

Abstract
A variety of hypotheses have been proposed to explain the association between trisomy and increasing maternal age in humans, virtually all of which assume that the underlying mechanisms involve meiotic errors. However, recently Hultén and colleagues [Hulten et al., 2010b] proposed a provocative model—the Oocyte Mosaicism Selection Model (OMSM)—that links age-dependent trisomy 21 to pre-meiotic errors in the ovary. Specifically, they propose that nondisjunctional events occur in a proportion of germ cells as they mitotically proliferate, resulting in mosaicism for trisomy 21. Assuming that the presence of an additional chromosome 21 delays meioticprogression, these cells would be ovulated later in reproductive life, resulting in an age-dependent increase in aneuploid eggs. Because this model has important clinical implications, we initiated studies to test it. We first analyzed oocytes from two trisomy 21 fetuses, combining immunostaining with FISH to determine the likelihood of detecting the additional chromosome 21 at different stages of meiosis. The detection of trisomy was enhanced during the earliest stage of prophase (leptotene), before homologs synapsed. Accordingly, in subsequent studies we examined the chromosome content of leptotene oocytes in seven second trimester female fetuses, analyzing three chromosomes commonly associated with human trisomies (i.e., 13, 16, and 21). In contrast to the prediction of the OMSM, we found no evidence of trisomy mosaicism for any chromosome. We conclude that errors in premeiotic germ cells are not a major contributor to human aneuploidy and do not provide an explanation for the age-related increase in trisomic conceptions.
Keywords: aneuploidy, meiosis, trisomy 21, mosaicism
INTRODUCTION
Aneuploidy (monosomy or trisomy) occurs with extraordinary frequency in humans, affecting 10–30% of fertilized eggs and typically resulting in either very early pregnancy loss, miscarriage, or live-born infants with developmental disabilities [Nagaoka et al., 2012]. Since the vast majority of aneuploidies derive from errors in oogenesis, considerable attention has been directed at identifying maternal risk factors. Although a number of factors have been suggested, the most convincingly demonstrated is increasing age of the mother [Hassold and Hunt, 2001]. The maternal age effect is remarkable, with women in their 20s having only a 2–3% chance of a clinically recognized trisomic pregnancy, while women in their forties have a risk exceeding 30% [Hassold and Chiu, 1985]. Recent changes in reproductive patterns have heightened the importance of this relationship. That is, in developed countries the mean maternal age at delivery has increased remarkably over the past 2–3 decades; indeed, in the United States the value in 2010 was 27.7 years, nearly 3 years higher than the 25.0 mean for 1980 [Martin et al., 2012]. This would be expected to increase the incidence of aneuploidy among clinically recognized pregnancies, and the increasing prevalence of trisomies 13, 18, and 21 in pregnancies in Europe suggests that this is the case [Loane et al., 2013].
Although the relationship between age and aneuploidy is well documented, the underlying basis of the effect remains obscure. Over the years, several different hypotheses have been proposed, most of which can be placed into one of four broad categories based on the developmental stage of oogenesis at which the precipitating event is postulated to occur.
The first category invokes events that occur in the fetal ovary when the oocyte enters meiotic prophase. In humans, oocytes initiate meiosis at about 10–13 weeks gestation. Homologous chromosomes pair, synapse, and undergo recombination and the cell then enters a period of dictyate arrest that lasts from late in gestation until just prior to ovulation in the adult ovary [Johnson, 2013]. Likely the most famous model invoking events at this developmental stage is the production line model originally proposed by Henderson and Edwards [1968]. In an analysis of female mice, they noted an increase in the frequency of univalents in metaphase I oocytes from older females and suggested that age-related decreases in recombination might be responsible for the elevated level of aneuploidy in older females. However, since recombination occurs in the fetal ovary in mammals, they were forced to propose a two-step model, that is, that the first oocytes to enter meiosis are the first to be ovulated, and oocytes that enter meiosis later form fewer chiasmata [Henderson and Edwards, 1968]. Consequently, oocytes ovulated at the end of the reproductive lifespan would have the fewest chiasmata and be most susceptible to nondisjunction. Subsequent evidence has supported the first tenet of this hypothesis, that is, that the first oocytes to initiate meiosis are ovulated first [Polani and Crolla, 1991]. However, genetic linkage data from studies of humans provide no evidence of an age-related decrease in recombination levels [Kong et al., 2004; Coop et al., 2008; Hussin et al., 2011]; thus the relevance of the production line model to age-related human aneuploidy is unclear.
A second category of hypotheses has focused on the long dictyate arrest that lasts from late in gestation until just prior to ovulation. Some researchers have suggested that, because the maternal age curve mimics that of an infectious process, a pathogenic agent may be involved in the genesis of abnormal oocytes [Hook, 1989]. Others have suggested that the long period of meiotic arrest allows for the accumulation of DNA damage, which has been shown to cause aneuploidy in model organisms [Wang and Hoog, 2006]. However, perhaps the most widely supported of these “meiotic arrest” models proposes that the crucial event is degradation of cohesin proteins [Hodges et al., 2005; Liu and Keefe, 2008; Vogt et al., 2008; Chiang et al., 2010; Lister et al., 2010; Tachibana-Konwalski et al., 2010; Jessberger, 2012]. In meiosis, the cohesin complex has dual functions: ensuring cohesion between sister chromatids, and maintaining inter-homolog associations distal to the sites of crossovers. Both activities are crucial in orchestrating the segregation of homologs at the first meiotic division. It is thought that there is little or no turnover of the proteins of the cohesin complex, and age-related degradation of cohesion established during fetal development has been postulated to lead to the premature separation of homologs and/or sister chromatids, resulting in aneuploidy [Chiang et al., 2010; Lister et al., 2010; Tachibana-Konwalski et al., 2010; Jessberger, 2012].
