

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) protein is a cAMP-regulated Cl− channel whose major function is to facilitate epithelial fluid secretion. Loss-of-function mutations in CFTR cause the genetic disease cystic fibrosis. CFTR is required for transepithelial fluid transport in certain secretory diarrheas, such as cholera, and for cyst expansion in autosomal dominant polycystic kidney disease. High-throughput screening has yielded CFTR inhibitors of the thiazolidinone, glycine hydrazide and quinoxalinedione chemical classes. The glycine hydrazides target the extracellular CFTR pore, whereas the thiazolidinones and quinoxalinediones act at the cytoplasmic surface. These inhibitors have been widely used in cystic fibrosis research to study CFTR function at the cell and organ levels. The most potent CFTR inhibitor has IC50 of approximately 4 nM. Studies in animal models support the development of CFTR inhibitors for antisecretory therapy of enterotoxin-mediated diarrheas and polycystic kidney disease.
Keywords: Chloride channels, cystic fibrosis, diarrhea, polycystic kidney disease, drug discovery
INTRODUCTION
The cystic fibrosis transmembrane conductance regulator (CFTR) gene encodes a cAMP-regulated Cl− channel that is expressed widely in mammalian epithelia in the airways and lung, gastrointestinal tract, sweat gland, and reproductive organs [1,2]. Loss-of-function mutations in the CFTR gene cause the genetic disease cystic fibrosis (CF), in which there is progressive deterioration of lung function, pancreatic insufficiency, infertility and other abnormalities [3,4]. Native (non-mutated) CFTR plays an important role in two human diseases: enterotoxin-mediated secretory diarrheas and autosomal dominant polycystic kidney disease (PKD). CFTR Cl− conductance is the rate-limiting step in transepithelial fluid secretion in epithelial cells lining the normal intestine (enterocytes) and lining renal cysts in PKD. There is compelling rationale, as discussed below, for application of CFTR inhibitors as antisecretory therapy for certain diarrheas such as cholera and for autosomal dominant PKD. CFTR inhibitors also have broad applications in basic research in delineating the functional involvement of CFTR in various cellular and physiological processes, and for pharmacological creation of the CF phenotype at the cell, tissue and animal levels.
Here, we review the currently available CFTR inhibitors, including their discovery, optimization and biological testing. We also review evidence supporting the potential utility of CFTR inhibitors for antisecretory therapy of diarrheas and PKD. The reader is referred elsewhere for discussion of the biology and pharmacology of other mammalian Cl− channels, including Ca2+-activated, voltage-gated, volume-sensitive and ligand-gated Cl− channels [5].
CFTR STRUCTURE AND FUNCTION
The CFTR gene was identified in 1989 as the genetic basis of CF. There are now more than 1500 distinct loss-of-function mutations in CFTR that cause CF. The most common CF-causing mutation is ΔF508 (deletion of phenylalanine at position 508), with ~90 % of CF subjects having the ΔF508 mutation as one or both CFTR gene alleles. How do the loss-of-function mutations in CFTR cause CF pathology is the subject of ongoing research. As CFTR is involved in transepithelial ion and fluid transport, one set of explanations postulates that defective CFTR anion (Cl− and HCO3−) transport in airway epithelial cells results in a relatively dehydrated, hyperacidic surface liquid layer that impairs mucociliary clearance and promotes bacterial colonization. Defective transepithelial fluid secretion in CF is also postulated to account for the other major clinical manifestations of CF, including pancreatic and biliary insufficiency, meconium ileus and male infertility.
CFTR is a member of the ATP-binding cassette transporter (ABC) superfamily. Members of the ABC superfamily contain two homologous nucleotide binding domains (NBDs) and two membrane spanning domains (each composed of six membrane-spanning alpha helices) [6]. CFTR also contains a regulatory domain (R) between the first membrane spanning and second nucleotide binding domains, whereas other ABC transporters lack a R domain. The two NBDs (NBD1 and NBD2) are oriented in a head-to-tail manner. CFTR activation involves cAMP-dependent phosphorylation of multiple residues in the R domain, as well as ATP binding and hydrolysis at the NBDs, the details of which have been the subject of extensive research.
There is limited information about CFTR structure. Early structural work utilizing electron-diffraction crystallography on 2-dimensional crystals of full-length CFTR provided a low-resolution structure of the entire protein [7]. Subsequently, cryo-electron microscopy gave low-resolution three-dimensional structures of CFTR in its phosphorylated and non-phosphorylated states [8]. More recently, an improved higher-resolution structure of full-length CFTR was obtained by electron crystallography on 2-dimensional crystals of CFTR [9], with the electron density data fit into a high-resolution x-ray crystal structure of Sav1866, a well-characterized ABC protein from Staphylococcus aureus [10]. High-resolution x-ray crystal structures have also been determined on the isolated cytoplasmic NBD domains of CFTR, both in monomeric and head-to-tail dimeric forms [11]. Also, several homology models of full-length CFTR have been reported based on high-resolution structures of homologous templates such as bacterial Sav1866 and MsbA [12,13].
