despite this evidence, it is difficult to understand how Keap1 redox interactions and/or Nrf2 phosphorylation solely are responsible for enhanced Nrf2 nuclear tenure following a stimulus. Cellular Nrf2 levels are generally quite low under basal conditions, making it difficult to account for the large and rapid stress-induced increases in nuclear Nrf2 ostensibly coming from an existing pool of Keap1-tethered Nrf2. More specifically, Eggler et al. found that mutation of critical cysteine residues was insufficient to release Nrf2 from Keap1 [13], therefore calling into question the exclusive redox-dependent nature of the Keap1/Nrf2 complex. The question remains whether other potentially significant pathways are involved in enhancing nuclear Nrf2 levels after a stimulus. In this regard, the recent work by Kong and coworkers is instructive, as this group showed that Nrf2 mRNA has an internal ribosome entry sequence (IRES) [14], suggesting that Nrf2 translation would continue even under high stress insults that typically halt global translation. IRESs are present in genes vital for stress response and survival, and they allow the translational machinery to bypass the 7-methyl-guanosine cap at the 5′ end of mRNAs by providing an internal site for ribosomal binding, which is known as “cap-independent translation.” Previous research on Nrf2 nuclear accumulation has elucidated the mechanism of its proteasomal degradation, but little is known about how Nrf2 translation is regulated. Thus, the full mechanism involved in stress-mediated Nrf2 nuclear accumulation has yet to be divulged.
Because of these significant gaps in knowledge, the current study was undertaken to determine whether Nrf2 protein synthesis is part of the stimulus-induced mechanism associated with nuclear Nrf2 accumulation. Using the HepG2 liver cell line, this report shows that (R)-α-lipoic acid (LA), a dithiol redox-active compound known to induce Phase II detoxification enzymes in an Nrf2-dependent manner, elicits nuclear accumulation of Nrf2 by 1) increasing the half-life of Nrf2 protein and 2) markedly enhancing translation of Nrf2 from its existing hepatic mRNA pools. Synthesis of Nrf2 protein at the basal level was blocked by rapamycin, an inhibitor of cap-dependent translational initiation, but LA-induced Nrf2 synthesis was rapamycin-independent. Use of a bicistronic construct containing the IRES of Nrf2 confirmed that LA was able to activate cap-independent translation of Nrf2. These results thus represent a novel and potentially significant addition to the existing knowledge of how Nrf2 functions to meet environmental challenges.
2. Materials & Methods
2.1. Cell culture
Cell culture-approved chemicals were obtained from Sigma-Aldrich (St. Louis, MO, USA) except where noted. The human hepatic carcinoma cell line, HepG2, was maintained with EMEM (ATCC), 10% FBS, 2 mM l-glutamine, and antibiotics. Cells were maintained in a 37 °C incubator with 5% CO2. Media was changed at the end of each day, at least 12 hours before an experiment, to eliminate artifactual stimulation from fresh media components.
2.2. Chemical treatments
(R)-α-lipoic acid was obtained from MAK Wood, Inc. (Grafton, WI, USA). A 50 mM solution was made in dimethyl formamide (DMF) vehicle, and LA was added to cell culture media at 1:1000 for a concentration of 50 μM. Cycloheximide (CHX) was solubilized in DMF and used at 50 μg/ml. Rapamycin (Calbiochem, IC50 = 50 pM) was used to inhibit mTOR complex 1 (mTORC1) and was supplied at 10 nM final concentration to cells at least 2 h prior to LA treatment. Vehicle treatments were used as controls. PS341 (bortezomib), a boronic acid that acts as a very potent proteasome inhibitor [15], was a gift of Millennium Pharmaceuticals (Cambridge, MA, USA) and was solubilized in DMF and provided to cells at 50 nM final concentration.
2.3. Whole cell lysates and nuclear extracts
For whole cell lysates, cells were harvested by scraping, washed in phosphate buffered saline (PBS), and sonicated in TNSEV buffer (50 mM Tris, pH 7.5, 1% NP-40 (v/v), 100 mM NaCl, 2 mM EDTA, 2 mM sodium orthovanadate) containing protease and phosphatase inhibitors. For nuclear extracts, cells were harvested, washed, and cell pellets were allowed to swell for 15 minutes in Hypotonic Buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, plus freshly added protease and phosphatase inhibitors). Plasma membranes were disrupted by adding 0.6% (v/v) Igepal and vortexing, then nuclei were pelleted by centrifugation at 12,000 rpm for 1 min. at 4°C. The cytosolic fraction was removed and nuclei were washed and resuspended in the buffer with Igepal. The method is described in detail in Shay et al. [16].
2.4. Immunoblotting
Lysates were prepared as described above, sonicated, and proteins were solubilized for PAGE in Laemmli loading buffer containing SDS. Samples were heat-denatured for 5 min. at 100°C. Normalized amounts of protein (30 μg/lane) were run on SDS-PAGE and transferred to PVDF membranes with a semi-dry blotter. Membranes were blocked in PBS containing 1% Tween-20 with either 5% nonfat dry milk or 3% BSA, incubated with primary antibodies for 2 h at room temperature, washed, and incubated with secondary antibodies for 1 h at room temperature, washed, incubated with chemiluminescence reagents, exposed to film, and developed. Antibodies made to the following proteins were used: Nrf2 (H-300) and Keap1 (C-20) (Santa Cruz Biotech, USA), 4EBP1, Phospho-T-37/46-4EBP1, Lamin A/C, and LDHA (Cell Signaling Technology, Beverly, MA, USA), and Actin (Sigma-Aldrich). The primary antibodies were diluted in PBS-Tween for blotting. Blots were densitometrically analyzed with ImageJ software from NIH.
