

Abstract
Dihydropyrimidine dehydrogenase (DPD) is the initial and rate limiting enzyme of the uracil catabolic pathway, being critically important for inactivation of the commonly prescribed anti-cancer drug 5-fluorouracil (5-FU). DPD impairment leads to increased exposure to 5-FU and, in turn, increased anabolism of 5-FU to cytotoxic nucleotides, resulting in more severe clinical adverse effects. Numerous variants within the gene coding for DPD, DPYD, have been described, although only a few have been demonstrated to reduce DPD enzyme activity. To identify DPYD variants that alter enzyme function, we expressed 80 protein-coding variants in an isogenic mammalian system and measured their capacities to convert 5-FU to dihydrofluorouracil, the product of DPD catabolism. The M166V, E828K, K861R, and P1023T variants exhibited significantly higher enzyme activity than wildtype DPD (120%, P=0.025; 116%, P=0.049; 130%, P=0.0077; 138%, P=0.048; respectively). Consistent with clinical association studies of 5-FU toxicity, the D949V substitution reduced enzyme activity by 41% (P=0.0031). Enzyme activity was also significantly reduced for 30 additional variants, 19 of which had <25% activity. None of those 30 variants have been previously reported to associate with 5-FU toxicity in clinical association studies, which have been conducted primarily in populations of European ancestry. Using publicly available genotype databases, we confirmed the rarity of these variants in European populations, but showed that they are detected at appreciable frequencies in other populations. These data strongly suggest that testing for the reported deficient DPYD variations could dramatically improve predictive genetic tests for 5-FU sensitivity, especially in individuals of non-European descent.
Keywords: Dihydropyrimidine dehydrogenase, enzyme assays, 5-fluorouracil, individualized medicine, genetic polymorphism
Introduction
Genetic polymorphisms in the dihydropyrimidine dehydrogenase (DPD) gene (DPYD) have emerged as predictive risk alleles for developing severe toxicity to the commonly prescribed anti-cancer drug 5-fluorouracil (5-FU). Three DPYD variants have consistently been reported to be associated with 5-FU toxicity and impaired DPD enzyme activity. The most studied of the DPYD variants, *2A (rs3918290; also known as IVS14+1G>A) interrupts a splice accepter sequence and causes the in-frame deletion of amino acids corresponding to exon 14 (1). Carriers of *2A have significantly reduced DPD enzyme levels resulting in prolonged clearance times for 5-FU (2) and, as such, are more likely to develop adverse toxicity following treatment with the drug (3, 4). The second well-accepted DPD deficiency-associated variant, I560S (rs55886062), is exceptionally rare in the general population, but has been consistently linked to reduced DPD activity (5) and increased incidence of 5-FU toxicity (6, 7). Clinical studies have also consistently shown association between a third variant, D949V, and severe toxicity following chemotherapy that included 5-FU (4, 7).
More than 100 additional missense variants have been reported for DPYD, many from large scale sequencing efforts utilizing individuals from various racial groups (8, 9). Few of these additional variants have been evaluated in case-control studies of 5-FU toxicity, and those that have been studied have yielded unclear or conflicting results. For instance, the M166V variant was shown to strongly associate with grade III and IV toxicity in a cohort of patients with breast, gastroesophageal, or colorectal cancer treated with 5-FU-based therapy (10); however additional reports failed to confirm a link between the variant and 5-FU toxicity (4, 11). Further studies have suggested that M166V might be protective against specific 5-FU-related toxicities in women (12).
To aid in developing predictive genetic tests of 5-FU toxicity, we determined the contributions of many of these additional DPYD variants to DPD activity. Our lab previously demonstrated the utility of a recombinant system of protein expression to measure the enzyme activity of a small set of DPD protein variants using human cells (13). We hypothesized that additional DPYD variants may contribute to DPD deficiency, especially in populations not of European ancestry that have been under-represented in large case-control clinical association studies of 5-FU toxicity. In all, 80 DPD variants were expressed in mammalian cells and the enzyme activity of each variant measured. Thirteen variants (9 missense, 2 stop-gained, 1 frame-shift, and 1 in-frame insertion) had less than 12.5% enzymatic activity and were classified as *2A-like. Six variants had enzyme activities similar to I560S (12.5%-25%), and the enzyme activities of 11 variants were similar to that of D949V (>25%, but significantly lower than wildtype). Four variants showed enzyme activities that were significantly higher than wildtype, similar to our previous findings for C29R and S534N (13). Consistent with our hypothesis, these newly classified DPYD deficiency variants were present at higher frequencies in non-European populations.
Materials and Methods
In silico functional prediction
A list of missense DPYD variants was compiled using the NCBI dbSNP (14), the 1000 Genomes Project (9), and the NIH Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) (8) databases. The PolyPhen-2 web server version 2.2.2 was used to predict the impact of amino acid changes on protein function (15) using the translated product of DPYD transcript ENST00000370192 from UniProtKB/UniRef100 release 2011_12 as the reference protein sequence. PolyPhen-2 predictions rely upon a naïve Bayes classifier model trained using machine-learning algorithms applied to publicly available datasets. In silico data presented in Figure 1 were determined using the prediction model trained with the HimDiv dataset (15). The estimated false positive rate (FPR) for each variant is calculated by the software as the fraction of benign variants incorrectly classified as damaging for a given threshold of naïve Bayes probabilistic scores (15). Qualitative predictors (“benign,” “possibly damaging,” and “probably damaging”) are reported by the software based on the thresholds determined from estimated FPR values. Complete details regarding the algorithms and outputs of the PolyPhen-2 software have been detailed by the software’s developers (15, 16). Additional predictions were performed using PolyPhen-2 trained with the HumVar dataset, PROVEAN version 1.1.3 Mutation Assessor web server (17), SIFT version 4.0.3 (18), and the SNAP webserver (19) using the default settings.