ORIGINAL CFTR INHIBITORS
Prior to small molecule screening, several non-selective and relatively low-affinity inhibitors of CFTR Cl− conductance were available, including glibenclamide, diphenylamine-2-carboxylate and 5-nitro-2-(3-phenylpropyl-amino)benzoate (Fig. 1). These compounds inhibit Cl− transport by CFTR as well as other Cl− channels and transporters with IC50 generally >100 μM. One of the more widely used Cl− channel inhibitors, glibenclamide, was initially discovered and primarily used as an oral antidiabetic drug targeting an ATP-sensitive K+ channel in pancreatic islet beta cells. An initial study reported α-aminoazaheterocyclic-methylglyoxal adducts as CFTR inhibitors with low picomolar potency [14]; however, subsequent studies using multiple independent CFTR assays done by independent labs showed that the reported adducts did not inhibit CFTR at concentrations up to 100 μM [15]. The availability of potent and selective inhibitors of Cl− channels has remarkably lagged that of cation channels.
Fig. (1).

Chemical structures of small-molecule CFTR inhibitors. Structure shown of older CFTR inhibitors (DPC, NPPB, glibenclamide), the thiazolidinone CFTRinh-172, the hydrazides GlyH-101 and MalH-PEG and the PPQ/BPO inhibitors PPQ-102 and BPO-27.
HIGH-THROUGHPUT SCREENING FOR CFTR INHIBITORS
Various assays have been applied to measure anion transport across cell membranes. Early assays, which are not easily adaptable to high-throughput screening, involve measurement of 36Cl− or 131I− cellular uptake or efflux. Indirect assays based on measurement of cell membrane potential or volume have also been used; however, the caveat in these indirect measurements is the multiple determinants of membrane potential and cell volume such as the activities of non-CFTR membrane transporters. Small-molecule (chemical) Cl− sensors such as SPQ and MQAE have been used widely in cell culture and tissue measurements [16], though their relatively dim blue fluorescence and need for cell loading and repeated washing limit their utility for high-throughput screening applications. Another concern is the sensitivity of quinolinium-based indicators to non-Cl− cellular anions. A yellow-fluorescent I−-selective chemical sensor (LZQ) [17] was developed for screening applications that is substantially brighter than the quinolinium-based indicators, though it has not been used in screening applications because better, genetically encoded halide sensors were developed soon thereafter. Several halides are conducted by CFTR, including Cl−, I− and Br−, and, to a lesser extent, HCO3−.
Genetically encoded fluorescent sensors generated by mutation of green fluorescent protein (GFP) have been of great utility in Cl− channel drug discovery. GFP is a fluorescent protein of ~30 kdalton molecular size that can be stably expressed in cytoplasm or targeted to specified organellar compartments. The original GFP variants are sensitive to pH but not to halides. Halide sensitivity was conferred to GFP using a rational mutagenesis strategy based upon crystallographic data, in which several point mutations allowed halide access near the GFP chromophore [18]. The fluorescence of the resultant ‘yellow fluorescent protein’ (YFP) is red-shifted by ~20 nm (to 528 nm) compared to GFP, and is sensitive to halide concentration. The original halide-sensing YFP, YFP-H148Q, is 50 % quenched by ~100 mM Cl− or 20 mM I− [19]. Targeted mutagenesis of YFP-H148Q yielded YFP-based sensors with improved halide quenching efficiency and brightness [20]. YFP-H148Q/I152L has the highest I− sensitivity of the YFP sensors, with 50% fluorescence quenching at ~3 mM I−. The halide-sensing mechanism of YFPs involves a shift in pKa with altered halide concentration. The pKa of YFP-H148Q/I152L increases from ~7 to 8 with increasing halide concentration, such that at constant pH YFP fluorescence is reduced with increasing halide concentration. YFP-H148Q/I152L fluorescence responds in <100 ms to changes in halide concentration or pH. For screening applications or transport measurements in which YFPs are stably expressed in cytoplasm, it is important to acknowledge that YFP fluorescence is sensitive to changes in both halide concentration and pH. For ratio imaging, dual-wavelength YFP-based sensors have been developed by genetically fusing YFPs with red fluorescent proteins [21].