2.5. RT-qPCR
For quantitative analysis of mRNA in cells, total RNA was isolated from HepG2 cells using the RNAeasy Mini Kit (Qiagen, Valencia, CA, USA). RNA (2 μg) was reverse transcribed using the RETROscript kit (Ambion, Austin, TX, USA) and the provided polyadenylate primers. TaqMan primers specific to NQO1, GCLC, NFE2L2 and β-actin were obtained from Applied Biosystems (Austin, TX, USA). PCR amplification reactions were assembled using the TaqMan Gene Expression Array Master Mix per the manufacturer’s instructions. Reactions were run on an MJ Research DNA Opticon 2 (Bio-Rad, Hercules, CA, USA) for 40 cycles. Messenger RNA levels were estimated by the ΔΔCT method (where CT is cycle threshold) from the β-actin internal standard and are represented as percent change from untreated cells.
2.6. Transfection of bicistronic vector and measurement of luciferase activity
A bicistronic vector containing human Nrf2’s 5′ UTR and part of the ORF (−83 through +45) in the intercistronic region was used as described in Li et al. [14]. As a control to rule out ribosomal read through, an additional vector with a hairpin structure upstream of the Nrf2 5′ UTR was used, and cryptic promoter activity was also ruled out [14]. HepG2 cells were transfected in 96-well plates using Jet-PEI (Polyplus-transfection SA, Illkirch, France) according to the manufacturer’s instructions. The Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) was used to measure the activities of Renilla luciferase (Rluc) and firefly luciferase (Fluc). Eighteen hours post-transfection, cells were washed in HBSS and lysed with 20 μl passive lysis buffer. A SpectraMax L luminometer (Molecular Devices, Sunnyvale, CA) was used to measure the Fluc and Rluc intensities. To measure the Fluc activities, 20 μl lysate was mixed with 100 μl Luciferase Assay Reagent II to generate a stabilized luminescent signal. After the Fluc luminescence was quantified, the Fluc reaction was quenched and the Rluc was simultaneously initiated by adding 100 μl Stop & Glo Reagent. Rluc/Fluc activities were normalized by total protein concentration.
2.7. Statistics
Statistical analysis was performed with GraphPad Prism 5.0 software (GraphPad, San Diego, USA). At least 3 replicates were performed for each experiment. Student’s t-test, one-way ANOVA, and two-way ANOVA were used to determine statistical significance, with Bonferroni’s or Tukey’s post hoc test. Results were considered significant if the P value was <0.05.
3. Results
3.1. Nrf2 accumulates in both cytoplasmic and nuclear compartments under proteasome inhibition
We used a relevant cell model for oxidative stress response and detoxification, the transformed hepatocyte cell line HepG2, to define the characteristics of steady-state nuclear Nrf2 homeostasis during quiescence and under stimulated conditions. While it is generally accepted that Nrf2 responds to a variety stresses by accumulating in the nucleus, relatively little is known about Nrf2 homeostasis under basal conditions, except that it is subject to rapid proteasomal degradation [8, 17, 18]. We therefore determined baseline Nrf2 turnover characteristics by treating cells with PS341 or cycloheximide (CHX) to inhibit Nrf2 degradation or synthesis, respectively. Treating cells for 4 hrs with 50 nM PS341 resulted in a 3- and 17-fold increase in cytoplasmic and nuclear Nrf2 levels, respectively, relative to vehicle treatment (Figure 1A). Keap1 is also subject to proteasomal degradation, but 4 h PS341 did not produce a noticeable increase in either cytoplasmic or nuclear Keap1 (Figure 1A) under these specific conditions. The enrichment in Nrf2 nuclear accumulation suggests that no inducer of Nrf2 is necessary to initiate nuclear Nrf2 translocation, which corroborates the observation that Nrf2 has a constitutively active nuclear localization signal [2]. Further, it is notable that Nrf2 also accumulates in the cytoplasm when degradation is inhibited, though the ways in which this pool differs from the nuclear pool remain unknown. The nuclear Nrf2 that accumulates in the nucleus under PS341 is active, as evidenced by increased transcript profiles of gclc and nqo1, two quintessential Nrf2-mediated Phase II detoxification genes (Figure 1B).
Figure 1. Nrf2 enters the nucleus without a stimulus but has a short half-life.