Figure 1. Predicted impact of missense DPYD variations on DPD protein function.
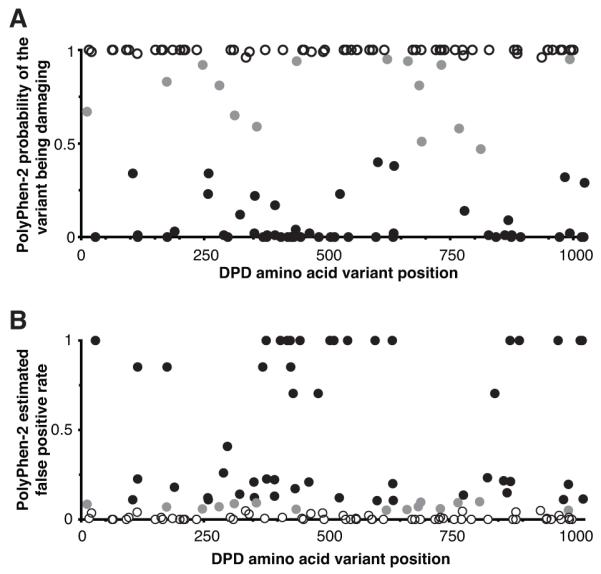
A, the probability of each variation being damaging to protein function or structure was determined using PolyPhen-2. B, for each probability reported in panel A, the estimated FPR was calculated by the software. Each mark represents an individual amino acid substitution. Shading indicates the qualitative classifier predicted by PolyPhen-2 (black, benign; gray, possibly damaging; white, probably damaging).
Vector construction
Human DPYD variant expression vectors were prepared as previously described (13) and confirmed by the Mayo Clinic Gene Analysis Shared Resource (Rochester, MN). Site-directed mutagenesis primers are listed in Supplementary Table S1.
Experimental design
DPYD variants were randomly divided into groups of 6 for functional evaluation. Each experiment consisted of a group of 6 variants tested in parallel with a positive control (wildtype DPYD) and a negative control (exon 14 deletion mimicking the *2A transcript). Experimental groups were tested in triplicate (technical replicates) with all three replicates being transfected, processed, and assayed in parallel. The results for all three technical replicates were pooled to constitute a biological replicate. At least three independent biological replicates were performed for each variant.
Cells
Low passage HEK293T/c17 cells (culture CRL-11268) were obtained from ATCC (Manassas, VA) and cultured as previously described (13). Aliquots of low passage cell stocks were prepared within two weeks of receipt. Cells were maintained in culture for no more than 10 passages or two months. Cell lines were periodically monitored for mycoplasma by Hoechst staining (Sigma-Aldrich, St. Louis, MO). Culture identity and health were monitored by microscopy. Population doubling times were determined by cell counting and compared to those for the original cell stock at time of receipt.
For transfection, cells were seeded at 106 cells per well in 6 well plates. After incubation for 16 hours, 70-80% confluent cultures were transfected with 1 μg plasmid using 3 μl X-tremeGENE HP (Roche Applied Science, Indianapolis, IN) per manufacturer’s instructions. 48 hours after transfection, cells were washed with PBS, trypsinized (0.05% Trypsin and 0.53 mM EDTA; Mediatech, Manassas, VA), pelleted, and an equivalent volume of 0.1 mm diameter glass beads (Next Advance, Averill Park, NY) added. Cells were resuspended in buffer consisting of 35 mmol L−1 potassium phosphate at pH 7.4 supplemented with 2.5 mmol L−1 MgCl2, 0.035% 2-mercaptoethanol, and Complete EDTA-free protease inhibitor cocktail (Roche). Cells were disrupted using a Bullet Blender Storm homogenizer (Next Advance) at 4° C. Total protein concentration was determined for supernatants using the BioRad Protein Assay (BioRad, Hercules, CA).
Dot blotting
Protein lysates were mixed 5:1 with reducing buffer (62.5 mmol L−1 Tris pH 6.8, 2% SDS, 5%, 2-mercapoethanol) in a final volume of 50 μl and incubated at 98° C for 10 minutes. Serial dilutions were blotted to nitrocellulose using a BioDot microfiltration apparatus (BioRad). Membranes were blocked using Odyssey blocking buffer (LI-COR, Lincoln, NE). Blots were probed with primary antibodies against DPD (ab54797, AbCam, Cambridge, MA) and alpha-tubulin (ab4074, AbCam) and subsequent secondary IRDye800CW conjugated goat anti-mouse (92632210, LI-COR) and IRDye 680 conjugated goat anti-rabbit antibodies (96268071, LICOR). Blots were scanned and dot intensities quantified using the LI-COR Odyssey Infrared Imaging system. The background signal was determined by repeating quantification using a larger diameter for each dot and subtracting the original intensity value. Relative intensity was calculated as (SdInt / SdArea) – [(LdInt – SdInt) / ( LdArea – SdArea)], where SdInt is the total intensity of the small dot, SdArea is the area of the small dot, LdInt is the total intensity of the large dot, and LdArea is the area of the large dot.