A third group of hypotheses invoke events that occur at or around the time of the meiotic cell divisions. For example, age related abnormalities may involve internal factors such as hormonal variation, including changes in the levels of FSH [Roberts et al., 2005] or LH [Crowley et al., 1979] or the ability of the oocyte to respond to those signals [Hodges et al., 2005], or age-related changes in the microcirculation of the ovary that disrupt formation of the meiotic spindle [Gaulden, 1992]. Other hypotheses relate to the ability of the oocyte to monitor errors. Cell cycle checkpoint proteins such as MAD2 and BUB1 delay the cell cycle until bipolar attachment of homologs is complete, and if protein levels decrease with age, normal functioning of the spindle assembly checkpoint may be impaired [Steuerwald et al., 2001]. Recent studies have shown that cell cycle control mechanisms differ between mammalian oocytes and spermatocytes, operating in a less stringent manner in the former [Nagaoka et al., 2011]. Thus, it may be that errors occur at a similar frequency in male and female gametes but that the inability to sense and eliminate them in eggs results in a higher frequency of maternally-derived aneuploidies.
Finally, a fourth category of hypotheses suggest that aneuploidy may be attributable to a combination of “hits” that occur at different stages in meiosis. For example, Sherman and colleagues have suggested that abnormalities in meiotic recombination in the fetal stages of oogenesis render oocytes susceptible to nondisjunction, and that a second “hit” that occurs in the aging ovary increases the likelihood that such oocytes will, indeed, nondisjoin [Hassold and Sherman, 2000]. Consistent with this idea, the incidence of cases of trisomy 21 attributable to achiasmate chromosomes 21 appears to increase with maternal age [Lamb et al., 2005; Oliver et al., 2008].
While the above models differ in the way that they explain the maternal age effect, they are united by a common assumption, namely, that the age effect is attributable to events occurring during meiosis. Recently, however, Hultén and colleagues proposed a new type of model—the Oocyte Mosaicism Selection Model (OMSM)—that casts doubt on the assumption that the age effect is meiotic in origin [Hulten et al., 2008]. In a study of surface spread human fetal oocytes from eight female fetuses, they performed FISH for two subtelomeric loci on chromosome 21. They found that roughly 1 in 200 cells contained three chromosome 21 FISH signals, suggesting that pre-meiotic mitotic nondisjunction generates a surprisingly high proportion of primary oocytes that carry an extra chromosome 21. If these trisomic oocytes are equally as likely to be ovulated as chromosomally normal oocytes, these observations suggest that the vast majority of cases of trisomy 21 derive from mitotic, not meiotic, errors. Further, if as suggested by Hultén and colleagues, the progression of trisomic oocytes through meiotic prophase is delayed, resulting in their ovulation later in reproductive life [Hulten et al., 2008], one does not have to invoke meiotic defects in the genesis of the maternal age effect.
The implications of the OMSM are wide-ranging. Importantly, it suggests that further analyses of meiosis and meiotic abnormalities are unlikely to provide important insights on the etiology of maternally-derived human aneuploidy. Further, it implies that therapeutic strategies to prevent the occurrence of trisomy are doomed to failure, since intervention would have to occur in the first trimester of pregnancy. Thus, if confirmed, the OMSM fundamentally changes our understanding of the origin of human aneuploidy, and re-directs our research efforts.
Unfortunately, only one subsequent study has attempted to repeat the study. In an analysis of eight samples and 51,146 cells, Morris and colleagues were unable to replicate the original observations [Morris et al., 2012]. However, neither the original study nor the study of Morris and colleagues analyzed different stages of meiotic prophase, and Hulten et al. [2008] did not analyze chromosomes other than 21. Thus, we decided to re-examine the OMSM, analyzing levels of clinically relevant trisomies 13, 16, and 21 in oocytes at the earliest stages of prophase. We found no evidence of trisomy mosaicism for any of the three chromosomes, suggesting that factors other than pre-meiotic nondisjunction are responsible for the age-related increase in human trisomies.
MATERIALS AND METHODS
Study Population
Material for this study consisted of prophase stage oocytes from ovarian samples of nine second trimester female fetuses, two with trisomy 21, and the other seven presumed to be chromosomally normal. Ovaries were collected either at the University of Washington Medical Center in Seattle, Washington or the San Francisco General Hospital's Women's Options Center in San Francisco, California from therapeutic, or elective terminations of pregnancies. Immediately following the surgical procedures, whole ovarian tissue samples were shipped by overnight delivery to Washington State University (WSU) for processing. All procedures were approved by the University of California, San Francisco's Human Research Protection Program or the University of Washington Institutional Review Board, and by the WSU Institutional Review Board, and informed consent was obtained from all study participants.
Processing
Upon receipt at WSU, ovaries were processed using a standard surface spreading technique [Peters et al., 1997]. Briefly, ovaries were cleaned of excess tissue, placed in a watch glass, covered in hypo-extraction buffer (600 mM TRIS, 500 mM sucrose, 500 mM EDTA, 500 mM DTT, 170 mM sodium citrate, and 100 mM PMSF), incubated at room temperature for about 45 min, suspended in 100 mM sucrose and macerated with 26 gauge needles. Following maceration, 10 μl aliquots of cell suspension were spread on slides coated with 2% PFA and placed in a humid chamber overnight. The following morning, slides were air dried, washed in a 0.04% Photoflo solution for 2 min, and stored at 4°C prior to staining.
