

Abstract
Alterations of DNA methylation occur during the course of both stem cell development and tumorigenesis. We present a novel strategy that can be used to stratify glioblastoma multiforme (GBM) patients through the epigenetic states of genes associated with human embryonic stem cell (hESC) identity in order to 1) assess linkages between the methylation signatures of these stem cell genes and survival of GBM patients, and 2) delineate putative mechanisms leading to poor prognosis in some patient subgroups. A DNA methylation signature was established for stratifying GBM patients into several hESC methylator subgroups. The hESC methylator-negative phenotype has demonstrated poor survival and upregulation of glioma stem cell (GSC) markers, and is enriched in one of the previously defined transcriptomic phenotypes—the mesenchymal phenotype. We further identified a refined signature of 36 genes as the gene panel, including SOX2, POU3F2, FGFR2, GAP43, NTRK2, NTRK3, and NKX2-2, which are highly enriched in the nervous system. Both signatures outperformed the O6-methylguanine-DNA methyltransferase (MGMT) methylation test in predicting patient's outcome. These findings were also validated through an independent dataset of patients. Furthermore, through statistical analyses, both signatures were examined significantly. Hypomethylation of hESC-associated genes predicted poorer clinical outcome in GBM, supporting the idea that epigenetic activation of stem cell genes contributes to GBM aggression. The gene panel presented herein may be developed into clinical assays for patient stratification and future personalized medicine interventions.
Introduction
DNA cytosine methylation plays an essential role in genome regulation, development, and disease (Holliday and Pugh, 1975; Riggs, 1975). Feinberg and Vogelstein (1983) reported that alterations in DNA methylation occurred in cancer, including hypomethylation of oncogenes and hypermethylation of tumor suppressor genes (Feinberg and Vogelstein, 1983). Prior studies focused mainly on specific genes-of-interest and regions assumed to be functionally important, such as promoters and CpG islands (Baylin and Ohm, 2006; Feinberg and Tycko, 2004). Currently, methylation changes are generally understood to occur in cancerous tissues concurrently with changes in normal tissue differentiation. This is consistent with the epigenetic progenitor model of cancer, which proposes that epigenetic alterations affecting tissue-specific differentiation are the predominant mechanism causing cancer (Feinberg et al., 2006; Irizarry et al., 2009). Recent genome-wide studies have demonstrated distinct patterns of DNA methylation occur in cancerous tissues compared with their normal counterparts (Figueroa et al., 2009; Noushmehr et al., 2010; Ordway et al., 2007; Rauch et al., 2008). Toyota et al. (1999) first described a CpG island methylator phenotype (CIMP) as a cancer-specific CpG island hypermethylation of a subset of genes in a subpopulation of colorectal cancer patients (Toyota et al., 1999). Despite the central role of DNA methylation alterations, which is related to both stem cells and cancer, the correlation of clinical phenotypes with methylation status of genes specific to human embryonic stem cell (hESC) have not been systematically analyzed.
Cancer stem cells (CSCs, a.k.a. tumor initiating cells) are believed to have greater potential of cancer initiation and repopulation. CSCs in malignant gliomas (e.g., glioblastoma stem cells, GSC) have been identified and characterized by several researchers (Bao et al., 2006a, 2006b, 2008; Galli et al., 2004; Hemmati et al., 2003; Lee et al., 2006a; Li et al., 2009; Singh et al., 2004). These studies demonstrated that GSCs displayed much greater tumorigenic potential than matched non-stem tumor cells when xenotransplanted into the brain of immunocompromised rodents (Bao et al., 2006a; Calabrese et al., 2007; Galli et al., 2004; Hemmati et al., 2003; Lee et al., 2006a; Li et al., 2009; Singh et al., 2004). The developmental hierarchy of GSCs may be regulated at both genetic and epigenetic levels and may contribute to the heterogeneity of GBM cell populations. Hence, we hypothesize that the epigenetic status of stem cell-related genes constitutes an excellent biomarker that could be successfully exploited to stratify and characterize GBMs.
Several recent studies employed large-scale mRNA expression profiling on GBM (Gravendeel et al., 2009; Murat et al., 2008; The Cancer Genome Atlas Research Network 2008; Parsons et al., 2008; Phillips et al., 2006) have defined four molecular subtypes of GBM—proneural, neural, classical, and mesenchymal (Verhaak et al., 2010). Relatively few studies have investigated global epigenetic alterations in GBM. The TCGA project conducted DNA methylation profiling of 272 GBM tumor samples and identified a distinct GBM CpG island methylator phenotype (G-CIMP) with hypermethylation at a large number of loci (Noushmehr et al., 2010). Crossed comparison with the four transcriptomic subtypes showed significant overlaps of the G-CIMP-positive population with the proneural GBM subtype and the G-CIMP-negative population with the classical and mesenchymal GBM subtypes. Thus, the G-CIMP-positive subgroup of patients represented a subclass within the proneural population that presented a better prognostic outcome, was tightly associated with IDH1 somatic mutations, and displayed a distinctive profile of copy-number alterations. Moreover, an eight-gene methylation signature has been identified with the proneural G-CIMP-positive group more frequently in lower grade (WHO grade II/III) gliomas associated with IDH1 mutations and has better clinical outcomes based on an independent dataset. However, the molecular mechanism underlying the distinct survival differences among these subgroups of patients remains uncertain; a clinical need exists to develop a method to identify the more aggressive subgroup of GBM for impedance match with therapy. The current study investigated whether the methylation and gene expression status of stem cell-related genes contribute to the different methylator phenotypes of GBM.