DPD enzyme activity assay
Enzyme activity was determined using a method described earlier by our lab (13). Briefly, lysates were incubated with 200 μmol L−1 NADPH (Sigma-Aldrich) and 8.2 μmol L−1 [6-C14]-5-Fluorouracil ([6-C14]-5-FU; Moravek Biochemicals, Brea, CA) for 30 minutes at 37° C. Conversion of [6-C14]-5-FU to [6-C14]-5-Dihydrofluorouracil ([6-C14]-5-DHFU) was determined using a reverse-phase C18 HPLC column (Grace, Columbia, MD) connected to a PerkinElmer Radiomatic 625TR flow scintillation analyzer (Waltham, MA). DPD activity was calculated by measuring the percent region of interest as the area under the curve for ([6-C14]-5-DHFU) / ([6-C14]-5-FU + [6-C14]-5-DHFU) using ProFSA software (PerkinElmer). Validation that ([6-C14]-5-FU and NADPH were not limiting in reactions is presented in Supplementary Figure S1.
Calculation of enzyme activity for DPD variants
DPD enzyme activity for a given variant was normalized by relative input amount of DPD as measured by dot blot. Data for each biological replicate was centered and standardized scores (Z-scores) calculated by subtracting the mean of all data points in the replicate from each individual data point and dividing by the standard deviation of all data points in the replicate. Unpaired two-tailed Student’s t-tests (assuming equal variance) were used to compare results for a given variant to the matched positive control (wildtype). For data presentation purposes, normalized results for each experiment were rescaled such that the mean of the negative control was equal to 0, and that of the positive control was equal to 1.
Amino acid alignment and protein modeling
Amino acid sequences for DPD were retrieved from NCBI for human (Homo sapiens, NP_000101.2), pig (Sus scrofa, NP_999209.1), mouse (Mus musculus, NP_740748.1), chicken (Gallus gallus, XP_426639.3), and zebrafish (Danio rerio, NP_998058.1). Multiple sequence alignment was performed using Clustal Omega version 1.2.0. Percent identities between amino acid sequences were calculated using NCBI BLASTP version 2.2.28+. Protein modeling was performed using UCSF Chimera version 1.8 (20) and Modeller version 9.12 (21). For homology modeling, the human DPD protein (NP_000101.2) was used as a query sequence and the pig crystal structure corresponding to substrate-bound DPD with closed active site loop (PDB ID 1GTH) was used as the template structure. All 3 dimensional protein images were prepared using UCSF Chimera.
Allele frequency determination
Allele frequency data was obtained from the NHLBI ESP (8) and the 1000 Genomes Project (9) databases. The NHLBI ESP dataset is comprised of whole exome sequence data from 2,203 African American and 4,300 European American individuals. 1000 Genomes Project data was from 246 individuals of West African ancestry (AFR), 181 of American ancestry (AMR), 286 of East Asian ancestry (ASN), and 379 of European ancestry (EUR). Additional details regarding the specific racial groups included in 1000 Genomes Project populations can be found on the project’s website (9).
Statistical tests
All data analyses and transformations were performed using JMP 9.0.3 (SAS Institute Inc.), unless otherwise noted. Additional tests and software algorithms used are described in relevant sections above.
Results
In silico prediction of DPD variant effects
A total of 128 missense DPYD variants were compiled from NCBI dbSNP (14), 1000 Genomes Project (9), and NHLBI ESP (8) databases. A complete list of variants and allele frequencies is presented in Supplementary Table S2. To ascertain which variants were most likely to affect enzyme activity, the PolyPhen-2 software program (15) was used to predict the impact of each amino acid substitution on the structure and function of the DPD protein. The predicted probability that a given variant is damaging to the protein is presented in Figure 1A. The estimated FPR for each variant as calculated by PolyPhen-2 is presented in Figure 1B. Of the 128 missense DPYD variants, 63 were predicted to be probably damaging (<5% estimated FPR), 15 possibly damaging (5-10% estimated FPR), and 50 benign (>10% estimated FPR). Qualitative predictions for each variant are presented in Supplementary Table S2.
Functional study of variations
Functional studies were performed to directly measure the enzyme activity of 75 transgenically expressed missense DPYD variants using a mammalian system of protein production that we described previously (13). The selected variant pool was enriched for those that had been previously reported in clinical-case control studies and/or case reports relating to 5-FU toxicity or DPD deficiency. 37 of the selected variants were predicted to be probably damaging, 7 were predicted to be possibly damaging, and 31 were predicted to be benign. Five additional variants (2 truncation, 1 frame-shift, 1 in-frame insertion, and 1 somatic missense mutation) were also studied. Enzyme activity was corrected for differences in expression as determined by quantitative dot blot (Fig. 2A-B), which yielded comparable results to western blotting (data not shown). A comparative summary of DPD enzyme activities for selected variants is presented in Figure 2C-2F; numerical values and statistics for missense variants are also detailed in Supplementary Table S2.
Figure 2. In vitro enzyme activity of DPD variants.