Immunofluorescence (IF) and Fluorescence In Situ Hybridization (FISH)
Slides were immunostained using similar methodology to that published previously [Cheng et al., 2009]. Slides were pre-incubated for 60 min in 1X antibody dilution buffer (ADB) composed of 10 ml normal donkey serum (Jackson ImmunoResearch), 3 g BSA (Sigma–Aldrich), 50 μl Triton X-100 (Alfa Aesar),and 990 ml PBS. Sixty microliter of 1X ADB containing kinetochore-associated CREST antisera (1:500 dilution, Fisher, rabbit anti-human) was applied and the slides were incubated overnight at 37°C. Sixty microliter of 1X ADB containing an antibody against the synaptonemal complex protein SYCP3 (1:150 dilution; Novus, rabbit anti-human) was then added to the slides and incubated for 2 hr at 37°C. Slides were then washed twice in 1X ADB for 20 min, 60 μl of ADB containing ADAH (1:100 dilution, Jackson ImmunoResearch) was added, and slides were incubated overnight at 37°C. The following morning 60 μl of 1× ADB containing RDAR (1:100 dilution, Jackson ImmunoResearch) was added and slides incubated for 45 min at 37°C. Slides were washed twice in 1X PBS for 30 min, twice more in 1X PBS for 1 min, and coverslipped with 40 μl Prolong Gold Antifade Reagent (Invitrogen) and stored at 4°C. Images were captured on a Zeiss fluorescence microscope and coordinates recorded or slides were scanned using the ASI Scanview Case Data Manager.
After initial examination of IF images, we used chromosomespecific FISH to identify individual chromosomes. For the two fetuses with trisomy 21, we used a Vysis LS1 21 probe. For the other seven fetuses, we used Cytocell Aquarius Satellite Enumeration point probes for chromosome 16 (Green) and a probe detecting both chromosomes 13 and 21 (Red Dual Probe). Previously immunostained slides were dehydrated in an ethanol series (75%, 90%, and 100%) at room temperature. Slides and probe mixtures (6 μl each probe and 8 μl hybridization solution) were both pre-warmed at 37°C for 5 min and the probe was applied to the slide and coverslipped. Preparations were denatured at 75°C for 5 min and incubated overnight at 37°C in a humid chamber. The following morning, slides were washed in 0.4X SSC at 72°C for 30 sec, washed in 2X SSC/0.5% Tween-20 at room temperature for 2 min, and then coverslipped with 40 μl prolong Gold Antifade Reagent with DAPI (Invitrogen) and stored at 4°C until analysis. Slides were evaluated in the same manner as after IF, on either the Zeiss fluorescence microscope or scanned using the ASI Scanview Case Data Manager software. Cells from IF were located, captured for FISH signals, and scored.
Scoring
Cells were analyzed for the number of FISH signals displayed. To be classified as a signal, a focus had to be punctuate, round to elliptical in shape, and regular in appearance. For the two trisomy 21 fetuses, we simply counted the number of chromosome 21 FISH signals. For the other seven, presumably euploid, fetuses, cells that contained an abnormal number of FISH signals were overlaid with their corresponding IF images to determine whether the FISH signals co-localized with centromeres (detected by CREST). FISH signals that localized to the centromere-specific CREST signal were classified as bona fide, while those that did not localize to a CREST signal were classified as false positive signals.
RESULTS AND DISCUSSION
Rationale for the Analytic Approach
Conceptually, the use of FISH to detect trisomy in mitotic cells is a simple exercise. A chromosome-specific probe localizes to each copy of the target chromosome, thereby enabling detection of the number of chromosomes per nucleus. The reliability of the approach depends on a number of factors, including the quality of the cytological preparation, the type of cell being analyzed, and the hybridization efficiency; nevertheless under optimal conditions the analysis is straightforward. However, the same does not apply to meiotic cells. Because prophase involves the pairing of homologous chromosomes and, ultimately, their intimate association, the number of expected signals for a chromosome pair depends on the developmental stage of the oocyte being examined. For example, in the first stage of prophase (leptotene) homologs are largely unpaired; thus in a normal meiocyte two FISH signals per cell should be evident, while the presence of three chromosome-specific signals would indicate a trisomic cell (Fig. 1A,D). However, as cells progress to zygotene, the homologous chromosomes begin to synapse and, depending on whether the probe hybridizes to a synapsed or unsynapsed region, a normal cell could have one or two signals, and a trisomic cell could have one, two, or three signals. (Fig. 1B,E). Finally, because the subsequent pachytene stage is characterized by complete synapsis of homologs, both normal and trisomic cells should exhibit a single large FISH signal, although synaptic defects associated with trisomy may lead to the presence of two or three signals (Fig. 1C,F).
FIG. 1.

The expected number of FISH signals per chromosome is dependent on the stage of meiotic prophase. In euploid oocytes (A–C), homologs may be completely separated at leptotene, allowing for detection of both chromosomes. At zygotene, homologs begin to synapse, so that either one or two FISH signals may be apparent. At pachytene, homologs are completely synapsed; thus, typically one signal should be evident. In trisomic oocytes (D–F), three signals should be readily apparent at leptotene but, depending on the extent of synapsis at zygotene and pachytene, one, two, or three FISH signals may be observed.
A Pilot Study: FISH Studies of Oocytes in Trisomy 21 Fetuses
The basic meiotic principles detailed above complicate assessments of the level of trisomy in prophase stage oocytes. Importantly, the inclusion of cells at zygotene and pachytene should generate underestimates of the real values of aneuploidy. Thus, in contrast to the original analyses of Hulten et al. [2008]—in which no attempt was made to discriminate by stage of prophase—we reasoned that careful staging of oocytes and a focus on the analysis of leptotene stage cells would generate the most accurate determinations of trisomy mosaicism. To test this, we conducted a pilot study of prophase stage oocytes in trisomy 21 fetuses, since this situation provides the maximal likelihood of identifying three FISH signals. We examined 37 prophase cells from two fetal ovarian samples, using an antibody to SYCP3 to identify the synaptonemal complex and a distal chromosome 21 FISH probe to determine the number of chromosomes 21 per cell (Fig. 2). We visualized three chromosome 21 signals in both of two leptotene cells examined, but rarely identified three signals in either zygotene or pachytene cells (Fig. 2; Table I). These observations fit our expectations vis a vis the likelihood of detecting three chromosome 21 signals in trisomic cells and are consistent with previous cytological studies of meiocytes from trisomy 21 cases (Table I). Indeed, taken together, the results from four recent studies of trisomy 21 female fetuses (including the present report) indicate that the majority of leptotene stage cells but only 29–39% of zygotene and 0–7% of pachytene cells exhibit three chromosome 21 signals [Cheng et al., 1998; Barlow et al., 2002; Robles et al., 2007].