YFP-H148Q/I152L has been used extensively for chloride channel drug discovery in identification of activators and inhibitors of wildtype and mutant CFTRs [22-26] and of Ca2+-activated Cl− channels [27-30]. (Fig. 2A) shows one strategy used for identification of CFTR inhibitors. The assay utilizes Fischer rat thyroid (FRT) cells, an epithelial cell line that was found to grow rapidly and adhere well to plastic multi-well plates. Importantly, FRT cells have low intrinsic halide permeability that is CFTR-independent, and allow strong, stable expression of both CFTR and the YFP sensor [31]. FRT cells form electrically tight monolayers, allowing efficient short-circuit current measurements in secondary screening. The original CFTR inhibitor screening assay was implemented in a 96-well format utilizing an automated screening platform [23]. CFTR halide conductance in FRT cells stably coexpressing human wildtype CFTR and YFP-H148Q/I152L was activated by an agonist mixture targeting different CFTR activation pathways, reasoning that active compounds would likely target CFTR itself. Following incubation for 10 min with test compounds, fluorescence was measured for 2 seconds before, and for 12 seconds after rapid addition of I− to the extracellular solution by automated injection into each well. I− rather than Cl− was chosen because of the strong and selective YFP-H148Q/I152L quenching by I−, the high CFTR I− conductance, and the minimal CFTR-independent I− leak in FRT cells. As shown in (Fig. 2B), fluorescence following I− addition was stable in the absence of agonists, indicating minimal I− leak, but declined rapidly with agonists; CFTR inhibitors are identified from the reduced negative slope. The assay is sensitive and robust, allowing high-throughput identification of CFTR inhibitors with few if any false positives.
Fig. (2).

Thiazolidinone CFTR inhibitors identified by high-throughput screening. A. Screening protocol in which cells co-expressing CFTR and a halide-sensitive YFP fluorescent protein were cultured in 96-well microplates. After 24-48 hours, CFTR was activated by an agonist mixture (forskolin, apigenin, IBMX). After addition of test compound an I− containing solution was added. B. Original fluorescence data from individual wells showing controls (no activators, no test compound) and test wells. C. (top) Structure of the thiazolidinone CFTRinh-172. (center) Concentration-dependence of CFTR inhibition. (bottom) Inhibition of short-circuit current in permeabilized FRT cells expressing human wildtype CFTR after stimulation by 100 μM CPT-cAMP. Adapted from ref. 23.
SMALL-MOLELCULE CFTR INHIBITORS
Thiazolidinones
The first high-throughout screen identified thiazolidinone-class CFTR inhibitors, which, after analog screening, yielded the compound CFTRinh-172, 3-[(3-trifluoromethyl)phenyl]-5-[(4-carboxyphenyl)methylene]-2-thioxo-4-thiazolidinone (Fig. 2C) [23]. In short-circuit current measurements, CFTRinh-172 inhibits CFTR Cl− conductance with IC50 of ~0.2-4 μM, depending on cell type. (Fig. 2C) show concentration-dependent CFTR inhibition in transfected FRT cells as measured by short-circuit current analysis. CFTRinh-172 has a single negative charge at physiological pH (carboxylic acid), and thus its partitioning into cytoplasm is dependent on cell plasma membrane potential as well as possible facilitated transport by organic anion transporters. The strongly interior-negative membrane potential of epithelial cells thus can reduce CFTRinh-172 inhibition potency. Nonetheless, CFTRinh-172 has been used extensively as a presumed selective inhibitor to identify CFTR currents in various cell types and to investigate the functional role of CFTR at the cell and organ levels [32,33]. As described below, thiazolidinones have shown antisecretory activity in rodent models of cholera and polycystic kidney disease.
Single-channel patch-clamp analysis indicated that CFTRinh-172 is a closed-channel stabilizer in which channel closed time is prolonged following CFTRinh-172 addition [34]. The current-voltage relationship for CFTR Cl− current remains linear after CFTRinh-172, but reduced in slope. A more extensive patch-clamp study proposed a six-state model in which CFTRinh-172 binds to both the open and closed states of CFTR [35]. Mutations of arginine-347 in CFTR greatly reduced CFTRinh-172 inhibition potency with little effect on CFTR channel function [36], suggesting that CFTRinh-172 binds at or near arginine-347 on the intracellular surface of CFTR without direct pore occlusion. The large variance in CFTRinh-172 inhibition potency for CFTRs from different species [37] may be related to CFTR sequence difference at or near arginine-347.
In pharmacological studies, 14C-labeled CFTRinh-172 was cleared primarily by renal glomerular filtration with minimal chemical modification [38]. CFTRinh-172 accumulated in liver within a few minutes after intravenous injection in mice, and was concentrated ~5-fold in bile over blood, suggesting enterohepatic recirulation. At later times, CFTRinh-172 accumulated mainly in liver, intestine and kidney, with little detectable in brain, heart, skeletal muscle or lung. Pharmacokinetic analysis in rats following intravenous bolus infusion showed a distribution volume of 770 ml with redistribution and elimination half-times of 0.14 and 10.3 h, respectively. CFTRinh-172 was quite stable in hepatic microsomes and showed no measurable toxicity at concentrations more than 100-fold its EC50 for inhibition of secretory diarrheas.