Dot blots were used to calculate relative expression of DPD (A) and alpha tubulin (B). Relative DPD enzyme activity was determined for selected variants predicted to be probably damaging (C), possibly damaging (D), and benign (E). F, enzyme activity was determined for R21X, E386X, P633[FS], F100[FS], and the somatic mutation G252V. For panels C-F, each “x” represents a single biological replicate, which was the average of 3 technical replicates. The mean of biological replicates is presented as a horizontal bar ± standard deviation. The mean relative activity and standard deviation for wildtype DPD are presented as horizontal gray and dashed lines, respectively. Variants with activities that are significantly different from wildtype are color coded according to the scale on the right: red, *2A-like (<12.5% activity); yellow, I560S-like (12.5%-25% activity); green, D949V-like (>25% activity); and blue, significantly greater than wildtype.
Within the probably damaging group of missense variants, 19 (51%) showed significantly decreased enzyme activity compared to wildtype DPD (Fig. 2C). Of these, 8 variants had little to no residual enzymatic activity (<12.5%), including K958E (P=1.3×10−5), S201R (P=3.0×10−7), V995F (P=6.9×10−5), G593R (P=3.9×10−5), G880V (P=3.5×10−4), G764D (P=5.7×10−9), H978R (P=9.6×10−4), and R592W (P=4.0×10−5), and were thus classified as *2A-like (Fig. 2C). Dramatic reductions in activity (12.5-25% of wildtype, similar to our previous report for I560S) (13) were noted for 6 variants: R235W (P=9.2×10−7), Y211C (P=0.0021), D495G (P=0.0017), R592Q (P=2.6×10−5), D342N (P=1.9×10−6), and S492L (P=2.3×10−4; Fig. 2C). D949V, which had previously not been tested using this assay, showed significantly decreased enzyme function compared to wildtype (59% activity, P=0.0031; Fig. 2C). Four additional variants had enzyme activities that were significantly lower than wildtype but greater than 25%: T760I (P=0.0035), P92A (P=0.045), Y304H (P=0.026), and Y186C (P=0.027; Fig. 2C). One variant, M166V, had significantly higher enzyme activity than wildtype DPD (120% activity, P=0.025; Fig. 2C). The highest average activity in this group was noted for L310S (133%), however this result was not significantly different from wildtype due to high variability (P=0.076; Fig. 2C).
Fewer variants showed significant reductions in enzyme activity in the possibly damaging and benign prediction groups. Of the 7 possibly damaging variants, 2 (29%) had significantly impaired enzyme function (Fig. 2D). F438L retained 47% of enzymatic activity (P=0.0042) and D687A was 75% active (P=0.011; Fig. 2D). Of the 31 amino acid changes predicted to not affect function, 5 (16%) had enzyme activities significantly lower than wildtype (Fig. 2E). R353C completely lacked enzymatic activity (P=2.0×10−5; Fig. 2E). The enzyme activities of K290E, T983I, L352V, and N893S were 45%, 73%, 77%, and 80%, respectively (P=0.0019, P=0.023, P=0.0085, and P=0.018; Fig. 2E). Three variants in the predicted benign group showed significantly higher enzyme activities than wildtype DPD. E828K was 16% hyperactive (P=0.049), K861R was 30% hyperactive (P=0.0077), and P1023T was 38% hyperactive (P=0.048; Fig. 2E).
We tested 4 additional variants that affected more than 1 amino acid. These included the truncation variants R21X (rs72549310) and E386X (rs78060119), the P633[FS] frame-shift variation (rs72549303), and the F100[FS] in-frame 3-nucleotide insertion (rs72549301). Additionally, G252V (NM_000101.2-c.755G>T, COSM74430), a somatic missense mutation contained in the Catalogue of Somatic Mutations in Cancer database (22, 23) that was originally identified in an ovarian tumor specimen analyzed as part of The Cancer Genome Atlas project (24), was also assayed. As expected, neither truncation variant, R21X or E386X, yielded detectible enzyme activity (Fig. 2F). Protein fragments corresponding to the predicted sizes were detectible for both variants by western blotting using an anti-DPD antibody, however both were expressed at lower levels than wildtype DPD (data not shown). Both P633[FS] and F100[FS] also lacked detectible activity (Fig. 2F). The enzyme activity of the somatic G252V mutation was significantly lower than wildtype DPD (51% activity, P=1.2×10−3; Fig. 2F) suggesting that de novo tumor mutations can affect catabolism of 5-FU within cancerous cells.
Performance of in silico prediction tools
To assess the utility of in silico tools for predicting the effects of missense DPYD variations on enzyme activity, functional predictions were compared to the actual enzyme activity results for the DPYD variants tested (Supplementary Table S3). PolyPhen-2 trained with the HumDiv dataset showed a sensitivity of 81% and a specificity of 53%. Tradeoffs between sensitivity and specificity, as well as between negative predictive value (NPV) and positive predictive value (PPV), are noted when low confidence possibly damaging predictions were treated as benign predictions and/or the threshold used to classify variants based on enzyme activity was adjusted (Supplementary Table S3). Overall, balanced accuracy was highest when low confidence predictions were excluded from the deleterious set and only variants with <25% activity were considered deficient. It is notable that while these adjustments to the prediction criteria generally increased sensitivity, specificity, and NPV, as a consequence, PPV was reduced to 38%. Similar performance was noted for PolyPhen-2 trained with the HumVar dataset, and a comparison with predictions from the PROVEAN (17), SIFT (18), and SNAP (19) software programs is also presented (Supplemental Table S3).