FIG. 2.
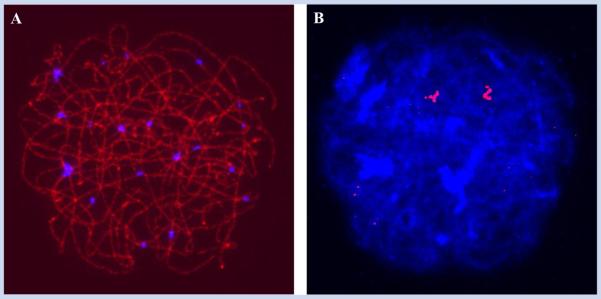
Prophase stage oocytes from trisomy 21 fetuses seldom exhibit three chromosome 21 signals. Representative immunostaining (A) and FISH (B) images of a late zygotene oocyte from a trisomy 21 fetus. Due to partial synapsis between the three homologs, only two chromosome 21 signals are evident.
TABLE I.
Detection of Chromosome 21 Signals by Stage of Prophase in Fetuses With Trisomy 21
| Stage | No. of cells | No. of signals |
|||
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| Present Study | Leptotene | 2 | 0 (0%) | 0 (0%) | 2 (100%) |
| Zygotene | 21 | 14 (67%) | 6 (29%) | 1 (5%) | |
| Pachytene | 14 | 8 (57%) | 5 (36%) | 1 (7%) | |
| Cheng et al. [1998] | Leptotene | 3,069 | 430 (14%) | 966 (31%) | 1,673 (55%) |
| Zygotene | 662 | 316 (48%) | 257 (39%) | 89 (13%) | |
| Pachytene | 2,067 | 1,351 (65%) | 616 (30%) | 100 (5%) | |
| Robles et al. [2007] a | Leptotene | 3 | 0 (0%) | 0 (0%) | 3 (100%) |
| Zygotene | 27 | 11 (41%) | 9 (33%) | 7 (26%) | |
| Pachytene | 84 | 55 (65%) | 28 (33%) | 1 (1%) | |
| Barlow et al. [2002] a | Pachytene | 88 | 37 (42%) | 51 (58%) | 0 (0%) |
Original scoring was based on pairing configurations and has been converted assuming that cells with 3 univalents = 3 signals; cells with bivalent + univalent = 2 signals; and cells with trivalent/partial trivalent = 1 signal.
The implication of these observations is straightforward: accurate estimates of the frequency of trisomic oocytes cannot be obtained by pooling data from cells at different stages of prophase. The concern about accuracy would be minimized if leptotene cells were the predominant cell type identified in fetal oocytes. However, this is not the case, as previous analyses of late first and second trimester female fetuses indicate that, at least among conceptuses of 15 weeks gestation or more, the majority of oocytes have completed leptotene [Roig et al., 2005; Cheng et al., 2009] and progressed to zygotene, pachytene or beyond.
Analysis of Trisomy Mosaicism in Euploid Fetuses
We concluded that an accurate assessment of trisomy mosaicism would require analysis of cells at leptotene, as this is the only stage at which a trisomic oocyte would reliably display three signals. Accordingly, we initiated studies of trisomy mosaicism in leptotene stage cells of seven female fetuses, with gestational ages ranging from 16 to 23 weeks. We did not karyotype the samples. However, given the low rate of chromosome abnormality in elective pregnancy terminations [e.g., Burgoyne et al., 1991], it seems reasonable to assume that all were karyotypically normal.
For each sample we first immunostained the preparations, using SYCP3 to detect synaptonemal complexes, and CREST to identify centromeric regions. Subsequently, the slides were denatured and a FISH probe recognizing the pericentromeric region of chromo-some 16 and one detecting the pericentromeric regions of chromosomes 13 and 21 were applied. In total, we examined 1,405 leptotene stage oocytes from the seven samples; of these 1,034 were informative for the chromosome 16 probe and 1,206 for the probe detecting both chromosomes 13 and 21 (Table II). For chromosome 16, approximately two-thirds of the cells (686/1,034 = 66.3%) contained two signals, with almost all of the remainder (339/1,034 = 32.8%) exhibiting a single signal. An additional nine cells (9/1,034 = 0.9%) had three signals, consistent with trisomy 16. However, when we merged the immunostained and FISH images, it was clear that, in each of these nine cells, one of the three FISH signals did not co-localize with a CREST signal (Fig. 3). Thus, these are clearly non-specific signals, and when removed from the data set, the results provide no evidence for any trisomy 16 cells.
TABLE II.
Number of FISH Signals per Chromosome in Leptotene Oocytes from Seven Female Fetuses
| No. of signals |
No. of false positivesb | ||||||
|---|---|---|---|---|---|---|---|
| Chromosomea | No. Cells | 1 | 2 | 3 | 4 | 5 | |
| 16 | 1,034 | 339 | 686 | 0 | 0 | 0 | 9 |
| 13/21 | 1,206 | 1 | 222 | 220 | 758 | 0 | 5 |
For chromosome 16, the presence of three signals would indicate a trisomic cell. The probe used for chromosomes 13 and 21 detects both chromosomes; thus the presence of five signals would indicate a trisomy for either chromosome 13 or 21.
Cells in which the additional FISH signal failed to co-localize with a CREST signal.
FIG. 3.