We hypothesize that methylation states of stem cell-related genes may provide clues for undrstanding the stem cell origin of cancer, and for predicting clinical outcomes. We present here a novel strategy that can be used to stratify GBM patients using the epigenetic states of genes associated with hESC identity to 1) assess linkages between the methylation signatures of these stem cell genes and the survival of GBM patients, and 2) delineate possible mechanisms leading to the poor prognosis observed in some subgroups of patients.
Methods
Study population
DNA methylation and gene expression profiling data were obtained from the TCGA website. The training dataset contained 181 tumor samples and three controls. DNA methylation data was generated on the HumanMethylation27 BeadChip (Illumina, Inc.) to include 27,578 CpG dinucleotides spanning ∼14,000 genes. The probe information was available on the Illumina website, whereas the clinical information was downloaded from the TCGA Data Portal. An independent dataset of 71 tumor samples used to validate the analysis was also obtained from the TCGA Data Portal, which used the Infinium HumanMethylation450 platform (Illumina, Inc.) to assess methylation status of more than 480,000 cytosines distributed over the entire genome.
Generation of hESC-related gene sets
We compiled a hESC-related gene panel as previously reported (Ben-Porath et al., 2008; Sperger et al., 2003) including ESC overexpressed genes (Assou et al., 2007), Nanog, Oct4, and Sox2 targets (Boyer et al., 2005). Polycomb targets in hESCs (Lee et al., 2006b) and Myc targets (Fernandez et al., 2003; Li et al., 2003) were also used for subsequent analysis. This hESC-specific gene panel is enriched in poorly differentiated tumors (Ben-Porath et al., 2008). We limited our primary analysis to the common gene set between this hESC dataset and the Infinium platform—3,800 genes in total—for subsequent analysis.
Statistical analysis
Kaplan-Meier analysis was used to generate survival curves and log-rank test to determine univariate differences between phenotypes. Bootstrapping was used to evaluate the robustness of our model: two groups of samples, representing 25 and 15 patients—the numbers of samples grouped in the hESC methylator-negative and -positive phenotype, respectively—were resampled from patients of training data with the original features. F-score was used for measuring the performance for each of the 1000 resampling sets. To determine the significance of the gene panel further, a hypothesis was established that assumed the 36 randomly selected genes could distinguish the two phenotypes significantly in the same way as the identified gene panel did by evaluating their log-rank test; a t-test was used for examining the significance of the gene panel when compared to the 36 randomly selected genes. We randomly resampled 36 genes from the hESC-related genes for 1000 iterations to generate a p value. Finally, the performances of the classifier were further assessed by using receiver operating characteristic (ROC) curves and calculating the area under curve (AUC). The AUC in our study was an added measure of how the consistent features and the gene panel can distinguish between two diagnostic groups (hESC methylator-positive/hESC methylator-negative). The ROC curve is an excellent tool for use in machine learning and data mining research. Also, the ROC curve is a basic tool used for the performance of the diagnosis of a test or the ability and accuracy of a test to discriminate between two states, such as “normal” vs. “abnormal” or “positive” vs. “negative” case (Metz, 1978; Zweig and Campbell, 1993). Here, ROC curves and AUC were used for comparing the performance of the features and the gene panel clustered patients' consistency. In a ROC curve, the true positive rate (Sensitivity) was plotted as a function of the false positive rate (100-Specificity) for different classified performances of biomarker candidates.
Results
Identification of distinct DNA methylation subtypes
We analyzed the data to determine whether the methylation status of hESC-related genes can stratify GBM patients and provide clues for understanding GBM pathogenesis. In this study, a strategy was devised to correlate the DNA methylation profiles with clinical information. We first applied Pearson's Correlation Coefficient through DNA methylation intensity of the hESC-related genes to measure the differences shown in the patient samples when compared to data from nontumor controls. We then selected the three most normal-like GBM samples (surrogate 1) and the three most normal-deviant samples (surrogate 2) (Fig. 1).
FIG. 1.

The process for surrogate samples extraction. Pearson Correlation Coefficient was used to obtain two kinds of typical surrogate samples.
The signature genes were extracted from the hESC-related genes that separated the two surrogate groups most effectively. The methylation level (beta value) at each locus was determined to be between 0 and 1, based on the proportion of the methylated vs. unmethylated probes in the Infinium Methylation Assay platform. When compared to the gene methylation status of control samples, the beta value of the surrogate tumor samples with an opposite value would be selected (beta value of 0.5 was used to be the reference). This condition ensures that the hypermethylated genes in surrogate 1 are hypomethylated in the control samples (and vice versa) in order to be considered as candidates for subsequent clustering analysis. After setting the beta difference between surrogate 1 and the control samples to be greater than 0.4, a set of 250 genes was obtained. This set of genes was referred to as features and was used to assign tumors to subclasses via two-step k-means clustering analysis.
Two distinct groups showing the most self-similarity containing their corresponding three surrogate patients were first clustered; each was designated as belonging to either the hESC methylator-negative or -positive group. Each hESC methylator group was then further clustered into three subgroups after making k equal to three. Six hESC methylation clusters were identified with the two sets of three surrogate samples each clustered in distinct subgroups—25 samples were grouped together as the hESC methylator-negative phenotype and 15 samples were grouped together as the hESC methylator-positive phenotype (Fig. 2). Table 1 provides the clinical characteristics of the patients whose data were used in the analysis. Kaplan-Meier plot demonstrated the hESC methylator-positive patients had significantly more favorable outcomes than the hESC methylator-negative patients (Fig. 3a).