Modeling the human DPD protein structure
DPD is highly conserved through vertebrates, with 93% identity between the pig and human amino acid sequences (Fig. 3). The crystal structure of human DPD has not been solved, however the structure of pig DPD has been reported (25). To identify structural elements that may be disrupted in dysfunctional DPD variants, we generated a theoretical homology model of human DPD using the crystal structure of pig DPD with NADPH and 5-iodouracil as a template. The predicted human structure contained 47 alpha helices and 31 beta sheets (Fig. 3). Of the 27 variants with reduced enzyme activity (Fig. 2C-2F), 14 were located in predicted secondary structural elements (Fig. 3). 11 deficient variants were located in alpha helical domains (Y186C, S201R, D342N, L352V, R353C, S492L, D495G, G880V, N893S, K958E, and V995F), and 3 deficient variants were in beta sheets (Y304H, R592Q, and R592W).
Figure 3. Location of variants relative to structural features of DPD.

An alignment of vertebrate DPD amino acid sequences was prepared using Clustal Omega. Sequence consensus is denoted below the alignment (asterisk, fully conserved residue; colon, strongly similar properties; period, weakly similar properties; no symbol, dissimilar amino acids). Predicted secondary structural features are indicated by colored bars above the alignment (red, alpha helices; blue, beta turns). Differences between the predicted human structure and the published pig structure are denoted by horizontal (not present in predicted structure) and vertical (not present in published structure) white lines overlaid on the secondary structural prediction. Boxes indicate functional elements (4Fe-4S, iron-sulfur coordinating domains; FAD, FAD binding domain; NADP, NADP binding domain; FMN, FMN binding domain; UBC, uracil binding domain; ASL, active site loop; F, FAD interacting residue; N, NADP interacting residue; F2, FMN interacting residue; PA, pyrimidine associated residue). Amino acid changes that significantly reduced enzyme activity are colored as described for Figure 2. In instances where multiple amino acid variations have been reported for a given residue (i.e. R592W and R592Q), the color indicates the more severe phenotype.
Location of deficient variants on the DPD structure
The DPD monomer consists of five distinct structural domains, each of which contains a subset of the prosthetic groups and co-factors necessary for enzyme function (26). Domain I contains two iron-sulfur (Fe-S) clusters (residues 27-172). The P92A variant was shown to be located at a conserved residue adjacent to the Fe-S coordinating cysteine at position 91 (Fig. 3), and constituted the only deleterious variant located in this domain. Domain II (residues 173-286 and 442-524) and domain III (residues 287-441) are closely intertwined and bind FAD and NADPH, respectively (Fig. 4B). 13 of 29 variations tested in the combined domains II and III significantly impaired enzyme function. R235W disrupts a residue important for FAD binding (Fig. 3). D342N and F438L are both located within NADPH coordinating regions (Fig. 3 and 4B). Domain IV contains the uracil-binding site and consists of residues 525-847 (Fig. 4C). G674D is located within the active loop structure, and D687A is located adjacent to the loop (Fig. 3). In all, 4 of 18 variant tested in this region had significantly reduced activity. Domain V (residues 1-26 and 848-1025) contains two additional Fe-S clusters (Fig. 4D). 7 of 16 variants in this region were deficient. Notably, K958E, H978R, and V995F are all located within Fe-S cluster coordinating domains (Fig. 3).
Figure 4. Location of deficient variants relative to major structural domains of DPD.

The position of damaging amino acid variants are depicted within the N-terminal Fe-S cluster containing alpha-helical domain I (A), the FAD binding domain II and NADPH binding domain III (B), the pyrimidine binding domain IV (C), and the C terminal Fe-S cluster containing domain V (D). Deleterious amino acids changes are colored as described for Figure 2. The position of I560S, which was not evaluated in this study, is shown in panel C for the benefit of the reader.
Allele frequencies of deficient alleles
Allele frequencies for deficient variants were obtained from the NHLBI ESP (8) and the 1000 Genomes Project (9) databases and are summarized in Figure 5 (full data is presented in Supplementary Table S2). Within the NHLBI ESP dataset, the allele frequencies for the 5-FU toxicity-associated DPYD variants *2A, I560S, and D949V were 0.09%, 0.00%, and 0.09%, respectively, in African American individuals. Collectively, variants showing less than 12.5% enzyme activity (denoted as *2A-like) had an allele frequency of 0.43% in the African American population. The additive allele frequencies for I560S-like and D949V-like variants were 0.02% and 2.22%, respectively, suggesting that the newly classified variants may be significant contributors to DPD deficiency in African Americans. Within the European American cohort of the NHLBI ESP dataset, the additive allele frequency of newly classified deficient variants was 0.08%, whereas the frequency of *2A, I560S, and D949V was 0.58%, 0.06%, and 0.54%, respectively.
Figure 5. Frequencies of deleterious variants in publicly available population data.