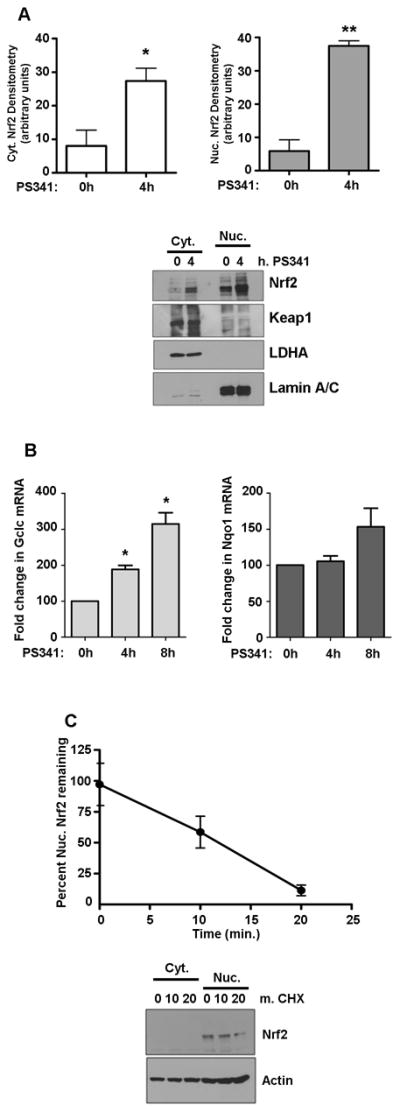
A) Cytoplasmic and nuclear Nrf2 were assessed by western blot using protein from HepG2 cells incubated with 50 nM PS341 for the indicated times. Keap1 levels were also determined by western blot under these conditions. Nrf2 graphs show the means ± SE of 3 experiments. *P<0.04, **P<0.0002. B) RT-PCR analysis for gclc and nqo1 mRNA following PS341 time course, N=5 for each. *P<0.0001 vs. time 0. C) Cells were treated with 50 μg/ml cycloheximide (CHX) for the indicated times. Nrf2 levels were determined by western blot and densitometry. Graph of nuclear Nrf2 shows the mean ± SE of 3 experiments.
Vehicle-treated cells displayed very little Nrf2, so we sought to determine the half-life of Nrf2 in our model. Monitoring the rate of Nrf2 loss over time under CHX treatment demonstrated that nuclear Nrf2 has a nominal half-life in HepG2 cells of ~12 min. as shown in the graph in Figure 1C, which is in accordance with the whole-cell Nrf2 half-life in previous studies [8]. Cytoplasmic Nrf2 was below detection levels in the accompanying western blot. Based on these results using inhibitors of protein synthesis and degradation, it appears that Nrf2 is continuously produced and will spontaneously accumulate in the nucleus if it is not degraded.
3.2. LA-mediated Nrf2 nuclear accumulation characteristics
For cell stimulation, we used LA, a dithiol compound that initiates Nrf2-mediated expression of glutathione synthesizing genes [19, 20]. A low, physiological dose of LA (50 μM) [21] resulted in a 2.5-fold (P<0.05) increase in protein levels of Nrf2 in the nuclei of HepG2 cells within 1 hr after treatment (Figure 2A). Nrf2 levels remained elevated even 24 h following LA treatment in HepG2 cells, but did not result in changes to Nrf2 transcript levels up to 8 hrs after the stimulus relative to vehicle treatment (Figure 2B). This shows that LA elicits a rapid, sustained increase in nuclear Nrf2 levels, which is not from an induction of Nrf2 gene expression.
Figure 2. LA stimulates nuclear accumulation of Nrf2 without changes in its mRNA.

A) HepG2 cells (N=3) were treated with 50 μM LA for up to 24 h prior to harvest. Nrf2 levels were determined by western blot and densitometry. The graph of nuclear Nrf2 shows the mean ± SE. *P<0.05 vs. time 0. B) RT-PCR analysis for nrf2, gclc, and nqo1 mRNA following an LA time course, N=5 for each. *P<0.05 vs. time 0.
LA-induced nuclear Nrf2 accumulation was sufficient to increase transcript profiles of the Nrf2-regulated genes gclc and nqo1 (Figure 2B), showing that LA did indeed induce sufficient Nrf2 to perform its transcriptional function. Thus, conditions were defined for both steady-state nuclear homeostasis under both basal conditions and stemming from a physiologically relevant stimulus.
3.3. LA-induced Nrf2 nuclear accumulation requires new protein synthesis
Next, we asked whether the Nrf2 protein found in the nucleus in both quiescent and LA-stimulated conditions stemmed from alterations in Nrf2 half-life (reflecting less interaction with Keap1), newly translated protein, or both. For monitoring Nrf2 half-life under a stimulus, cells were incubated with LA (50 μM) for 1 hr prior to adding CHX, after which samples were taken at the times indicated. Under these conditions, Nrf2 T1/2 was approximately 55 min. (Figure 3A), which was more than 4 times longer than that observed under quiescent conditions (see Fig. 1C); however, it must be noted that the prolonged stimulus-induced T1/2 alone could not account for the sustained nuclear Nrf2 tenure observed in Figure 2A.
Figure 3. Nuclear accumulation of Nrf2 under LA requires new protein synthesis.