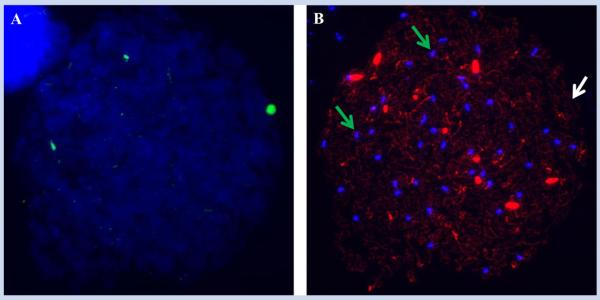
Putative trisomic cells may contain an artifactual FISH signal. We identified 14 leptotene cells with an additional FISH signal, but in each instance one of the three signals failed to co-localize with a CREST signal, indicating a false positive result. Shown here are FISH (A) and immunostained (B) images of the same cell, in which we identified three chromosome 16 FISH signals but on immunostaining, CREST signals overlapped the FISH signals in only two of the three instances (i.e., the green arrows indicate where FISH signals overlap CREST signals, while the white arrow indicates where an artifactual FISH signal is present).
Similar results were observed for chromosomes 13 and 21, for which the presence of four signals would be consistent with euploidy for these chromosomes, five signals with trisomy for one of the two chromosomes and six signals with trisomy for both chromosomes. In the majority of cells (758/1,206 = 62.9%) we identified four signals, but in five cells a fifth signal was observed. However, as with chromosome 16, in each of these cells one of the three signals did not co-localize with a CREST signal, indicating a false positive result. Thus, taken together, our analyses provide no evidence for trisomy mosaicism for either chromosomes 13, 16, or 21 in fetal ovarian preparations.
Putting it Into Perspective: Is Germ Cell Mosaicism an Important Source of Human Aneuploidy?
There is a wealth of data linking mosaicism for trisomy 21 to the etiology of Down syndrome. For example, a number of case studies of recurrent Down syndrome have identified mosaicism for trisomy 21 in gonadal, skin or blood samples of one of the parents [Harris et al., 1982; Uchida and Freeman, 1985; Sachs et al., 1990; Pangalos et al., 1992; Tseng et al., 1994; Bruyere et al., 2000; Frias et al., 2002]. Further, in studies of 374 singleton Down syndrome patients, Uchida and Freeman identified parental mosaicism in nearly 3% of the families [Uchida and Freeman, 1985], and in a review of 221 cases, Harris et al. [1982] reported parental mosaicism in 1.6% of the families. Thus, the importance of parental mosaicism to a small, but non-trivial, proportion of cases of Down syndrome is well established.
However, the OMSM model proposed by Hultén and colleagues suggests a much more important role for mosaicism in the genesis of human aneuploidy [Hulten et al., 2008]. Specifically, it suggests that trisomy mosaicism is a major source of trisomy 21 and the cause of the maternal age effect on trisomy 21, as well as a likely contributor to other human trisomies [Hulten et al., 2010a,b]. Several aspects of the OMSM are appealing. First, it provides a unifying model for the genesis of trisomy 21, and unlike many recent reports, requires only a single “hit” (i.e., pre-meiotic mitotic nondisjunction) to explain the origin of aneuploidy. Second, by assuming that trisomic oocytes are developmentally delayed and consequently among the last to be ovulated, the model provides a mechanism for generating a maternal age-dependent increase in the incidence of disomic oocytes. Finally, by invoking different segregation modes for the various meiotic configurations that three chromosomes can adopt in prophase, the OMSM is able to account for the well-known—if complicated—association between meiotic recombination errors and trisomy 21 [Oliver et al., 2008].
However, the OMSM also has at least two important shortcomings. First, Hultén and colleagues suggest that it provides a straightforward alternative to more complex models of maternal age-dependent trisomy that invoke a variety of biological and environmental factors [Hulten et al., 2010b]. Unfortunately, as appealing as it would be to have only one major contributor to age-related aneuploidy, mounting evidence indicates that the origin of the age effect is, indeed, complicated [Nagaoka et al., 2012]. Studies of model organisms have reported a number of routes to age-related aneuploidy, including abnormalities in formation of the synaptonemal complex [Yuan et al., 2002; Wang and Hoog, 2006], in maintenance of the sister chromatid cohesion complex [Chiang et al., 2010; Lister et al., 2010; Jessberger, 2012] and in the function of cell cycle checkpoint controls [Vogt et al., 2008; Nagaoka et al., 2011]. Further, analyses of humans also provide evidence of multiple, chromosome-specific mechanisms of nondisjunction and, presumably, of maternal age-related trisomy. For example, it is now clear that the parent and meiotic stage of origin varies depending on the chromosome involved [Hassold et al., 1991; Bugge et al., 1998, 2007; Hassold and Hunt, 2001; Hall et al., 2007a,b], that there are different maternal age curves for trisomies involving different chromosomes [Hassold and Chiu, 1985] (and even for different meiotic origins involving the same chromosome [Yoon et al., 1996]), and that the effects of abnormal meiotic recombination depend on maternal age and on the chromosome involved [Lamb et al., 2005; Sherman et al., 2006]. Further, recent direct analyses of prophase stage fetal oocytes provide compelling evidence that abnormalities in pairing or recombination—and not the presence of three chromosomes—are common in the human female, and that they vary depending on the specific chromosome [Cheng et al., 2009]. Thus, there is now ample evidence that no one mechanism is likely to explain all, or even a substantial proportion of, cases of human aneuploidy.