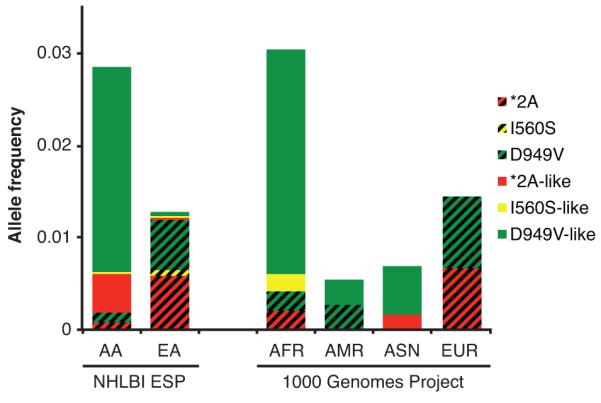
Allele frequency data is presented for the NHLBI ESP African American (AA) and European American (EU) populations, and for the 1000 Genomes populations of West African (AFR), American (AMR), East Asian (ASN), and European (EUR) ancestry. The allele frequencies for *2A, I560S, and D949V are presented as red, yellow, and green bars containing diagonal black lines. Additional variants that resulted in significant reductions in DPD enzyme activity are clustered by similarity to known deficient variants (as depicted in Fig. 2) and are represented as solid colored bars.
While the 1000 Genomes Project database has information from fewer individuals than the NHLBI ESP dataset, it contains genotyping information from additional racial groups. Within the AFR 1000 Genomes population, the addition of newly classified variants increases the cumulative allele frequency of deleterious variants to 2.64%, up from 0.41% when only *2A, I560S, and D949V were considered (Fig. 5). The allele frequency of deleterious variants in the AMR population doubled from 0.28% to 0.56% when newly classified variants were added. *2A, I560S, or D949V were not detected in the ASN population; however, the allele frequencies of newly classified *2A-like and D949V-like variants were 0.17% and 0.52%, respectively. Lastly, it was noted that the EUR population did not carry any of the newly identified deficient DPYD variants.
Discussion
128 missense variants have been reported within the DPYD gene, which alter 119 (12%) of the 1,025 amino acids that comprise DPD (in 9 instances, variations affect the same codon). Relatively few of these variants have been evaluated in the clinical context, and far fewer have been directly studied using functional tests. Three DPYD variants (*2A, I560S, and D949V) are generally considered to be deficiency-associated alleles, whereas many of the remaining variants have been dismissed as being too low-penetrance to be of pharmacological importance. Contrary to this opinion, our results indicate that rare DPYD variants that perturb function of the translated DPD protein are collectively present at sufficiently high frequencies in non-European populations to be considered as candidate risk alleles for developing severe adverse toxicity to 5-FU-based treatments.
The extensively studied *2A, I560S, and D949V variants are most likely to be detected in individuals of European ancestry, but are far less penetrant in other racial groups (Fig. 5). Using a system of in vitro DPD enzyme activity measurement, we systematically classified 80 reported variations in DPYD that alter the amino acid sequence of the encoded protein. We show that the enzyme activity of D949V is significantly impaired, but not to the extent observed previously for *2A or I560S (13). 30 of the additional variants tested in the present study were shown to be deficient; 19 of which were severely deficient with enzyme activities similar to those of *2A or I560S (less than 25% of wildtype, Fig. 3). Based on publicly available allele frequency data, these deficient variants are expected to be exceedingly rare or non-existent in European populations, but are collectively carried by an appreciable fraction of non-European individuals (Fig. 5). For instance, the cumulative allele frequency for *2A, I560S, and D949V in the ESP African American population was less than 0.2%. In contrast, the cumulative allele frequency of the newly classified deficient variants was approximately 2.7% in that population. Additionally, within the ASN population of the 1000 Genomes Project, no individuals carried *2A, I560S, or D949V; however, newly classified deficient variants had an additive allele frequency of 0.7% (Fig. 5). While the population sizes for racial groups in the 1000 Genomes Project are relatively small, these findings suggest that the newly classified deficient variants may account for a significant fraction of DPD deficiency, particularly in individuals with non-European ancestry. Additional studies to ascertain the allele frequencies of DPYD variants in other under-represented populations are underway in our laboratory.
Recently, we examined the enzyme activity of common DPYD variants, C29R, I543V, S534N, and V732I (13). None of these variants showed decreased enzymatic activity relative to wildtype DPD. Surprisingly though, C29R and S534N showed increased 5-FU catabolism, establishing a new class of hyperactive DPYD variations. In the present study, 4 additional variants had significantly higher enzyme activity than wildtype: M166V, E828K, K861R, and P1023T. M166V was initially reported in two individuals with partial DPD deficiencies, one of whom also carried D949V (27). A subsequent study of M166V in an extended family containing DPD deficiency showed that the variant did not contribute to the disorder (6). One clinical case-control study with a limited number of patients suggested that M166V may contribute to 5-FU related toxicities in gastroesophageal and breast cancers, but not in colorectal cancer (10). Three additional clinical association studies failed to establish any link between M166V and 5-FU-related toxicities (4, 11, 28). A fourth study suggested that M166V may protect against hematological toxicity and neutropenia following administration of 5-FU; however, the association was observed only in women (12). The P1023T variant is rare in European populations and is more common in individuals of African ancestry (Supplementary Table S2). The hyperactive phenotype of P1023T is supported by a previous study from our lab in which individuals carrying P1023T, but not the deficiency allele Y186C, had 22% higher DPD activity than those that did not carry P1023T (5). To our knowledge the hyperactive variants E828K and K861R have not been reported in any clinical or case studies.