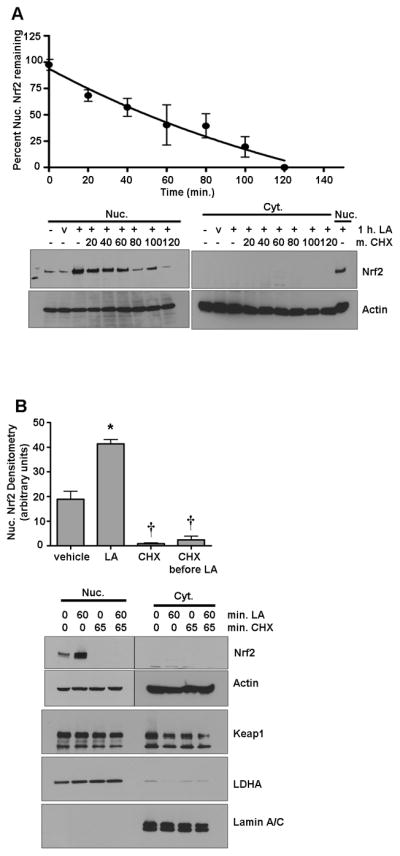
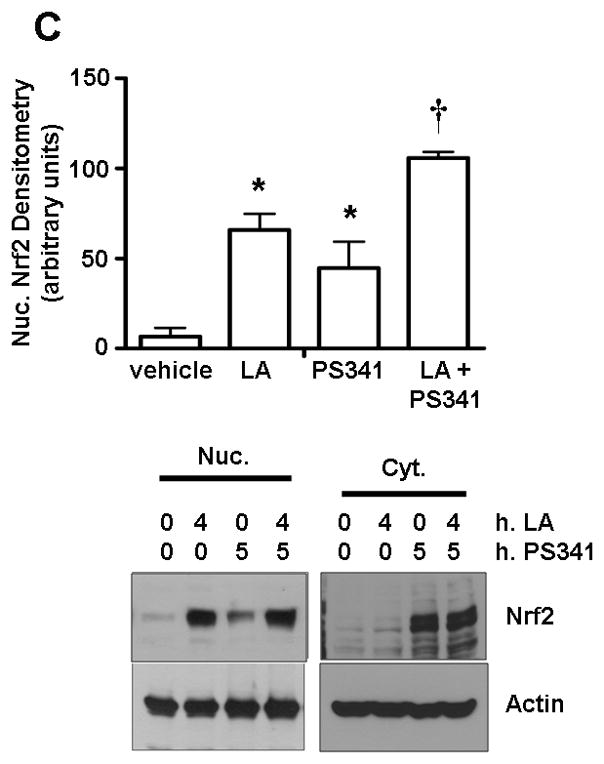
A) HepG2 cells were treated with 50 μg/ml CHX for the indicated times after preincubation for 1 h with 50 μM LA. Nuclear extracts were separated by SDS-PAGE, transferred to membranes, and probed for Nrf2, Keap1, and loading controls. The graph shows the mean ± SE of 3 experiments. B) Cells were treated with vehicle only, 50 μM LA for 1 h, or pretreated with 50 μg/ml CHX for 5 min. before 1 h treatment with LA or additional vehicle. Cell extracts were separated by SDS-PAGE, transferred to membranes, and probed for Nrf2. The graph of nuclear Nrf2 shows the mean ± SE of 4 experiments. *P<0.0001 vs. vehicle and †; †P<0.0001 vs. vehicle and *. C) Cells were preincubated with 50 nM PS341 for 1 h prior to a 4 h bolus of 50 μM LA or vehicle. Cell extracts were run on SDS-PAGE and western blotting was performed for Nrf2. The graph of nuclear Nrf2 shows the means ± SE of 3 experiments. *P<0.05 vs. vehicle and †; †P<0.05 vs. vehicle and *.
To determine whether the Nrf2 accumulation resulted from sequestered cytoplasmic pools or from newly synthesized protein stemming from the LA stimulus, CHX was added prior to LA stimulation. If pre-existing Nrf2 translocates to the nucleus upon LA treatment, there would be an elevation of nuclear Nrf2 levels despite the addition of CHX. In contrast, results showed that no Nrf2 accumulated in the nucleus (Figure 3B) under CHX interdict (P<0.0001), nor was any increase in Nrf2 observed in the cytoplasm after LA. Interestingly, an overall loss of nuclear Nrf2 is observed under CHX vs. vehicle alone (P<0.0001). This was likely because of proteasomal degradation where basal levels of Nrf2 are being degraded and are not being renewed. These results show that LA, in addition to increasing nuclear Nrf2 half-life from ~12 to ~55 min., also surprisingly induces new Nrf2 protein synthesis. Keap1, in contrast, was not changed with LA treatment (Figure 3B). To further examine the implications of these results, the combination of proteasome inhibition and LA stimulation was tested. When HepG2 cells were treated with both LA and PS341, significantly (P<0.05) more Nrf2 nuclear accumulation occurred vs. either treatment alone (Figure 3C), confirming that LA plays a dual role in increasing Nrf2 protein levels.
3.4. LA overcomes translational inhibition by rapamycin and induces use of Nrf2’s IRES
To further assess a potential LA-dependent translational mechanism, we determined whether inhibiting mTORC1, the serine/threonine kinase that mediates cap-dependent translation, modulates LA-stimulated nuclear Nrf2 levels. Using the macrolide rapamycin to inhibit mTORC1, we observed a steady decrease in whole-cell Nrf2 levels over time (Figure 4A). This indicates that Nrf2 translation in unstimulated cells, Nrf2 translation is cap-dependent and regulated by mTORC1. However, pretreating cells with rapamycin to inhibit mTORC1 did not prevent an LA-induced Nrf2 protein increase (Figure 4A), which shows that LA overcomes mTORC1 inhibition to increase Nrf2 levels.
Figure 4. LA overcomes translational inhibition of Nrf2 by rapamycin.