FIG. 2.

250 features were obtained from the proposed method to identify six DNA methylation subtypes. The control samples are included and contribute to the unsupervised clustering. Samples of each cluster are labeled to be consistent to the previously defined gene expression clusters (proneural, neural, classical, and mesenchymal) (Verhaak et al., 2010).
Table 1.
Characteristics of Patients Whose Data Were Used in the Analysis
| hESC methylator-negative | hESC methylator-positive | Total | |
|---|---|---|---|
| No. of patients | 25 | 15 | 40 |
| TCGA patient phenotype | |||
| age, years | |||
| Median (LQ, UQ) | 56.3 (51.3, 67.0) | 32 (28.4, 38.8) | 53.6 (41.8, 58.3) |
| No. ≤40 years old | 1 | 12 | 13 |
| Survival (weeks) | |||
| Mediana (CI) | 44.0 (28.6, 50.7) | 141.0 (80.4, 325.1) | 60.5 (44.7, 90.0) |
| Sex | |||
| Female | 18 | 8 | 26 |
| Male | 7 | 7 | 14 |
CI, confidence interval; LQ, lower quartile; UQ, upper quartile.
Median survival and corresponding confidence intervals were estimated from the Kaplan-Meier curve
FIG. 3.

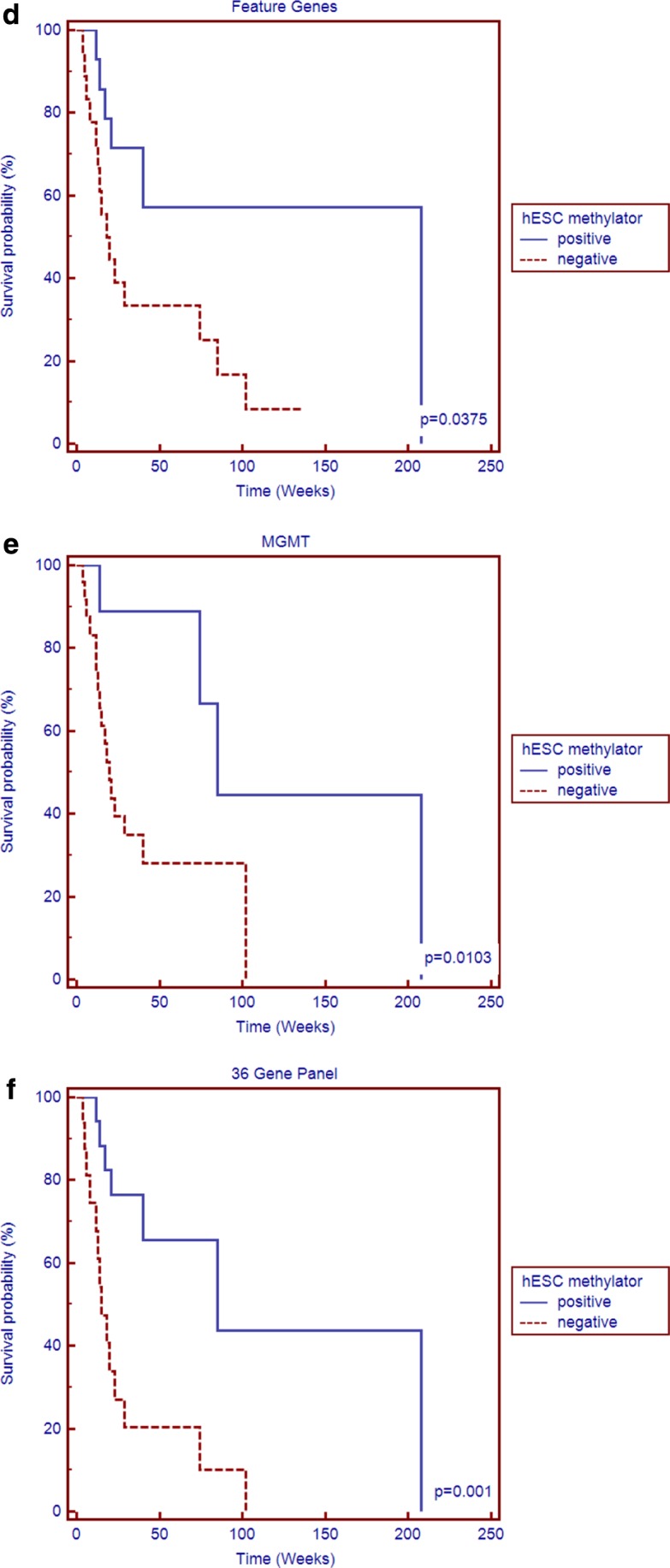
Kaplan-Meier plots between hESC methylator-positive and hESC methylator-negative on the experimental and validation datasets: (a) cases in the experimental testing dataset showed the features clustered phenotypes had significantly more favorable outcomes for hESC methylator-positive patients than for hESC methylator-negative patients; (b) phenotypes clustered by MGMT methylation status in the experiment; (c) phenotypes clustered by the 36 gene panel in the experiment; (d) validation dataset phenotypes clustered by the features; (e) phenotypes clustered by MGMT in the validation dataset; (f) phenotypes clustered by the 36 gene panel in the validation dataset.