Numerous case reports support our findings for many of the variants classified as deficient in this study. In early studies of severe DPD deficiency in children with delayed motor skills development, van Kuilenburg and colleagues identified H978R (29) and subsequently S201R, Y211C, and S492L (30) as potential contributors to DPD deficiency. The truncation variant E386X was reported in a DPD deficient Japanese individual (31), and R592W was detected in a Korean individual who experienced grade 4 toxicity after receiving 5-FU (32). R21X was detected in an individual also carrying *2A who experienced severe toxicity and died 21 days following 5-FU treatment (33). R235W and the frame-shift variant P633[FS] were detected in a compound heterozygous state in a severely DPD deficient individual (34). V995F was likewise reported in an individual with DPD deficiency (35).
In a previous report, we showed that in a cohort of healthy African American individuals, DPD enzyme activity in peripheral blood mononuclear cells (PBMCs) from Y186C carriers was 46% lower than that in non-carriers (P=2.9×10−4), suggesting that the amino acid change impaired the catalytic activity of the enzyme (5). In the present manuscript, we observed that DPD containing the Y186C amino acid substitution was reduced by approximately 15% relative to wildtype. This reduction is less than we expected based on the data from healthy volunteers, but is similar to a previously reported study focused on this variant (36). We hypothesized that Y186C may affect dimerization, as the residue is located on the monomer surface, and previous reports have suggested that tyrosine to cysteine substitutions could cause aberrant dimer crosslinking (37). Multiple additional missense variants have been detected in individuals carrying Y186C (5); the potential implication of co-expressed DPYD variants to overall DPD activity needs further clarification.
To date, predictive tests of 5-FU toxicity have had limited value, since carriers of known DPD deficiency-associated alleles (*2A, I560S, and D949V) constitute a relatively small percentage of toxicity cases (38). In a previous study, we confirmed that *2A and I560S were deleterious DPYD variants with dramatically impaired DPD enzyme activity (13). In the present study, we confirmed D949V as a deleterious variant, and presented comparative data showing that at least 30 additional DPYD variants impair DPD function. Some of these variants have been documented in case reports; however, the rarity of these alleles has prevented the determination of their statistical significance as predictive markers of 5-FU toxicity. The results presented in this paper should address that gap and provide guidance for the individualization of 5-FU therapy for carriers of rare, damaging DPYD variants. Our findings also highlight the importance of performing genetic analyses that are unbiased by previous studies conducted in populations of limited diversity.
Supplementary Material
Acknowledgements
The authors wish to thank the Mayo Clinic Center for Clinical and Translational Science (supported by NCATS UL1 TR000135) for statistical support. We also thank the Mayo Clinic Cancer Center Gene Analysis Shared Resource (supported by NCI 5P30 CA15083-37) for sequencing services.
Financial support: This study was funded by NIH grant CA116964 (R.B.D.). S.M.O. is funded in part by a grant from the Mayo Clinic Center for Individualized Medicine.
Footnotes
Conflict of interest statement: The authors have no potential conflicts of interest to declare.
References
- 1.Meinsma R, Fernandez-Salguero P, Van Kuilenburg AB, Van Gennip AH, Gonzalez FJ. Human polymorphism in drug metabolism: mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol. 1995;14:1–6. doi: 10.1089/dna.1995.14.1. [DOI] [PubMed] [Google Scholar]
- 2.van Kuilenburg AB, Hausler P, Schalhorn A, Tanck MW, Proost JH, Terborg C, et al. Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.1905+1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clinical pharmacokinetics. 2012;51:163–74. doi: 10.1007/BF03257473. [DOI] [PubMed] [Google Scholar]
- 3.Johnson MR, Hageboutros A, Wang K, High L, Smith JB, Diasio RB. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res. 1999;5:2006–11. [PubMed] [Google Scholar]
- 4.Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J, et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131–8. doi: 10.1200/JCO.2006.10.4182. [DOI] [PubMed] [Google Scholar]
- 5.Offer SM, Lee AM, Mattison LK, Fossum C, Wegner NJ, Diasio RB. A DPYD Variant (Y186C) in Individuals of African Ancestry Is Associated With Reduced DPD Enzyme Activity. Clinical pharmacology and therapeutics. 2013;94:158–66. doi: 10.1038/clpt.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson MR, Wang K, Diasio RB. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin Cancer Res. 2002;8:768–74. [PubMed] [Google Scholar]
- 7.Morel A, Boisdron-Celle M, Fey L, Soulie P, Craipeau MC, Traore S, et al. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol Cancer Ther. 2006;5:2895–904. doi: 10.1158/1535-7163.MCT-06-0327. [DOI] [PubMed] [Google Scholar]
- 8.Exome Variant Server . NHLBI GO Exome Sequencing Project (ESP); Seattle (WA): [cited 2013 April 25]. (EVS data release ESP6500SI-V2) Available from: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 9.The 1000 Genomes Browser . The European Bioinformatics Institute (EBI); Hinxton, Cambridgeshire (UK): [cited 2013 April 25]. (1000 Genomes release 13) Available from: http://browser.1000genomes.org/index.html. [Google Scholar]