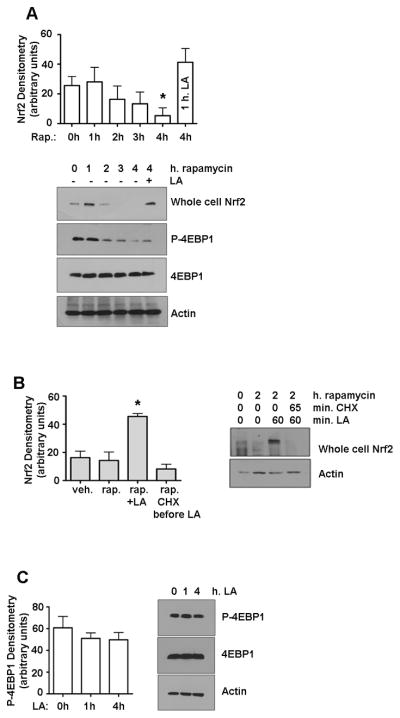
A) Nrf2 was measured by western blot in whole cell extracts from HepG2 cells treated with 10 nM rapamycin. *P<0.05 vs. time 0. B) Nrf2 in whole cell extracts from cells pretreated with 10 nM rapamycin and then given CHX (50 μg/ml) or vehicle before the addition of 50 μM LA. *P<0.0015 vs. time 0. C) Phospho-4EBP1 and total 4EBP1 were measured by western blot in cells treated up to 4 h with 50 μM LA, P>0.05. All graphs show the means ± SE of 3 experiments.
In light of these results and those showing that Nrf2 half-life was lengthened following LA treatment (Figure 3A), further experiments were performed to determine whether the LA-induced Nrf2 seen after pretreatment with rapamycin was pre-existing. Results showed that adding CHX first kept LA from overcoming rapamycin inhibition (Figure 4B). Thus, we conclude that LA-induced Nrf2 accumulation largely resulted from newly synthesized Nrf2.
4EBP1 is a key protein for regulating cap-independent translation; when hypophosphorylated, it sequesters eIF4E from binding to the 5′ cap of mRNAs. Thus, phosphorylation of 4EBP1 promotes cap-dependent translation, while failure to phosphorylate it (as under stress conditions) decreases global protein translation and only cap-independent translation of stress-response and survival-related proteins can proceed. As shown in Figure 4A, within 2 hrs, rapamycin reduced phosphorylation of T37/464EBP1 dramatically but not completely, which is in accordance with Yip et al. who state that this is a function of mTOR’s structure [22]. In spite of the continued presence of Nrf2 under rapamycin + LA, LA was not able to overcome rapamycin’s reduction of 4EBP1 phosphorylation (Figure 4A), indicating that LA does not promote cap-dependent translation of Nrf2 by facilitating the re-phosphorylation of 4EBP1. LA alone caused no significant change in phospho-4EBP1 (Figure 4C), and thus would not appear to affect the amount of eIF4E available to bind the 5′ caps of mRNAs [23].
In the same time course of up to 4 hrs, LA stimulated nuclear accumulation of Nrf2 even in the presence of rapamycin (Figure 5A). Although the results in Figure 4 show that this is largely due to new Nrf2 protein synthesis and not solely reduced turnover, we sought to separate these mechanisms. Because a recent report by Li et al. [14] showed that Nrf2 has an IRES, we sought to determine whether this IRES is utilized to maintain Nrf2 translation under LA treatment. For this determination, HepG2 cells were transfected with a bicistronic vector in which the 5′ UTR of wild type Nrf2 was subcloned into the intercistronic region, resulting in the production of Firefly Luciferase when the IRES is used (Figure 5B). Renilla Luciferase is produced in response to cap-dependent translation. Transfected cells were treated with rapamycin and/or LA in the same manner as performed in Figure 5A. LA treatment markedly stimulated appearance of Firefly Luciferase (Figure 5C), which qualitatively suggests that LA’s actions on translation were mediated through the IRES of Nrf2. Interestingly, there was a small but significant (P<0.05) decrease in Renilla Luciferase under not only rapamycin, but also LA or the combination of the two compounds. This suggests that both modes of translation may operate, to some degree, depending on the stimulus. Figure 5C confirms that the Nrf2 IRES is active in response to LA in spite of the presence of rapamycin. These results further suggest that LA-dependent translational changes to Nrf2 can occur even when mTORC1 activity is impaired. Taken together, these data indicate that LA promotes cap-independent translation of Nrf2.
Figure 5. Nrf2 IRES is induced by LA during rapamycin treatment.