Features extracted from surrogate samples provide confidence for subsequent clustering
The original samples were randomly partitioned into three equally size subgroups. First, 120 samples were randomly selected with the original extracted features for clustering into six clusters (k=6). These were compared to the clustered hESC methylator-positive and hESC methylator-negative with the same phenotypes clustered in the original analysis to obtain TP, TN, FP, and FN. The same procedures were repeated fifty times. The performance of the classifier was assessed by ROC curves and the AUC equals as 0.788 is shown in Figure 4.
FIG. 4.
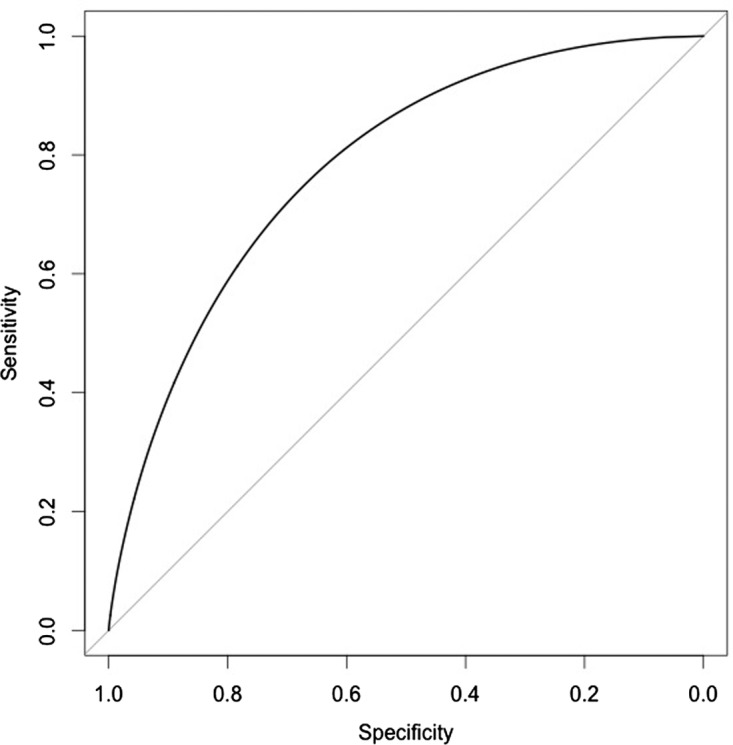
The performance examined for the classifier. The experiment performing on the 50 randomly partitioned sets was assessed using ROC and the AUC was assessed as 0.788.
GO analysis of the features showed enrichment in some important Gene Ontology (GO) terms, such as neuron differentiation, neuron-related development, axonogenesis, embryonic morphogenesis, and cell activity-related terms (Supplementary Table S3; supplementary material is available online at www.liebertpub.com), indicating that these features were functionally relevant for further clustering. To further validate the significance of the association between the features we obtained and the enriched GO term categories have been established; we then randomly selected 30 sets of 250 genes out of the hESC-related genes as features to observe whether these important GO terms we obtained from original features are significant. The statistical significance was assessed as p<2.3E-05.
These analyses strongly supported the idea that the features derived from the surrogates provided high confidence for subsequent clustering that was strongly statistically significant.
Identification of nervous system-specific gene panel
We reasoned that nervous system-specific genes would serve as better GBM biomarkers since they reflect tissue origin. We compiled genes with specific enrichment patterns in the nervous system according to distinct datasets available in GeneNote (bioinfo2.weizmann.ac.il) for significantly overexpressed genes in the brain and spinal cord. DNA methylation status was correlated with corresponding gene expression profiles to identify hypomethylated and upregulated genes in the hESC methylator-negative phenotype when compared to the hESC methylator-positive phenotype. Figure 5 shows the 36 hypomethylated and upregulated brain-specific candidates among the 486 of nervous system-specific genes. The gene panel was also significantly enriched in some important GO terms, such as neuron differentiation, regulation related to neurogenesis, nervous system development, and cell development (Supplementary Table S4). Through the gene panel we were successfully able to separate the hESC methylator-positive and -negative phenotypes in the training dataset (Fig. 3c). In the gene panel, some of these genes have been reported to be involved in nervous system functions and all 36 genes have been shown to belong to the target category of stem cell-related gene sets (Ben-Porath et al., 2008) (Supplementary Table S2). Interestingly, SOX2 (Avilion et al., 2003; Graham et al., 2003; Miyagi et al., 2008; Takahashi et al., 2007; Takahashi and Yamanaka, 2006), POU3F2 (Hagino-Yamagishi et al., 1997; He et al., 1989; Nakai et al., 1995; Schonemann et al., 1995; Sugitani et al., 2002; Vierbuchen et al., 2010), FGFR2 (Ever et al., 2008; Gutin et al., 2006; Maric et al., 2007; Mason 2007; Paek et al., 2009), GAP43 (Aigner et al., 1995; Benowitz and Routtenberg, 1997; Dent and Meiri, 1998), NTRK2 (Klein et al., 1990a; 1990b; 1993; Rudiger Klein et al., 1989), NTRK3 (Klein et al., 1994; Lamballe et al., 1991), and NKX2-2 (Briscoe et al., 1999; Chiang et al., 1996; Ericson et al., 1995; Fuccillo et al., 2006; Marti et al., 1995; Roelink et al., 1995; Vokes et al., 2007; Xu et al., 2005, 2010) are thought to play a significant role in developmental events in the central nervous system and axonal growth. Although most of the above genes have not been reported to be related to the GBM, they could potentially be of interest in future research.
FIG. 5.