There are two, 4-step synthetic routes to CFTRinh-172, both utilizing 3-triflouromethylaniline as starting material (Fig. 3A) [23]. The first route utilizes carbon disulfide to give the dithiocabamate, giving the carboxylic acid upon reaction with bromoacetic acid which cyclizes with catalytic acid to the thiazolidinone. Alternatively, this intermediate can be made in a one-pot reaction (shown as steps e-g) in higher yields. The thiazolidinone intermediate then condenses with 4-formylbenzoic acid using catalytic piperidine to give CFTRinh-172. For SAR analysis, sixty-nine CFTRinh-172 analogs were synthesized, exploring modifications throughout the structure [39]. One goal of the SAR analysis was to identify thiazolidinones with improved aqueous solubility compared to CFTRinh-172 (aqueous solubility < 20 μM). (Fig. 3B) summarizes the structural determinants for CFTR inhibition by thiazolidinones. Ring A was derivatized widely with various substituents, but none were as good as the 3-trifloromethyl moiety for CFTR inhibition. The thiazolidinone ring B was replaced by various other 5 member rings including thiazlidinedione, aminothiadiazole, maleimide, succinimide thiazole, thiadiazole, and 1,2,3-triazole. Ring C was replaced with pyridine, pyridine N-oxide, tetrazole, furan and substituted carboxylic acids, esters, amides, hydroxyl, methoxy and sulfonate, all in an attempt to increase the polarity and H-bonding capacity. Linker 1 was lengthened, and linker 2 was lengthened, shortened, saturated, substituted with nitrogen, sulfur, a thioamide, and removed completely. The greatest CFTR inhibition potency was found for 3-CF3 and polar group-substituted-phenyl rings, and a thiazolidinone core. Two compounds with submicromolar CFTR inhibition potency and solubility >180 μM were identified: tetrazolo-172, containing 4-tetrazolophenyl in place of 4-carboxyphenyl, and Oxo-172, containing thiazolidinedione in place of thiazolidinone.
Fig. (3).

Synthesis of CFTRinh-172 and thiazolidinone SAR. A. Reagents; (a) CS2 TEA, EtOAc, rt, 12 h; (b) BrCH2COOH, NaHCO3 rt, 2 h; (c) HCl, reflux, 2 h; (d) 4-formylbenzoic acid, piperidine, EtOH, reflux 2 h; (e) CSCl2 TEA; (f) HSCH2COOH, Et3N, 4h; (g) HCl, reflux, 2 h. B. SAR showing requirements for CFTR inhibition activity. Adapted from ref. 39.
Glycine and Malonic Acid Hydrazides
Additional screening designed to identify rapidly acting CFTR inhibitors yielded the glycine hydrazides class of inhibitors [40]. The structure of N-(2-naphthalenyl)-[(3,5-dibromo-2,4-dihydroxyphenyl) methylene]glycine hydrazide (GlyH-101) is shown in (Fig. 1). GlyH-101 contains a hydrophobic napthalene linked by a glycine hydrazide to a dibromodihydroxyphenyl moiety that has a single negative charge at physiological pH. GlyH-101 has good water solubility and rapidly inhibits CFTR Cl− conductance with low micromolar IC50. Whole-cell current measurements revealed that GlyH-101 produces a voltage-dependent CFTR block, altering the CFTR current-voltage relationship from linear to strongly inwardly rectifying such that IC50 was increased from 1.4 μM at +60 mV to 5.6 μM at −60 mV (Fig. 4A). The potency for GlyH-101 inhibition of CFTR Cl− current was decreased by reducing extracellular Cl− concentration. Single-channel analysis indicated fast channel closures within bursts of channel openings (Fig. 4B), with a reduction in mean channel open time from 264 to 13 ms at −60 mV at 5 μM GlyH-101. The electrophysiological measurements suggested a pore blocking mechanism in which GlyH-101 binds within the CFTR pore near its extracellular entrance. Recent computational chemistry suggested the site of GlyH-101 binding to CFTR [41]. Molecular dynamics was used to equilibrate a homology model of CFTR, which was validated by site-directed mutation of residues at the suspected binding site. It was concluded that GlyH-101 binds in the membrane-spanning pore region of CFTR (Fig. 4C). The unique extracellular site of action of GlyH-101 was proven by showing that a macromolecular polyethyleneglycol (PEG) conjugate of GlyH-101 inhibited CFTR Cl− current when added from the extracellular surface [42].
Fig. (4).

CFTR inhibition by GlyH-101. A. (left) Whole-cell membrane currents evoked by voltages from −100 to +100 mV (in 20 mV steps) in CFTR-expressing FRT cells after CFTR activation by 5 μM forskolin. 10 μM GlyH-101 added where indicated. (right) Current-voltage relationships in the absence of inhibitors (control, open circles), after addition of 10 μM (filled squares) and 30 μM (open squares) GlyH-101, after washout of 10 μM GlyH-101 (recovery, shaded circles) and after addition of 5 μM CFTRinh-172 (filled circles). B. Single channel analysis of CFTR inhibition by GlyH-101. Representative traces (left) and corresponding amplitude histograms (right) obtained from a cell-attached patch. CFTR was activated by forskolin (5 μM) in the absence and presence of GlyH-101 at indicated concentrations. Dashed lines show zero current level (channels closed) with downward deflections indicating channel openings. Adapted from ref. 36. C. GlyH-101 bound to a homology model of CFTR and validated by mutation studies based on ref. 41.