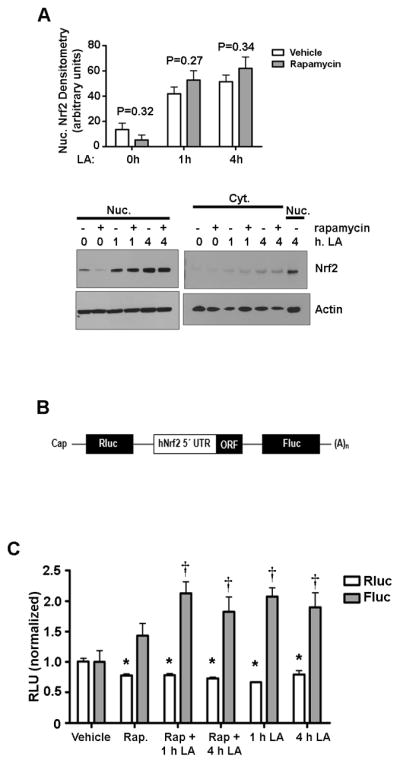
A) HepG2 cells were preincubated with vehicle or 10 nM rapamycin before a time course of 50 μM LA. Nrf2 protein was assessed by western blot. B) Adapted from Li et al. (14): wild-type 5′-UTR of human Nrf2 mRNA was subcloned into the intercistronic region of a bicistronic vector containing a Rluc and a Fluc. 5′-UTR: 5′-untranslated region; (A)n: poly(A) tail; ORF: open reading frame. C) Cells were transfected with the bicistronic vector in which the 5′UTR of Nrf2 is accessed only via its IRES and Firefly Luciferase (Fluc) activity indicates cap-independent translation. Renilla Luciferase (Rluc) activity indicates cap-dependent translation. Cells were treated as in (A) above, and dual luciferase activity was monitored by photon counting on a SpectraMax luminescence plate reader. The graphs show the means ± SE of 3 experiments. *P<0.05 compared to vehicle in Rluc samples. †P<0.05 compared to vehicle in Fluc samples.
4. Discussion
Herein, we describe a novel means of explaining a stimulus-induced increase in Nrf2 levels that must be considered an addition to the well-studied mechanism of Keap1/Nrf2 sequestration. In our model, rather than a large pool of existing Nrf2 being translocated to the nucleus upon LA treatment, the majority of Nrf2 was newly synthesized via translational mechanisms, without a concomitant increase in mRNA. This system of highly controlled protein synthesis indicates that Nrf2 is an important molecule for stress response; the cell can induce Nrf2 translation upon receiving a stress signal without having to induce transcription of Nrf2. Furthermore, Nrf2 can still be synthesized by cap-independent translation if the mTORC1 system is repressed. As a stimulus, LA also facilitates a lengthening of Nrf2 half-life. Keap1, which is associated with Nrf2 degradation, is thought to be the limiting factor in the Nrf2/Keap1 system [24, 25], so a surfeit of newly synthesized Nrf2 may overwhelm the ability of Keap1 molecules to bind it. In fact, our data show that nuclear Nrf2 half-life increases to approximately 55 min. after LA treatment (Figure 3A), which is notable, but is not long enough to account for Nrf2’s sustained nuclear tenure seen in Figure 2A. Thus, our data point to a translational control mechanism which should also be considered when evaluating the effects of other compounds that stimulate Nrf2 nuclear accumulation.
These findings must be placed in context with the known regulatory mechanisms that affect steady-state levels of Nrf2 in the nucleus. Nrf2 is constitutively expressed and its transcription is not affected by the stressors or antioxidants that so affect its protein levels. However, Nrf2 protein is subject to continual degradation by the Keap1-Cul3 ubiquitin ligase system, such that high amounts of cellular Nrf2 are not seen on a basal level. Nrf2 has nuclear localization signals (NLS) as well as nuclear export signals (NES) [2, 25–27]. Cells expressing an Nrf2 construct with a mutated NES display nuclear localization of Nrf2 even in the absence of a stimulus, while cells expressing an Nrf2 construct with a mutated NLS cannot transport Nrf2 into the nucleus even under conditions that induce nuclear accumulation of wild-type Nrf2 (i.e., 50 μM t-BHQ); instead, Nrf2 with a mutated NLS remains cytoplasmic [2]. Under basal conditions, most cells show both cytoplasmic and nuclear distribution of exogenously expressed wild-type Nrf2 [26].
Stresses shown to induce Nrf2 nuclear accumulation include the unfolded protein response [28], viral infection [29], and oxidative/genotoxic stress (reviewed in [30]), which can benefit from the upregulation of genes encoding Phase II enzymes. We have also shown that LA induces the nuclear accumulation of Nrf2 in rat liver tissue and increases the transcription of ARE-containing, Nrf2-driven genes [19]. As a dithiol electrophile, LA can be considered a typical sulfhydryl inducer of Nrf2. In the event of a stress signal such as treatment with LA, Nrf2 is localized in the nucleus. LA is a redox active compound, and thiol modification of Keap1 has been proposed as a mechanism for catalyzing the release of Nrf2 [6]. However, the precise mechanism(s) involved in Nrf2 nuclear tenure under a stimulus has not been fully elucidated. Eggler et al. [13] and Hong et al. [31] have proposed that thiol modification of Keap1 switches ubiquitylation from Nrf2 to Keap1, catalyzing not so much a “release” of Nrf2 as a lack of available Keap1 to tether it. Certainly, our results show that LA-induced nuclear accumulation of Nrf2 does not coincide with a rise in cytoplasmic Nrf2, but with de novo synthesis of Nrf2. It is not known, however, whether the thiol modification of Keap1 is enough to allow Nrf2 to localize to the nucleus. As Kobayashi et al. [7] found that mutation of Keap1 cysteines 273 and 288 was insufficient to cause Nrf2 release, although Nrf2 degradation was decreased and cytoplasmic accumulation was observed. In none of our experiments did we observe an LA-induced increase in cytoplasmic Nrf2. Further, the half-life of nuclear Nrf2 increased to 55 min. under LA stimulus (Figure 3A), which is still quite short. Jain and Jaiswal [32] showed that Nrf2 nuclear export is mediated by phosphorylation by Fyn at Y568, and that Nrf2 must be exported into the cytosol via its NES for Keap1-mediated degradation [2]. Although Keap1 occurs in the nucleus of HepG2 and NIH3T3 cells [33], where 26S proteasomes are also present [34], current evidence suggests that Nrf2 degradation takes place only in the cytoplasmic compartment [32, 35]. Our results make sense in this context, as Nrf2 may have translocated to the cytoplasm for Keap1-mediated degradation, explaining both its relatively short (<1 hr) half-life and its cytoplasmic levels typically being below detection limits.