The 36 gene panel. Thirty-six genes were separately methylated in hESC methylator-negative and compared with hESC methylator-positive. These genes, overexpressing in the nervous system from GeneNote, were all hypomethylated and significantly upregulated in hESC methylator-negative when compared with hESC methylator-positive.
Validation of features and gene panel in an independent dataset
We further validated the above findings using an independent dataset of 71 patients obtained from the TCGA Data Portal; none of these patients were included in the original training dataset of patients, through the features and the refined gene panel. The clinical characteristics of the patients are provided in Supplementary Table S1. Of note, this testing dataset was produced using the updated Infinium HumanMethylation450 platform. The same unsupervised strategy was used with the same features and the parameter k was set at six; this was done to determine if the features would allow the determination of the distinct phenotype by clustering the same number of clusters as was done with the training set. The hESC methylator clusters were able to be generated including hESC methylator-negative and hESC methylator-positive with their surrogate patients, by first making k equal to six. The significantly favorable clinical outcomes were again observed in patients with the hESC methylator-positive when compared to patients with the hESC methylator-negative phenotype (Fig. 3d). Finally, the significant distinction of the two phenotypes clustered by the gene panel was the same successfully implemented and shows in Figure 3f.
Evaluation of the features and the gene panel
To examine if the established cluster-gene association was within the confident interval, we used bootstrap resampling with replacement of 1000 iterations; the statistical significance was assessed as p<2.20E-16 (95% CI (confidence interval), 0.776 to 0.793). The 1000 sets of clustered hESC methylator-positive and hESC methylator-negative, which were clustered by these features, were compared to the original clustered phenotypes to obtain TP, TN, FP, and FN. To measure the reliability of the clustering performance in the resampling sets, the interval was calculated among the 1000 bootstrap resampling sets through an F-score with a 95% CI.
We next compared our hESC DNA methylation clusters to the four TCGA subtypes defined by gene expression clusters (Verhaak et al., 2010). The experimental result was consistent with the initial TCGA analysis; our hESC methylator-positive was identical to the proneural group characterized by TCGA, whereas our hESC methylator-negative was enriched in the mesenchymal GBM tumors (Fig. 2). In other words, none of the patients in other kinds of gene expression clustered phenotypes were clustered as were the patients with the hESC methylator-positive or hESC methylator-negative phenotype based on DNA methylation status. Thus, our strategy performed very well in stratifying GBM methylator populations.
MGMT is currently the best predictor for GBM treatment outcomes that has been characterized to date (Hegi et al., 2005). We next compared our biomarker panels (the features and the gene panel, respectively) to the promoter methylation status of MGMT. Except for the significantly distinct result from features in the validation dataset (Fig. 3e), better separation results (log-rank test) than MGMT were found in both training and validation datasets (Fig. 3).
The significance of the usefulness of the gene panel in distinguishing the hESC methylator-negative and -positive phenotypes was examined after 1000 iterations through randomly selected 36 genes from the hESC gene set by evaluating their log-rank test (p<2.20E-16). We further validated the performance of our gene panel by the ROC curve. The AUC was used here to assist in the determination of how many patients with a distinct phenotype clustered by features were found to cluster in the same specific phenotype compared with those clustered by markers. The gene panel gave rise to an AUC of 0.833 when compared to MGMT (0.800) in the training dataset (Fig. 6a). Of note, a much more dramatic difference was observed in the independent validation dataset (AUC=0.877 vs. 0.565 for MGMT), indicating the robustness of our gene panel (Fig. 6b). Even after adjusting for sex and age, our gene panel outperformed MGMT in the training dataset (AUC 0.887 vs. 0.756 in Fig. 6c; AUC 0.947 vs. 0.936 in Fig. 6d, respectively). As estimated by the odds ratio (OR), our 36 gene panel continued to be a better predictor of patient outcome (OR=97.36; p=0.0026) than MGMT (OR=74.54; p=0.0045).
FIG. 6.


ROC curves of the 36 gene panel and MGMT methylation profiles. (a) The ROC curves are based on training dataset methylation profiles on 36 gene panel and MGMT. The area under the curve (AUC) of MGMT is 0.800 (95% CI, 0.644 to 0.909), and 36 gene panel is better at predicting patient outcomes than MGMT in AUC of 0.833 (95% CI, 0.682 to 0.932). (b) For the validation dataset, the performance of 36 gene panel methylation profiles was assessed AUC equal to 0.877 (95% CI, 0.735 to 0.969) to be much better at predicting patient outcomes than that of MGMT 0.565 (95% CI, 0.388 to 0.731). (c) When the 36 gene panel was adjusted for gender (AUC=0.887, 95%CI, 0.746 to 0.965), it still has better performance at predicting patient outcomes than the adjusted MGMT profile (AUC=0.756, 95%CI, 0.595 to 0.878). (d) After adjusting for age, the 36 gene panel was assessed AUC equal to 0.947 based on the ROC curve training dataset (95% CI, 0.932 to 1.000) which was better than the AUC for MGMT (AUC=0.936, 95% CI, 0.811 to 0.989).
These results have demonstrated that our features and gene panel can be applied successfully to identify the two methylator phenotypes and that our method can robustly extract biologically meaningful biomarker candidates.