GlyH-101 is synthesized in 3 steps (Fig. 5A) in which 2-napthylamine is alkylated with ethyl iodoacetate to give N-2-napthylenylglycine ethyl ester, which is cleaved with hydrazine to give N-2-napthylenylglycine hydrazide, which condenses with 3,5-dibromo-2,4-dihydroxy-benzaldehyde to give the hydrazone GlyH-101. SAR of GlyH-101 was done in order to design active macromolecular conjugates [40] (Fig. 5B). Maximal inhibitory potency was achieved utilizing 2-naphthylamine and 3,5-dibromo-2,4-dihydroxybenzaldehyde condensed with an alpha substituted glycine hydrazide linker (N’-[(E)-(3,5-dibromo-2,4-dihydroxy-phenyl) methylidene] hydrazide). This structure proved remarkably tolerant to substitution alpha to the carbonyl (CH2 in GlyH-101). This site yielded active compounds when substituted or derivatized with a variety of groups including phenyl, carbonyl, esters, hydrazides, saturated carbon chains and those ending in alcohols. Analogs containing a napthylamide were synthesized by utilizing monoethyl oxalyl chloride in place of ethyl iodoacetate, some of which had better CFTR inhibition potency than their glycine-containing counterparts. Based on the tolerance of the alpha CH2 to substitution, non-absorbable malondihydrazides were synthesized by coupling 2,4-disulfobenzaldehyde, 4-sulfophenylisothiocyante, and PEG moieties to 2-naphthalenylamino-[(3,5-dibromo-2,4-dihydroxyphenyl)methylene] propanedioic acid dihydrazide [42,43], as discussed below.
Fig. (5).

Synthesis of GlyH-101 and SAR analysis. A. (a) Ethyl iodoacetate, NaOAc, H2O, 90 °C, 3 h, 65%; (b) EtOH, N2H4, reflux, 82%; (c) 3,5-dibromo-2,4-dihydroxybenzaldehyde, EtOH, 3 h, 78%. B. SAR showing requirements for CFTR inhibition activity. Adapted from ref. 40.
A series of multivalent MalH macromolecular conjugates was synthesized with the goal of improving CFTR inhibition potency and reducing CFTR dissociation rate. It was reasoned that externally acting, non-absorbable CFTR inhibitors could be useful for oral therapy of secretory diarrhea provided that their binding to CFTR is sufficiently tight to resist convective washout by rapid fluid secretion in intestinal crypts. In one set of studies mono- and divalent macromolecular conjugates were synthesized by coupling MalH to PEGs of different molecular sizes (0.2-100 kDa) via the disulfonic stilbene linker DIDS [43]. DIDS has two isothiocyanate (SCN-R) groups at the 4 and 4′ ends of a rigid sulfonated stilbene molecule. Isothiocyanates are reactive species that form stable thiourea linkages on contact with amines or hydrazides. The sulfate groups greatly enhance polarity hence water solubility in the linker. IC50 for inhibition of CFTR Cl− conductance was 10-15 μM for the monovalent MalH-PEGs, but substantially lower for divalent MalH-PEG-MalH conjugates, decreasing from 1.5 to 0.3 μM with increasing PEG size, and showing positive cooperativity. It was proposed that the relatively high CFTR inhibition potency of the divalent MalH-PEG conjugates may be a consequence of the increased concentration at the external CFTR pore afforded by one-site / two-ligand binding. As found for GlyH-101, patch-clamp analysis showed voltage-dependent CFTR block with inward rectification, with shortened single channel openings. Though IC50 was improved remarkably for the divalent MalH-PEG conjugates compared to the original GlyH-101, inhibitor dissociation from CFTR remained rapid.
To further improve IC50 and slow inhibitor dissociation, various MalH-lectin conjugates were synthesized, reasoning that tetravalent binding of the lectin moiety to the dense extracellular glycocalyx might result in entrapment of inhibitor at the cell surface and reduce CFTR-inhibitor dissociation because of additive energetic effects of concurrent MalH binding to CFTR and lectin binding to the glycocalyx [44]. MalH was conjugated to several lectins via a highly polar disulfonic stilbene linker. The lectin conjugation remarkably reduced IC50 for inhibition of CFTR Cl− conductance to ~50 nM and resisted washout. Patch-clamp analysis indicated CFTR inhibition by an external pore occlusion mechanism as found for GlyH-101 and the MalH-PEG conjugates. In support of lectin-glycocalyx binding as the mechanism for the high potency of MalH-lectin conjugates, potent CFTR inhibition was not seen after MalH-lectin heat-denaturation, protease digestion, or competition by mannose. In suckling mice in vivo, fluorescently labeled MalH-lectin remained bound in the intestine for >6 hours after washout, whereas washout occurred in a few minutes without the lectin. As discussed below, the MalH-lectin conjugates was effective in mouse models of cholera.