A recent paper by Sun et al. [36] implicates the protein KPNA6 in the nuclear import of Keap1, thereby promoting the ubiquitination and degradation of Nrf2 and “resetting” the basal levels of Nrf2. Because of this, siRNA knockdown of KPNA6 increases nuclear Nrf2. While we did not test the effect of LA on the levels of KPNA6 itself, we did note that Keap1 half-life was not affected by LA treatment (Supplemental Figure 1A), nor did LA significantly increase nuclear levels of Keap1 (Supplemental Figure 1B) at the dose and times measured. More work will need to be done to determine the effect, if any, of LA on KPNA6-mediated Keap1 import.
Examination of Nrf2 subcellular localization under proteasome inhibition showed that Nrf2 levels build up in both cytoplasm and nucleus without any stimulus (Figure 1A). The combination of PS341 and LA treatment increased Nrf2 levels in comparison to either compound alone (Figure 3C), confirming that LA does not merely prevent degradation of existing Nrf2, which is in accordance with Nrf2 being newly translated in response to LA. This is interesting in light of the important work done by Purdom-Dickinson et al., who showed that H2O2 induces Nrf2 translation, but does not lengthen its half-life [37]. LA, by contrast, does both. It is notable, however, that LA results in sustained Nrf2 nuclear tenure for as long as 24 h in our model (Figure 2A), which cannot be explained solely by its increase in half-life. Thus it appears that the majority of Nrf2 that accumulates in the nucleus after LA treatment is newly translated.
Additionally, our data suggest that the mTORC1-mediated translational system is involved in Nrf2 steady-state levels (Figures 4 and 5), but inhibition of this complex by rapamycin cannot prevent translation of Nrf2 under LA-induced stimulation. These results are significant, as mTORC1 is a major point of control for protein translation. In its activated state, mTORC1 receives multiple input signals related to energy status and cellular stress conditions [22]. Under permissive conditions, this complex catalyzes phosphorylation of 4EBP1, a regulator of cap-dependent protein translation, whereupon 4EBP1 is released from eIF4E. Cap-dependent translation proceeds when unbound eIF4E binds the 7mGpppN cap on the 5′ end of mRNAs, and eIF4G forms a bridge between this factor and the 40S ribosomal subunit [38]. In the event of no available eIF4E, cap-independent translation can still occur in certain mRNAs via IRESs [38], which vary in their use of particular initiation factors, but do not require eIF4E [39]. These sequences are found in transcripts encoding proteins that are important for cell stress response and survival, and a switch to conditions favoring cap-independent translation results from various stress stimuli [40]. Kong and coworkers found that Nrf2 contains an IRES [14], which further classifies Nrf2 as a vital stress response gene. Our data fit with this interpretation as nuclear accumulation of Nrf2 resulted from an alteration of the mTOR pathway by LA. When Kong’s group used a dual luciferase vector to measure the use of Nrf2’s IRES, H2O2 induced translation via both mechanisms, but sulforaphane primarily used the IRES [14]. Our findings showed that usage of general cap-dependent translation did not increase after LA treatment, but there was a marked increase in cap-independent translation of Nrf2 via its IRES, as measured by Firefly Luciferase (Fig. 5C). A recent work by Zhang et al. [41] shows that the Sjögren Syndrome Antigen B (SSB; also called La autoantigen) protein binds to the 5′ UTR of Nrf2 and induces cap-independent translation under H2O2 treatment. SSB colocalizes with ribosomes in a redox-sensitive manner. While we did not test our samples for the presence of this binding protein, it is quite possible that LA, as a redox-active compound, could affect the localization of SSB to increase Nrf2 protein translation.
In summary, the LA-induced nuclear accumulation of Nrf2 via a translational mechanism, rather than that of an existing pool of cytoplasmic Nrf2, is a novel finding. The large amounts of Nrf2 observed in the nucleus after stress stimuli do not appear to correlate with the cytoplasmic levels of Nrf2 held by Keap1, and indeed, as posited by Pickett’s group, constitutive synthesis of such a large pool of Nrf2 only to degrade it immediately would represent an inefficient use of cellular energy [24]. Our data using LA provide evidence that new protein synthesis, in addition to the inhibition of degradation, is crucial to increasing the nuclear levels of Nrf2.
Supplementary Material
A) HepG2 cells were treated with 50 μg/ml CHX for the indicated times after preincubation for 1 h with 50 μM LA. Nuclear extracts were run on SDS-PAGE, transferred to membranes, and probed for Keap1 and loading controls. B) Nuclear Keap1 western blot after treatment with 50 μM LA for the indicated times.