Discussion
GBM is the most common and lethal type of primary brain tumor. Despite recent therapeutic advances in the treatment of other cancers, the treatment of GBM remains ineffective and essentially palliative. The failure of traditional treatments to cure GBM results from a number of causes, but several recent studies have demonstrated that GSCs, a highly tumorigenic subpopulation of cancer cells, displayed relatively high resistance to radiation and chemotherapy. GSCs also contributed to tumor growth through the stimulation of angiogenesis, which has been shown to be a useful therapeutic target in the treatment of recurrent or progressive malignant gliomas. Thus, understanding the stem cell characteristics of GSCs would contribute to understanding GBM pathogenesis and would potentially improve GBM diagnostics and therapies.
This study employed a unique strategy of interrogating epigenetic alterations of hESC-related and nervous system-specific molecular expressions to identify biomarkers for GBM clinical outcome prediction. hESC methylator-negative and -positive phenotypes were identified through the extracted features and the identified gene panel. From the statistical significance tests, significance examining for the bootstrap resampling test and the obtained important GO terms, showed confidence of the extracted features for the subsequent analysis. Furthermore, we were able to evaluate the performance of our gene panel through an independent testing dataset generated from a different platform by using a straight one-step unsupervised clustering strategy. The refined gene panel showed the improved statistical significance over the clinically validated marker MGMT, and the identified gene panel did not overlap with previously reported genes (Noushmehr et al., 2010).
Moreover, the significance of the gene panel in distinguishing the hESC methylator-negative and -positive phenotypes was also examined by evaluating their log-rank test after 1000 iterations with 36 randomly selected genes from the original gene set. The results showed that these genes represented ideal GBM biomarkers bearing both stem cell and tissue specificity. Among the 36 genes, some have previously been reported to be associated with neural development; thus their expression profiles may be tightly regulated by epigenetic mechanisms.
For example, SOX2, one of the key members of the SOX family of transcription factors, is highly expressed in embryonic stem cells (Avilion et al., 2003), and in conjunction with KLF4, OCT4, and c-Myc, whose overexpression can induce pluripotency in both mice and human somatic cells (Takahashi et al., 2007; Takahashi and Yamanaka, 2006). The role of SOX2 in central nervous system development and adult neurogenesis has been extensively investigated (Graham et al., 2003; Miyagi et al., 2008). SOX2 plays a significant role in generating the appropriate number of neural stem and progenitor cells in the developing brain (Miyagi et al., 2008). SOX2 and the closely related genes, Sox1 and Sox3, are widely expressed in proliferating neural progenitors, including neural stem cells, throughout development and adulthood in vertebrate central nervous system (Pevny and Rao, 2003; Uchikawa et al., 2003; Zappone et al., 2000), suggesting a role in the maintenance of neural progenitor fate. SOX2 functions to maintain neural progenitor identity. SOX2, and more generally SOXB1 factors, have both been shown to be necessary and sufficient for maintaining panneural properties of neural progenitor cells (Graham et al., 2003).
Another candidate gene, POU3F2, originally found to be strongly expressed in neuronal cells, is required for maintaining neural cell differentiation (Hagino-Yamagishi et al., 1997; He et al., 1989; Nakai et al., 1995; Vierbuchen et al., 2010). POU3F2 knockout results were shown in the loss of specific neuronal lineages in the endocrine hypothalamus and subsequent loss of the posterior pituitary gland (Nakai et al., 1995; Schonemann et al., 1995). POU3F2 also plays a crucial role in the production and positioning of neocortical neurons (Sugitani et al., 2002). As for the gene, GAP43 is involved in axonal guidance and its expression is correlated with neurite outgrowth during development and regeneration following nerve injuries (Dent and Meiri, 1998; Strittmatter et al., 1990). In addition, the overexpression of GAP43 in transgenic mice produced increased levels of nerve sprouting (Aigner et al., 1995). High GAP43 mRNA expression was also seen during periods of developmental and regenerative axonal growth (Benowitz and Routtenberg, 1997).
Among our gene panel, FGFR2 was significantly reported as well. The proliferation and differentiation of neural stem cells are regulated by various growth factors including bFGF. FGF signaling is transduced via a family of four transmembrance receptor tyrosine kinases (FGFR1-4), and FGFR1-3 are expressed in neural stem cells (NSCs) (Maric et al., 2007; Mason, 2007). Conditional deletion of FGFR2 produced a similarly significant reduction in presynaptic differentiation (Umemori et al., 2004). FGFR signaling is required at early neural plate stages for the formation of the telencephalon, thus apparently promoting telencephalic cell survival (Maric et al., 2007; Mason, 2007; Paek et al., 2009). Developmental upregulation of FGFR2 expression correlates positively with a shift of NSCs into a multipotential state or apoptosis (Maric et al., 2007). The disruption of FGFR1 and FGFR2 at early stages of telencephalic development resulted in altered proliferation and cell death, particularly in the middle regions (Ever et al., 2008; Gutin et al., 2006). Accumulating evidence shows that FGFR2 plays a critical role in the development of the central nervous system, and previous work may provide a foundation for future studies exploring this key epigenetic modification in human disease over GBM and the development.
Neurotrophic factors have been implicated in the proliferation and differentiation of neurons during embryonic development and in their growth and survival in the adult nervous system (Levi-Montalcini, 1987). Among the gene panel, NTRK expression is crucial for the normal development of the peripheral nervous system. In situ hybridization analysis has shown that the NTRK2 transcripts are localized in the central and peripheral nervous system (Klein et al., 1990a; 1990b; Rudiger Klein et al., 1989). NTRK2 also encodes a functional receptor for brain-derived neurotrophic factor (BDNF). Interestingly, another important gene, NTRK3, was identified as a member of the gene panel. NTRK3, the third member of the neurotrophin receptor tyrosine kinase family, was also a receptor for neurotrophin-3 (NT-3) (Lamballe et al., 1991). Knockout mice for NTRK2, NTRK3 displayed unique but overlapping patterns of abnormalities involving the central and peripheral nervous system (Klein et al., 1994), but the NTRK2 knockout have multiple central and peripheral nervous system neuronal deficiencies, and die soon after birth (Klein et al., 1993).