Second, and more important, the OMSM has yet to be replicated and, indeed, the available data contradict the basic tenet of the model; that is, the occurrence of trisomy 21 mosaicism in fetal oocytes. For example, the results of the present study provide no evidence for the presence of trisomic oocytes for any of the three chromosomes we analyzed. For chromosome 21 our results are highly significantly different from the 0.66% level of trisomy 21 mosaicism reported by Hulten et al. [2008] for prophase cells (χ2 = 7.95; P < 0.01) as well as for the 0.54% value identified for all ovarian cells (χ2 = 6.51; P = 0.01). Further, this almost certainly underestimates the real difference between the observations of Hulten et al. [2008] and ours, since their analysis included cells (i.e., zygotene and pachytene) with a low likelihood of trisomy detection.
Similarly, other analyses have failed to identify appreciable levels of trisomy 21 mosaicism. For example, Cheng and colleagues analyzed 3008 prophase stage cells (1,195 at leptotene, 511 at zygotene, and 1,302 at pachytene) from eight euploid fetuses and were unable to identify a single cell with three chromosome 21 signals [Cheng et al., 1998]. More recently, Robles and colleagues examined seven euploid fetuses and identified trisomy 21 in 1/425 (0.2%) leptotene cells but in none of 985 zygotene or pachytene cells [Robles et al., 2007]. Finally, in the only study conducted after that of Hultén and colleagues [Hulten et al., 2008], Morris and colleagues analyzed both fetal ovarian and skin samples, and observed extremely low levels of trisomy 21 mosaicism in each of the two tissue types (2/8,365 = 0.02% in ovarian cells and 5/8,245 = 0.06% in skin cells) [Morris et al., 2012]. Thus, taken together, the results of four different analyses suggest that—for reasons that are unclear—the original report of Hulten et al. [2008] markedly overestimated the incidence of trisomy 21 in fetal oocytes.
In summary, we and others have failed to replicate the observations of Hulten et al. [2008]; accordingly, we conclude that premeiotic mitotic nondisjunction is unlikely to cause the maternal age effect and probably only contributes to a small subset of human aneuploidies. This interpretation is consistent with the growing body of data from studies of aneuploidy in model organisms and direct analyses of meiosis in human oocytes, which indicate the existence of multiple sources of human aneuploidy, and multiple causes of the maternal age effect. Thus, we suggest that future investigations of aneuploidy focus on events occurring in meiosis, and not those that occur in germ cells before they enter the meiotic pathway.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01 HD21341 (to T.H.) and R01 ES013527 (to P.H.). We thank the staff and faculty at San Francisco General Hospital's Women's Options Center for assistance in the collection of tissues. We also thank Katie Stephenson, Dylan Atchley, and Cynthia Megloza for their assistance in recruitment and data collection.
Grant sponsor: NIH; Grant numbers: R01 HD21341, R01 ES013527.
Footnotes
Conflict of interest: none.
REFERENCES
- Barlow AL, Tease C, Hulten MA. Meiotic chromosome pairing in fetal oocytes of trisomy 21 human females. Cytogenet Genome Res. 2002;96:45–51. doi: 10.1159/000063045. [DOI] [PubMed] [Google Scholar]
- Bruyere H, Rupps R, Kuchinka BD, Friedman JM, Robinson WP. Recurrent trisomy 21 in a couple with a child presenting trisomy 21 mosaicism and maternal uniparental disomy for chromosome 21 in the euploid cell line. Am J Med Genet. 2000;94:35–41. doi: 10.1002/1096-8628(20000904)94:1<35::aid-ajmg8>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Bugge M, Collins A, Petersen MB, Fisher J, Brandt C, Hertz JM, Tranebjaerg L, de Lozier-Blanchet C, Nicolaides P, Brondum-Nielsen K, Morton N, Mikkelsen M. Non-disjunction of chromosome 18. Hum Mol Genet. 1998;7:661–669. doi: 10.1093/hmg/7.4.661. [DOI] [PubMed] [Google Scholar]
- Bugge M, Collins A, Hertz JM, Eiberg H, Lundsteen C, Brandt CA, Bak M, Hansen C, Delozier CD, Lespinasse J, Tranebjaerg L, Hahnemann JM, Rasmussen K, Bruun-Petersen G, Duprez L, Tommerup N, Petersen MB. Non-disjunction of chromosome 13. Hum Mol Genet. 2007;16:2004–2010. doi: 10.1093/hmg/ddm148. [DOI] [PubMed] [Google Scholar]
- Burgoyne PS, Holland K, Stephens R. Incidence of numerical chromosome anomalies in human pregnancy estimation from induced and spontaneous abortion data. Hum Reprod. 1991;6:555–565. doi: 10.1093/oxfordjournals.humrep.a137379. [DOI] [PubMed] [Google Scholar]
- Cheng EY, Chen YJ, Bonnet G, Gartler SM. An analysis of meiotic pairing in trisomy 21 oocytes using fluorescent in situ hybridization. Cytogenet Cell Genet. 1998;80:48–53. doi: 10.1159/000014956. [DOI] [PubMed] [Google Scholar]
- Cheng EY, Hunt PA, Naluai-Cecchini TA, Fligner CL, Fujimoto VY, Pasternack TL, Schwartz JM, Steinauer JE, Woodruff TJ, Cherry SM, Hansen TA, Vallente RU, Broman KW, Hassold TJ. Meiotic recombination in human oocytes. PLoS Genet. 2009;5:e1000661. doi: 10.1371/journal.pgen.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol. 2010;20:1522–1528. doi: 10.1016/j.cub.2010.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Wen X, Ober C, Pritchard JK, Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–1398. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- Crowley PH, Gulati DK, Hayden TL, Lopez P, Dyer R. A chiasmahormonal hypothesis relating down's syndrome and maternal age. Nature. 1979;280:417–418. doi: 10.1038/280417a0. [DOI] [PubMed] [Google Scholar]