Acknowledgments
This work was supported by grants from the National Institutes of Health R01 2AG17141 and P01 AT002034-01. KPS was supported by T32 AT002688-01.
References
- 1.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37:139–143. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 2.Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280:29158–29168. doi: 10.1074/jbc.M502083200. [DOI] [PubMed] [Google Scholar]
- 3.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol. 2006;26:221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J Biol Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 10.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 11.Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. 2003;278:44675–44682. doi: 10.1074/jbc.M307633200. [DOI] [PubMed] [Google Scholar]
- 12.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 13.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A. 2005;102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Thakor N, Xu EY, Huang Y, Chen C, Yu R, Holcik M, Kong AN. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010;38:778–788. doi: 10.1093/nar/gkp1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, Stein RL. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg Med Chem Lett. 1998;8:333–8. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 16.Shay KP, Smith EJ, Hagen TM. Transcription Factor Nrf2: Examination of Nuclear Protein Levels by Immunoblotting and Promoter Response Element Binding by Chromatin Immunoprecipitation (ChIP) Current Protocols in Toxicology. 2010;45:17.13.1–17.13.13. doi: 10.1002/0471140856.tx1713s45. [DOI] [PubMed] [Google Scholar]
- 17.Sekhar KR, Yan XX, Freeman ML. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene. 2002;21:6829–6834. doi: 10.1038/sj.onc.1205905. [DOI] [PubMed] [Google Scholar]
- 18.Stewart D, Killeen E, Naquin R, Alam S, Alam J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278:2396–2402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- 19.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogborne RM, Rushworth SA, O’Connell MA. Alpha-lipoic acid-induced heme oxygenase-1 expression is mediated by nuclear factor erythroid 2-related factor 2 and p38 mitogen-activated protein kinase in human monocytic cells. Arterioscler Thromb Vasc Biol. 2005;25:2100–2105. doi: 10.1161/01.ATV.0000183745.37161.6e. [DOI] [PubMed] [Google Scholar]
- 21.Petersen Shay K, Moreau RF, Smith EJ, Hagen TM. Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life. 2008;60:362–367. doi: 10.1002/iub.40. [DOI] [PubMed] [Google Scholar]
- 22.Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–774. doi: 10.1016/j.molcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheper GC, van Kollenburg B, Hu J, Luo Y, Goss DJ, Proud CG. Phosphorylation of eukaryotic initiation factor 4E markedly reduces its affinity for capped mRNA. J Biol Chem. 2002;277:3303–3309. doi: 10.1074/jbc.M103607200. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Jain MR, Chen C, Yue X, Hebbar V, Zhou R, Kong AN. Nrf2 Possesses a redox-insensitive nuclear export signal overlapping with the leucine zipper motif. J Biol Chem. 2005;280:28430–28438. doi: 10.1074/jbc.M410601200. [DOI] [PubMed] [Google Scholar]
- 26.Li W, Yu SW, Kong AN. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. J Biol Chem. 2006;281:27251–27263. doi: 10.1074/jbc.M602746200. [DOI] [PubMed] [Google Scholar]
- 27.Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–8994. doi: 10.1074/jbc.M709040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burdette D, Olivarez M, Waris G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J Gen Virol. 2010;91:681–690. doi: 10.1099/vir.0.014340-0. [DOI] [PubMed] [Google Scholar]
- 30.Osburn WO, Kensler TW. Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat Res. 2008;659:31–39. doi: 10.1016/j.mrrev.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J Biol Chem. 2005;280:31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 32.Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281:12132–12142. doi: 10.1074/jbc.M511198200. [DOI] [PubMed] [Google Scholar]
- 33.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–4513. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters JM, Franke WW, Kleinschmidt JA. Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J Biol Chem. 1994;269:7709–7718. [PubMed] [Google Scholar]
- 35.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. KPNA6 (Importin α7)-Mediated Nuclear Import of Keap1 Represses the Nrf2-Dependent Antioxidant Response. Mol Cell Biol. 2011;31:1800–1811. doi: 10.1128/MCB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol. 2007;72:1074–1081. doi: 10.1124/mol.107.035360. [DOI] [PubMed] [Google Scholar]
- 38.Hentze MW. eIF4G: a multipurpose ribosome adapter? Science. 1997;275:500–501. doi: 10.1126/science.275.5299.500. [DOI] [PubMed] [Google Scholar]
- 39.Hernandez G. Was the initiation of translation in early eukaryotes IRES-driven? Trends Biochem Sci. 2008;33:58–64. doi: 10.1016/j.tibs.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Holcik M, Sonenberg N, Korneluk RG. Internal ribosome initiation of translation and the control of cell death. Trends Genet. 2000;16:469–473. doi: 10.1016/s0168-9525(00)02106-5. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Dinh TN, Kappeler K, Tsaprailis G, Chen QM. La autoantigen mediates oxidant induced de novo Nrf2 protein translation. Mol Cell Proteomics. doi: 10.1074/mcp.M111.015032. (EPub 2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) HepG2 cells were treated with 50 μg/ml CHX for the indicated times after preincubation for 1 h with 50 μM LA. Nuclear extracts were run on SDS-PAGE, transferred to membranes, and probed for Keap1 and loading controls. B) Nuclear Keap1 western blot after treatment with 50 μM LA for the indicated times.