The final significant reported gene of our gene panel was NKX2-2. The Hedgehog signaling pathway plays an important role in establishing patterning of the central nervous system during the earliest phase of neural development (Ericson et al., 1995; Roelink et al., 1995; Xu et al., 2005, 2010). In the ventral neural tube, motor neuron and interneuron generation depends on the graded activity of the signaling protein Sonic hedgehog (Shh) (Chiang et al., 1996; Marti et al., 1995). NKX2-2 has a primary role in ventral neuronal patterning (Briscoe et al., 1999), and is a direct target gene of Shh signaling that upregulates NKX2-2 expression during neuronal development (Briscoe et al., 1999; Fuccillo et al., 2006; Vokes et al., 2007).
Our gene panel constitutes biological meaningful biomarkers with great performance in stratifying GBM, and is amenable for developing clinically useful testing.
Supplementary Material
Acknowledgments
We thank Drs. Xiaowei Yan and Karolina Wallenborg in the Cancer and Stem Cell Group at the Institute for Systems Biology for helpful discussions and assistance. This work was supported by Research Grants from the National Science Council, Taiwan (NSC 99-2627-B-006−013), the National Institutes of Health (NanoSystems Biology Cancer Center/U54 CA119347; NIGMS Center for Systems Biology/ P50 GM076547), and the University of Luxembourg.
Author Disclosure Statement
The authors declare that they have no competing interests.
References
- Aigner L, Arber S, Kapfhammer JP, et al. (1995). Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 83, 269–278 [DOI] [PubMed] [Google Scholar]
- Assou S, Le Carrour T, Tondeur S, et al. (2007). A meta-analysis of human embryonic stem cells Transcriptome integrated into a web-based expression atlas. Stem Cells 25, 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, and Lovell-Badge R. (2003). Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Devel 17, 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, Li Z, et al. (2008). Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res 68, 6043–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, Mclendon RE, et al. (2006a). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, et al. (2006b). Stem cell–like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66, 7843–7848 [DOI] [PubMed] [Google Scholar]
- Baylin SB, and Ohm JE. (2006). Epigenetic gene silencing in cancer. A mechanism for early oncogenic pathway addiction? Nat Rev Cancer 6, 107–116 [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, et al. (2008). An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, and Routtenberg A. (1997). GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20, 84–91 [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, et al. (1999). Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 398, 622–627 [DOI] [PubMed] [Google Scholar]
- Calabrese C, Poppleton H, Kocak M, et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82 [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, et al. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413 [DOI] [PubMed] [Google Scholar]
- Dent EW, and Meiri KF. (1998). Distribution of phosphorylated GAP-43 (neuromodulin) in growth cones directly reflects growth cone behavior. J Neurobiol 35, 287–299 [DOI] [PubMed] [Google Scholar]
- Ericson J, Muhr J, Placzek M, Lints T, Jessel TM, and Edlund T. (1995). Sonic hedgehog induces the differentiation of ventral forebrain neurons: A common signal for ventral patterning within the neural tube. Cell 81, 747–756 [DOI] [PubMed] [Google Scholar]
- Ever L, Zhao R, Eswarakumar VP, and Gaiano N. (2008). Fibroblast growth factor receptor 2 plays an essential role in telencephalic progenitors. Develop Neurosci 30, 306–318 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Ohlsson R, and Henikoff S. (2006). The epigenetic progenitor origin of human cancer. Nat Rev Genet 7, 21–33 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, and Tycko B. (2004). The history of cancer epigenetics. Nat Rev Cancer 4, 143–153 [DOI] [PubMed] [Google Scholar]
- Feinberg AP, and Vogelstein B. (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301, 89–92 [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, et al. (2003). Genomic targets of the human c-Myc protein. Genes Devel 17, 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Skrabanek L, Li Y, et al. (2009). MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 114, 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M, Joyner AL, and Fishell G. (2006). Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci 7, 772–783 [DOI] [PubMed] [Google Scholar]
- Galli R, Binda E, Orfanelli U, et al. (2004). Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 64, 7011–7021 [DOI] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, and Pevny L. (2003). SOX2 functions to maintain neural progenitor identity. Neuron 39, 749–765 [DOI] [PubMed] [Google Scholar]
- Gravendeel LaM, Kouwenhoven MCM, Gevaert O, et al. (2009). Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res 69, 9065–9072 [DOI] [PubMed] [Google Scholar]
- Gutin G, Fernandes M, Palazzolo L, et al. (2006). FGF signalling generates ventral telencephalic cells independently of SHH. Development 133, 2937–2946 [DOI] [PubMed] [Google Scholar]
- Hagino-Yamagishi K, Saijoh Y, Ikeda M, Ichikawa M, Minamikawa-Tachino R, and Hamada H. (1997). Predominant expression of Brn-2 in the postmitotic neurons of the developing mouse neocortex. Brain Res 752, 261–268 [DOI] [PubMed] [Google Scholar]
- He X, Treacy MN, Simmons DM, Ingraham HA, Swanson LW, and Rosenfeld MG. (1989). Expression of a large family of POU-domain regulatory genes in mammalian brain development. Nature 340, 35–42 [DOI] [PubMed] [Google Scholar]
- Hegi ME, Diserens A-C, Gorlia T, et al. (2005). MGMT gene silencing and benefit from temozolomide in glioblastoma. New Eng J Med 352, 997–1003 [DOI] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, et al. (2003). Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci 100, 15178–15183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R, and Pugh J. (1975). DNA modification mechanisms and gene activity during development. Science 187, 226–232 [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, et al. (2009). The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41, 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Parada LF, Coulier F, and Barbacid M. (1989). trkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J 8, 3701–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Conway D, Parada LF, and Barbacid M. (1990a). The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61, 647–656 [DOI] [PubMed] [Google Scholar]