Pyrimido-pyrrolo-quinoxalinediones
Large-scale screening yielded a third chemical class of small molecule CFTR inhibitors, the pyrimido-pyrrolo-quinoxalinediones (PPQs) [45]. Primary screening and follow-on testing of 347 commercially available PPQ analogs gave 7,9-dimethyl-11-phenyl-6-(5-methylfuran-2-yl)-5,6-dihydro-pyrimido[4′,5′-3,4]pyrrolo[1,2-a]quinoxaline-8,10-(7H,9H)-dione, (PPQ-102, Fig. 1) as the most potent compound. PPQ-102 fully inhibited CFTR Cl− current by stabilizing the channel closed state (Fig. 6A) with IC50 ~90 nM (Fig. 6B). Unlike the thiazolidinones and glycine hydrazides, PPQ-102 is uncharged at physiological pH and therefore not subject to membrane potential-dependent cellular partitioning or block efficiency. Initial testing in a neonatal kidney organ culture model of PKD, as discussed further below, demonstrated the efficacy of PPQ-102 in preventing cyst expansion and reducing the size of pre-formed cysts. Though PPQ-102 was the most potent CFTR inhibitor at the time of its identification, concerns for its further development included its low aqueous solubility (<5 μM) and poor metabolic stability, as it was found to undergo rapid hydroxylation and aromatization in hepatic microsome assays in vitro.
Fig. (6).

CFTR inhibition by PPQ / BPO compounds. A. Single-channel patch-clamp recordings in the cell-attached configuration in CFTR-expressing FRT cells. CFTR was activated by 10 μM forskolin and 100 μM IBMX (holding voltage +80 mV). B. Dose-response for inhibition of CFTR Cl− current in which CFTR was activated by 100 μM CPT-cAMP. C. CFTR inhibition by enantiopure (R)-BPO-27 shown by short-circuit current. Adapted from refs. 45 and 46. D. (left) Low resolution structure of CFTR. (right) (R)-BPO-27 docked into the ATP-binding site of a high-resolution crystal structure of the head-to-tail NBD1-NBD1 homodimer (pdb = 2PZE).
A targeted SAR study was done to identify compounds with improved potency, metabolic stability and aqueous solubility compared to PPQ-102 [46]. The best compound to emerge from the study was BPO-27, which fully inhibited CFTR with IC50 ~ 8 nM, and had >10-fold greater metabolic stability and aqueous solubility compared to PPQ-102. BPO-27 inhibition of CFTR is voltage-independent. BPO-27 was synthesized in 5 steps with an overall yield of 51%, starting with alkylation of 6-methyluracil with dimethylsulfate to give trimethyluracil, which was acylated with benzoyl chloride to generate the corresponding phenylketone (Fig. 7A). The phenylketone was then brominated and condensed with ethyl 3-amino-4-hydroxybenzoate yielding the pyrrole, which was condensed with 5-bromofufural and then saponified to give BPO-27. Compared to PPQ-102, BPO-27 contains a bromine substituent at the 5-position of the furan ring and replacement of the secondary amine with an ether bridge, which prevent hydroxylation and aromatization, respectively, as well as a carboxylation, which increases its polarity and aqueous solubility. The SAR analysis is summarized in (Fig. 7B).
Fig. (7).

Synthesis of BPO-27 and SAR analysis. A. (a) (CH3)2SO4, H2O, NaOH, 3 d, 98%; (b) ZnCl2, PhCl, PhCOCl, 66%; (c) DCM, Br2 100%; (d) ethyl 3-amino-4-hydoxybenzoate, ethanol, reflux 98.5%; (e) 5-bromofufural, CHCl3, HBr, AcOH, 3A sieves, 150 °C, 76.4%; (f) KOH, THF, H2O, 3 d, 83.2%. B. SAR showing requirements CFTR inhibition activity. Adapted from ref. 46.
As BPO-27 contains a single chiral center, in a follow-on study the enantiomers of BPO-27 were separated by chiral supercritical fluid chromatography and their absolute configuration and activity determined. Enantiomer R, as determined by x-ray crystallography, inhibited CFTR chloride conductance with IC50 ~ 4 nM (Fig. 6C), while enantiomer S was inactive, indicating absolute stereo-specificity. Both enantiomers were stable in vitro in hepatic microsomes, with <5 % metabolism in 4 h. Pharmacokinetics in mice showed t1/2 ~ 2 h for BPO-27 in serum following bolus intravenous administration, with good accumulation in kidney.