- 10.Gross E, Busse B, Riemenschneider M, Neubauer S, Seck K, Klein HG, et al. Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PloS one. 2008;3:e4003. doi: 10.1371/journal.pone.0004003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amstutz U, Farese S, Aebi S, Largiader CR. Dihydropyrimidine dehydrogenase gene variation and severe 5-fluorouracil toxicity: a haplotype assessment. Pharmacogenomics. 2009;10:931–44. doi: 10.2217/pgs.09.28. [DOI] [PubMed] [Google Scholar]
- 12.Kleibl Z, Fidlerova J, Kleiblova P, Kormunda S, Bilek M, Bouskova K, et al. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapy. Neoplasma. 2009;56:303–16. doi: 10.4149/neo_2009_04_303. [DOI] [PubMed] [Google Scholar]
- 13.Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73:1958–68. doi: 10.1158/0008-5472.CAN-12-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Database of Single Nucleotide Polymorphisms (dbSNP) National Center for Biotechnology Information, National Library of Medicine; Bethesda (MD): [cited 2013 April 25]. (dbSNP Build ID: 138) Available from: http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?showRare=on&chooseRs=coding&go=Go&locusId=1806. [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 doi: 10.1002/0471142905.hg0720s76. Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 19.Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35:3823–35. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 21.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of molecular biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 22.Catalogue of Somatic Mutations in Cancer (COSMIC) Cancer Genome Project, Wellcome Trust Sanger Institute; Hinxton, Cambridgeshire (UK): [cited 2013 April 25]. (COSMIC v64 release) Available from: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/ [Google Scholar]
- 23.Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91:355–8. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobritzsch D, Ricagno S, Schneider G, Schnackerz KD, Lindqvist Y. Crystal structure of the productive ternary complex of dihydropyrimidine dehydrogenase with NADPH and 5-iodouracil. Implications for mechanism of inhibition and electron transfer. J Biol Chem. 2002;277:13155–66. doi: 10.1074/jbc.M111877200. [DOI] [PubMed] [Google Scholar]
- 26.Dobritzsch D, Schneider G, Schnackerz KD, Lindqvist Y. Crystal structure of dihydropyrimidine dehydrogenase, a major determinant of the pharmacokinetics of the anti-cancer drug 5-fluorouracil. EMBO J. 2001;20:650–60. doi: 10.1093/emboj/20.4.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Kuilenburg AB, Haasjes J, Richel DJ, Zoetekouw L, Van Lenthe H, De Abreu RA, et al. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res. 2000;6:4705–12. [PubMed] [Google Scholar]
- 28.Deenen MJ, Tol J, Burylo AM, Doodeman VD, de Boer A, Vincent A, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17:3455–68. doi: 10.1158/1078-0432.CCR-10-2209. [DOI] [PubMed] [Google Scholar]
- 29.van Kuilenburg AB, Haasjes J, Meinsma R, Waterham HR, Vreken P, van Gennip AH. Dihydropyrimidine dehydrogenase (DPD) deficiency: novel mutations in the DPD gene. Adv Exp Med Biol. 2000;486:247–50. doi: 10.1007/0-306-46843-3_48. [DOI] [PubMed] [Google Scholar]
- 30.van Kuilenburg AB, Dobritzsch D, Meinsma R, Haasjes J, Waterham HR, Nowaczyk MJ, et al. Novel disease-causing mutations in the dihydropyrimidine dehydrogenase gene interpreted by analysis of the three-dimensional protein structure. Biochem J. 2002;364:157–63. doi: 10.1042/bj3640157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kouwaki M, Hamajima N, Sumi S, Nonaka M, Sasaki M, Dobashi K, et al. Identification of novel mutations in the dihydropyrimidine dehydrogenase gene in a Japanese patient with 5-fluorouracil toxicity. Clin Cancer Res. 1998;4:2999–3004. [PubMed] [Google Scholar]
- 32.Cho HJ, Park YS, Kang WK, Kim JW, Lee SY. Thymidylate synthase (TYMS) and dihydropyrimidine dehydrogenase (DPYD) polymorphisms in the Korean population for prediction of 5-fluorouracil-associated toxicity. Ther Drug Monit. 2007;29:190–6. doi: 10.1097/FTD.0b013e318040b1fe. [DOI] [PubMed] [Google Scholar]
- 33.van Kuilenburg AB, Baars JW, Meinsma R, van Gennip AH. Lethal 5-fluorouracil toxicity associated with a novel mutation in the dihydropyrimidine dehydrogenase gene. Ann Oncol. 2003;14:341–2. doi: 10.1093/annonc/mdg056. [DOI] [PubMed] [Google Scholar]
- 34.Vreken P, Van Kuilenburg AB, Meinsma R, van Gennip AH. Dihydropyrimidine dehydrogenase (DPD) deficiency: identification and expression of missense mutations C29R, R886H and R235W. Hum Genet. 1997;101:333–8. doi: 10.1007/s004390050637. [DOI] [PubMed] [Google Scholar]
- 35.Vreken P, van Kuilenburg AB, Meinsma R, van Gennip AH. Dihydropyrimidine dehydrogenase deficiency. Identification of two novel mutations and expression of missense mutations in E. coli. Adv Exp Med Biol. 1998;431:341–6. [PubMed] [Google Scholar]
- 36.Offer SM, Diasio RB. Response to “A Case of 5-FU-Related Severe Toxicity Associated With the P.Y186C DPYD Variant”. Clinical pharmacology and therapeutics. 2013 doi: 10.1038/clpt.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W, Freed CR. Tyrosine-to-cysteine modification of human alpha-synuclein enhances protein aggregation and cellular toxicity. J Biol Chem. 2004;279:10128–35. doi: 10.1074/jbc.M307563200. [DOI] [PubMed] [Google Scholar]
- 38.Caudle KE, Thorn CF, Klein TE, Swen JJ, McLeod HL, Diasio RB, et al. Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clinical pharmacology and therapeutics. 2013;94:640–5. doi: 10.1038/clpt.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.