- Frias S, Ramos S, Molina B, del Castillo V, Mayen DG. Detection of mosaicism in lymphocytes of parents of free trisomy 21 offspring. Mutat Res. 2002;520:25–37. doi: 10.1016/s1383-5718(02)00163-8. [DOI] [PubMed] [Google Scholar]
- Gaulden ME. Maternal age effect: The enigma of down syndrome and other trisomic conditions. Mutat Res. 1992;296:69–88. doi: 10.1016/0165-1110(92)90033-6. [DOI] [PubMed] [Google Scholar]
- Hall HE, Chan ER, Collins A, Judis L, Shirley S, Surti U, Hoffner L, Cockwell AE, Jacobs PA, Hassold TJ. The origin of trisomy 13. Am J Med Genet A. 2007a;143A:2242–2248. doi: 10.1002/ajmg.a.31913. [DOI] [PubMed] [Google Scholar]
- Hall HE, Surti U, Hoffner L, Shirley S, Feingold E, Hassold T. The origin of trisomy 22: Evidence for acrocentric chromosome-specific patterns of nondisjunction. Am J Med Genet A. 2007b;143A:2249–2255. doi: 10.1002/ajmg.a.31918. [DOI] [PubMed] [Google Scholar]
- Harris DJ, Begleiter ML, Chamberlin J, Hankins L, Magenis RE. Parental trisomy 21 mosaicism. Am J Hum Genet. 1982;34:125–133. [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Chiu D. Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum Genet. 1985;70:11–17. doi: 10.1007/BF00389450. [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P. To err (meiotically) is human: The genesis of human aneuploidy. Nat Rev Genet. 2001;2:280–291. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- Hassold T, Sherman S. Down syndrome: Genetic recombination and the origin of the extra chromosome 21. Clin Genet. 2000;57:95–100. doi: 10.1034/j.1399-0004.2000.570201.x. [DOI] [PubMed] [Google Scholar]
- Hassold TJ, Pettay D, Freeman SB, Grantham M, Takaesu N. Molecular studies of non-disjunction in trisomy 16. J Med Genet. 1991;28:159–162. doi: 10.1136/jmg.28.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson SA, Edwards RG. Chiasma frequency and maternal age in mammals. Nature. 1968;218:22–28. doi: 10.1038/218022a0. [DOI] [PubMed] [Google Scholar]
- Hodges CA, Revenkova E, Jessberger R, Hassold TJ, Hunt PA. Smc1beta-deficient female mice provide evidence that cohesins are a missing link in age-related nondisjunction. Nat Genet. 2005;37:1351–1355. doi: 10.1038/ng1672. [DOI] [PubMed] [Google Scholar]
- Hook EB. Issues pertaining to the impact and etiology of trisomy 21 and other aneuploidy in humans; a consideration of evolutionary implications, maternal age mechanisms, and other matters. Prog Clin Biol Res. 1989;311:1–27. [PubMed] [Google Scholar]
- Hulten MA, Patel SD, Tankimanova M, Westgren M, Papadogiannakis N, Jonsson AM, Iwarsson E. On the origin of trisomy 21 down syndrome. Mol Cytogenet. 2008;1:21. doi: 10.1186/1755-8166-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulten MA, Jonasson J, Nordgren A, Iwarsson E. Germinal and somatic trisomy 21 mosaicism: How common is it, what are the implications for individual carriers and how does it come about? Curr Genomics. 2010a;11:409–419. doi: 10.2174/138920210793176056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulten MA, Patel S, Jonasson J, Iwarsson E. On the origin of the maternal age effect in trisomy 21 down syndrome: The oocyte mosaicism selection model. Reproduction. 2010b;139:1–9. doi: 10.1530/REP-09-0088. [DOI] [PubMed] [Google Scholar]
- Hussin J, Roy-Gagnon MH, Gendron R, Andelfinger G, Awadalla P. Age-dependent recombination rates in human pedigrees. PLoS Genet. 2011;7:e1002251. doi: 10.1371/journal.pgen.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger R. Age-related aneuploidy through cohesion exhaustion. EMBO Rep. 2012;13:539–546. doi: 10.1038/embor.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MH. Essential reproduction. John Wiley & Sons; Chichester West Sussex: 2013. p. 392. [Google Scholar]
- Kong A, Barnard J, Gudbjartsson DF, Thorleifsson G, Jonsdottir G, Sigurdardottir S, Richardsson B, Jonsdottir J, Thorgeirsson T, Frigge ML, Lamb NE, Sherman S, Gulcher JR, Stefansson K. Recombination rate and reproductive success in humans. Nat Genet. 2004;36:1203–1206. doi: 10.1038/ng1445. [DOI] [PubMed] [Google Scholar]
- Lamb NE, Yu K, Shaffer J, Feingold E, Sherman SL. Association between maternal age and meiotic recombination for trisomy 21. Am J Hum Genet. 2005;76:91–99. doi: 10.1086/427266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, Nathan A, Floros V, Adelfalk C, Watanabe Y, Jessberger R, Kirkwood TB, Hoog C, Herbert M. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and sgo2. Curr Biol. 2010;20:1511–1521. doi: 10.1016/j.cub.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Liu L, Keefe DL. Defective cohesin is associated with age-dependent misaligned chromosomes in oocytes. Reprod Biomed Online. 2008;16:103–112. doi: 10.1016/s1472-6483(10)60562-7. [DOI] [PubMed] [Google Scholar]