We recently used computational modeling to identify a possible site of BPO-27 binding to CFTR. Fig. 6C shows a putative binding site for the active R enantiomer on a high-resolution x-ray crystal structure of the NBD1-NBD1 head-to-tail homodimer, a model of NBD1-NBD2 (PDB = 2PZE; ref. 7). The putative binding site is located at the site of the co-crystallized ATP molecule. Electrophysiological and mutagenesis analysis will be required to validate BPO-27 binding near the ATP binding site on NBD1.
POTENTIAL CLINICAL APPLICATIONS OF CFTR INHIBITORS
Secretory Diarrhea
Secretory diarrhea is a major cause of mortality globally, particularly in children, accounting for an estimated 15% of childhood deaths. In addition, repeated diarrheal episodes in childhood have been correlated with malnutrition, growth stunting, and impaired physical and mental development [47]. Secretory diarrheas are characterized by large losses of fluid and electrolytes from the intestine. Fluid secretion in secretory diarrheas occurs secondary to active chloride transport across the epithelium [48]. CFTR provides a major route for intestinal chloride secretion in the intestine, and hence pharmacological inhibition of CFTR Cl− conductance has potential therapeutic value in limiting diarrhea initiated by a variety of different environmental pathogens. The primary mechanism of diarrhea induction varies from pathogen to pathogen and includes: activation of chloride and fluid secretion, induction of the host inflammatory/ immune responses in the intestinal mucosa, and direct damage to the epithelium. Examples of diarrheas with a primary secretory component include cholera (Vibrio cholerae), Travelers’ diarrhea (Enterotoxigenic Escherichia coli) and possibly some viral diarrheas (rotavirus, norovirus). Other pathogens such as Shigella and Giardia lamblia have a more inflammatory or direct damaging mechanism. Similarly, non-infectious diarrheas associated with inflammatory bowel disease, AIDS and chemotherapy may have a lesser secretory component. The primary treatment for secretory diarrheas is oral rehydration solution (ORS), which treats the major consequence of intestinal fluid loss, dehydration, by replacing fluid and promoting intestinal fluid absorption. Despite its huge success, ORS therapy has limitations, including its availability and consistent use, as well as its incomplete efficacy in severe diarrheas in the young and elderly. Although ORS administration is the first-line therapy of diarrheal disease, anti-secretory drugs that reduce intestinal fluid loss are predicted to be useful as adjunctive therapy, or as primary therapy when ORS or intravenous fluid therapy are not available, such as during and just after a natural disaster.
(Fig. 8A) diagrams the cellular mechanisms of intestinal fluid secretion, showing the involvement of CFTR as a luminal membrane Cl− channel on enterocytes, which are the epithelial cells lining the intestinal wall in contact with the luminal fluid. The intestinal epithelium is comprised of long, finger-like projections (villi) and cylindrical glands (crypts), both of which are lined by enterocytes and contribute to intestinal fluid secretion. Fluid secretion is driven by active Cl− transport from the basolateral to the apical side of enterocytes, obligating paracellular secretion of the counter ion, Na+, which generates an osmotic gradient that drives transepithelial water secretion. Cl− is transported from blood into the cytoplasm by the Na+/K+/2Cl− cotransporter (NKCC1), which is driven by Na+ and Cl− concentration gradients produced by the Na+K+-ATPase and basolateral K+ channels. The interior-negative membrane potential and high cytoplasmic Cl− concentration drive Cl− secretion across the cell apical membrane into the intestinal lumen through CFTR and Ca2+-activated Cl− channels (CaCCs). Various stimuli can cause enterocyte Cl− secretion. Bacterial enterotoxins such as cholera toxin from Vibrio cholerae and heat-stable enterotoxin (STa) from Escherichia coli activate Cl− secretion primarily through CFTR, though CaCCs may be involved as well. Some viral enterotoxins, such as rotaviral NSP4, cause elevation in cytoplasmic Ca2+ and consequent activation of CaCCs [49,50]. In addition, there are complex inputs from neuronal pathways involving 5-hydroxytryptamine release from enterochromaffin cells, and from inflammatory pathways involving release of prostaglandins, interleukins, and other mediators that result in increased cellular cAMP and Ca2+ [51,52]
Fig. (8).

CFTR inhibitor application in secretory diarrheas. A. Intestinal chloride and fluid secretion mechanisms. Enterocyte showing major luminal (top) and basal (bottom) transporters, ion channels and second messengers involved in chloride secretion. EC–enterochromaffin, 5–HT-5-hydroxytriptamine, ACh–acetylcholine, VIP–vasoactive intestinal peptide, SP–substance P. B. Antidiarrheal properties of CFTRinh-172. (left) Closed intestinal loops were injected with 1 μg cholera toxin. Dose-response for inhibition of fluid accumulation. Mice were given single dose of CFTRinh-172 by intraperitoneal injection and loop weight measured at 6 hours. (center) Intestinal loops 6 hr after cholera toxin or saline injection. (right) Improved survival of suckling mice following gavage with cholera toxin without vs. with the lectin conjugate MalH-ConA (125 pmol). Adapted from refs. 44 and 55.