- Klein R, Martin-Zanca D, Barbacid M, and Parada LF. (1990b). Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development 109, 845–850 [DOI] [PubMed] [Google Scholar]
- Klein R, Silos-Santiago I, Smeyne RJ, et al. (1994). Disruption of the neurotrophin-3 receptor gene trkC eliminates la muscle afferents and results in abnormal movements. Nature 368, 249–251 [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, et al. (1993). Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75, 113–122 [PubMed] [Google Scholar]
- Lamballe F, Klein R, and Barbacid M. (1991). trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell 66, 967–979 [DOI] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, et al. (2006a). Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403 [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, et al. (2006b). Control of developmental regulators by polycomb in human embryonic stem cells. Cell 125, 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R. (1987). The nerve growth factor: Thirty-five years later. EMBO J 6, 1145–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, et al. (2009). Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, and Ren B. (2003). A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci 100, 8164–8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maric D, Fiorio Pla A, Chang YH, and Barker JL. (2007). Self-renewing and differentiating properties of cortical neural stem cells are selectively regulated by basic fibroblast growth factor (FGF) signaling via specific FGF receptors. J Neurosci 27, 1836–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti E, Bumcrot DA, Takada R, and McMahon AP. (1995). Requirement of 19K form of Sonic hedgehog for induction of distinct ventral cell types in CNS explants. Nature 375, 322–325 [DOI] [PubMed] [Google Scholar]
- Mason I. (2007). Initiation to end point: The multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 8, 583–596 [DOI] [PubMed] [Google Scholar]
- Metz CE. (1978). Basic principles of ROC analysis. Seminars Nuclear Med 8, 283–298 [DOI] [PubMed] [Google Scholar]
- Miyagi S, Masui S, Niwa H, Saito T, Shimazaki T, Okano H, et al. (2008). Consequence of the loss of Sox2 in the developing brain of the mouse. FEBS Lett 582, 2811–2815 [DOI] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, et al. (2008). Stem cell–related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 26, 3015–3024 [DOI] [PubMed] [Google Scholar]
- Nakai S, Kawano H, Yudate T, et al. (1995). The POU domain transcription factor Brn-2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes Devel 9, 3109–3121 [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway JM, Budiman MA, Korshunova Y, et al. (2007). Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS ONE 2, e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek H, Gutin G, and Hébert JM. (2009). FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development 136, 2457–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevny L, and Rao MS. (2003). The stem-cell menagerie. Trends Neurosci 26, 351–359 [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, et al. (2006). Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 [DOI] [PubMed] [Google Scholar]
- Rauch TA, Zhong X, Wu X, et al. (2008). High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci 105, 252–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs AD. (1975). X inactivation, differentiation, and DNA methylation. Cytogen Genome Res 14, 9–25 [DOI] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, et al. (1995). Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell 81, 445–455 [DOI] [PubMed] [Google Scholar]
- Schonemann MD, Ryan AK, McEvilly RJ, et al. (1995). Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes Devel 9, 3122–3135 [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, et al. (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401 [DOI] [PubMed] [Google Scholar]
- Sperger JM, Chen X, Draper JS, et al. (2003). Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci 100, 13350–13355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter SM, Valenzuela D, Kennedy TE, Neer EJ, and Fishman MC. (1990). G0 is a major growth cone protein subject to regulation by GAP-43. Nature 344, 836–841 [DOI] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, et al. (2002). Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Devel 16, 1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, and Issa J-PJ. (1999). CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci 96, 8681–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikawa M, Ishida Y, Takemoto T, Kamachi Y, and Kondoh H. (2003). Functional analysis of chicken Sox2 enhancers highlights an array of diverse regulatory elements that are conserved in mammals. Devel Cell 4, 509–519 [DOI] [PubMed] [Google Scholar]
- Umemori H, Linhoff MW, Ornitz DM, and Sanes JR. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270 [DOI] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley KA, Purdom E, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, and Wernig M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, Mccuine S, et al. (2007). Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development 134, 1977–1989 [DOI] [PubMed] [Google Scholar]
- Xu Q, Guo L, Moore H, Waclaw RR, Campbell K, and Anderson SA. (2010). Sonic Hedgehog signaling confers ventral telencephalic progenitors with distinct cortical interneuron fates. Neuron 65, 328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wonders CP, and Anderson SA. (2005). Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon. Development 132, 4987–4998 [DOI] [PubMed] [Google Scholar]
- Zappone MV, Galli R, Catena R, et al. (2000). Sox2 regulatory sequences direct expression of a (beta)-geo transgene to telencephalic neural stem cells and precursors of the mouse embryo, revealing regionalization of gene expression in CNS stem cells. Development 127, 2367–2382 [DOI] [PubMed] [Google Scholar]
- Zweig MH, and Campbell G. (1993). Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin Chem 39, 561–577 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.