- Loane M, Morris JK, Addor MC, Arriola L, Budd J, Doray B, Garne E, Gatt M, Haeusler M, Khoshnood B, Klungsoyr Melve K, Latos-Bielenska A, McDonnell B, Mullaney C, O'Mahony M, Queisser-Wahrendorf A, Rankin J, Rissmann A, Rounding C, Salvador J, Tucker D, Wellesley D, Yevtushok L, Dolk H. Twenty-year trends in the prevalence of down syndrome and other trisomies in europe: Impact of maternal age and prenatal screening. Eur J Hum Genet. 2013;21:27–33. doi: 10.1038/ejhg.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Ventura SJ, Osterman MJ, Wilson EC, Mathews TJ. Births: Final data for 2010. Natl Vital Stat Rep. 2012;61:1–71. [PubMed] [Google Scholar]
- Morris CR, Haigh S, Cuthbert G, Crosier M, Harding F, Wolstenholme J. Origin of trisomy: No evidence to support the ovarian mosaicism theory. Prenat Diagn. 2012;32:668–673. doi: 10.1002/pd.3885. [DOI] [PubMed] [Google Scholar]
- Nagaoka SI, Hodges CA, Albertini DF, Hunt PA. Oocyte-specific differences in cell-cycle control create an innate susceptibility to meiotic errors. Curr Biol. 2011;21:651–657. doi: 10.1016/j.cub.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat Rev Genet. 2012;13:493–504. doi: 10.1038/nrg3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TR, Feingold E, Yu K, Cheung V, Tinker S, Yadav-Shah M, Masse N, Sherman SL. New insights into human nondisjunction of chromo-some 21 in oocytes. PLoS Genet. 2008;4:e1000033. doi: 10.1371/journal.pgen.1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangalos CG, Talbot CC, Jr., Lewis JG, Adelsberger PA, Petersen MB, Serre JL, Rethore MO, de Blois MC, Parent P, Schinzel AA, Binkert F, Boue J, Corbin E, Croquette MF, Gilgenkrantz S, de Grouchy J, Bertheas MF, Prieur M, Raoul O, Serville F, Siffroi JP, Thepot F, Lejeune MJ, Antonarakis SE. DNA polymorphism analysis in families with recurrence of free trisomy 21. Am J Hum Genet. 1992;51:1015–1027. [PMC free article] [PubMed] [Google Scholar]
- Peters AH, Plug AW, van Vugt MJ, de Boer P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- Polani PE, Crolla JA. A test of the production line hypothesis of mammalian oogenesis. Hum Genet. 1991;88:64–70. doi: 10.1007/BF00204931. [DOI] [PubMed] [Google Scholar]
- Roberts R, Iatropoulou A, Ciantar D, Stark J, Becker DL, Franks S, Hardy K. Follicle-stimulating hormone affects metaphase I chromosome alignment and increases aneuploidy in mouse oocytes matured in vitro. Biol Reprod. 2005;72:107–118. doi: 10.1095/biolreprod.104.032003. [DOI] [PubMed] [Google Scholar]
- Robles P, Roig I, Garcia R, Ortega A, Egozcue J, Cabero LL, Garcia M. Pairing and synapsis in oocytes from female fetuses with euploid and aneuploid chromosome complements. Reproduction. 2007;133:899–907. doi: 10.1530/REP-06-0243. [DOI] [PubMed] [Google Scholar]
- Roig I, Robles P, Garcia R, Martin M, Egozcue J, Cabero L, Barambio S, Garcia M. Evolution of the meiotic prophase and of the chromo-some pairing process during human fetal ovarian development. Hum Reprod. 2005;20:2463–2469. doi: 10.1093/humrep/dei079. [DOI] [PubMed] [Google Scholar]
- Sachs ES, Jahoda MG, Los FJ, Pijpers L, Wladimiroff JW. Trisomy 21 mosaicism in gonads with unexpectedly high recurrence risks. Am J Med Genet. 1990;7:186–188. doi: 10.1002/ajmg.1320370737. [DOI] [PubMed] [Google Scholar]
- Sherman SL, Lamb NE, Feingold E. Relationship of recombination patterns and maternal age among non-disjoined chromosomes 21. Biochem Soc Trans. 2006;34:578–580. doi: 10.1042/BST0340578. [DOI] [PubMed] [Google Scholar]
- Steuerwald N, Cohen J, Herrera RJ, Sandalinas M, Brenner CA. Association between spindle assembly checkpoint expression and maternal age in human oocytes. Mol Hum Reprod. 2001;7:49–55. doi: 10.1093/molehr/7.1.49. [DOI] [PubMed] [Google Scholar]
- Tachibana-Konwalski K, Godwin J, van der Weyden L, Champion L, Kudo NR, Adams DJ, Nasmyth K. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev. 2010;24:2505–2516. doi: 10.1101/gad.605910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng LH, Chuang SM, Lee TY, Ko TM. Recurrent down's syndrome due to maternal ovarian trisomy 21 mosaicism. Arch Gynecol Obstet. 1994;255:213–216. doi: 10.1007/BF02335088. [DOI] [PubMed] [Google Scholar]
- Uchida IA, Freeman VC. Trisomy 21 down syndrome. Parental mosaicism. Hum Genet. 1985;70:246–248. doi: 10.1007/BF00273450. [DOI] [PubMed] [Google Scholar]
- Vogt E, Kirsch-Volders M, Parry J, Eichenlaub-Ritter U. Spindle formation, chromosome segregation and the spindle checkpoint in mammalian oocytes and susceptibility to meiotic error. Mutat Res. 2008;651:14–29. doi: 10.1016/j.mrgentox.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Wang H, Hoog C. Structural damage to meiotic chromosomes impairs DNA recombination and checkpoint control in mammalian oocytes. J Cell Biol. 2006;173:485–495. doi: 10.1083/jcb.200512077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon PW, Freeman SB, Sherman SL, Taft LF, Gu Y, Pettay D, Flanders WD, Khoury MJ, Hassold TJ. Advanced maternal age and the risk of down syndrome characterized by the meiotic stage of chromosomal error: A population-based study. Am J Hum Genet. 1996;58:628–633. [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Liu JG, Hoja MR, Wilbertz J, Nordqvist K, Hoog C. Female germ cell aneuploidy and embryo death in mice lacking the meiosisspecific protein scp3. Science. 2002;296:1115–1118. doi: 10.1126/science.1070594. [DOI] [PubMed] [Google Scholar]