There is strong evidence that CFTR is the major apical membrane Cl− pathway in secretory diarrheas caused by the bacterial enterotoxins released in cholera and Travelers’ diarrhea [51-54]. Both the small intestine and colon show robust cAMP-activated CFTR Cl− currents [55]. Intestinal Cl− and fluid secretion are absent in mice lacking CFTR, and in CF patients [56-58]. Colonic Cl− transport in human tissue is effectively blocked by CFTR inhibitors [55]. The alternative, CaCC-mediated pathway, may be involved as well in these diarrheas, and may represent the primary pathway for apical membrane Cl− secretion in some viral and drug-induced secretory diarrheas [59]. There is probably extensive cross-talk in cAMP and Ca2+ signalling mechanisms in the intestinal epithelium, in which cAMP elevation may increase cytoplasmic Ca2+ though the exchange protein directly activated by cAMP (Epac) and conversely Ca2+ elevation may increase cytoplasmic cAMP by Ca2+-sensitive adenylyl cyclases or directly signal CFTR via activation of protein kinase C [60-62]. At present, there is insufficient data in enterocytes in intact intestine to quantify the importance of these cross-talk pathways. Also, the molecular identity of intestinal CaCC(s) remains unknown, making it difficult to evaluate their relative contribution to different diarrheas. Notwithstanding these gaps in current knowledge of mechanisms of secretory diarrheas in humans it is likely that CFTR inhibition therapy would be beneficial in cholera, Travelers’ diarrhea, and other diarrheas associated with cAMP elevation, such as that caused by tumors that release vasoactive intestinal peptide.
CFTR inhibition therapy for diarrhea requires acheiving therapeutic drug concentrations in enterocytes for inhibitors targeting the intracellular surface of CFTR, or, for inhibitors targeting the lumen-facing external surface of the CFTR protein, within the fluid bathing the surface of crypts and villi. For treatment of secretory diarrheas in developing countries, the cost of therapy should be very low and the drug stable in a hot, humid environment. Since the bulk of fluid secretion of most enterotoxin-mediated secretory diarrheal episodes occurs over the course of 1-5 days, the duration of inhibitor therapy would be generally limited to a few days or less.
Notwithstanding the limitations of animal models of secretory diarrheas, thiazolidinone and glycine hydrazide CFTR inhibitors have shown efficacy in variety of diferent models across a number of different species. These CFTR inhibitors strongly reduce Cl− currents in vitro when applied directly to mouse, rat, rabbit and human intestinal tissue [23,55]. In an initial study in vivo, a single intraperitoneal injection of 200 μg CFTRinh-172 blocked intestinal fluid secretion in a rat closed-loop model by > 90% for cholera toxin (Fig. 8B, left) and > 70% for STa E. coli toxin [23]. In mice, 20 μg CFTRinh-172 inhibited cholera toxin-induced intestinal fluid secretion by 90%. The inhibition lasted for many hours with t1/2 ~10 h. In addition, an oral CFTRinh-172 preparation reduced fluid secretion by > 90% in a mouse open-loop cholera model [55].
The monovalent and divalent MalH-PEG conjugates were also shown to be effective in preventing intestinal fluid accumulation in a mouse closed-loop model of cholera in which cholera toxin together with inhibitor were injected into closed mid-jejunal loops [42,43]. A MalH-lectin conjugate, which, as mentioned above, resists intestinal washout because of entrapment in the enterocyte glycocalyx, was shown to reduce fluid accumulation in intestinal closed loops and improve survival in a suckling mouse model of cholera (Fig. 8B, right) [44].
Though the glycine hydrazides showed efficacy in various mouse models of secretory diarrhea, a largely ignored concern in the use of externally targeted channel inhibitors is washout due to convection caused by rapid fluid secretion. Unfortunately, convection effects cannot be addressed in animal models because of differences from human intestine in crypt-villus geometry and fluid secretion rates. Fluid convection is predicted to reduce inhibitor concentration at the enterocyte surface and hence reduce inhibitor efficacy. We recently developed a convection-diffusion model of inhibitor washout in an anatomically accurate 3-dimensional model of human intestine comprising cylindrical crypts and villi secreting fluid into a central lumen [63]. The model computed inhibitor efficacy as percentage inhibition of net secreted fluid. The model predicted marked reduction in inhibitor efficacy for high crypt fluid secretion as occurs in cholera, such that the antisecretory efficacy of a surface-targeted inhibitor requires both: (i) high inhibitor affinity (low nanomolar Kd) in order to obtain sufficiently high luminal inhibitor concentration (> 100-fold Kd); and (ii) sustained high luminal inhibitor concentration or slow inhibitor dissociation compared to oral administration frequency. Modeling of a systemically absorbed, externally acting CFTR inhibitor predicted a similar convective washout effect, as inhibitor entering intestinal crypts from blood would be rapidly washed out. It was concluded that surface-targeted small molecules with micromolar Kd are unlikely to be efficacious for therapy of secretory diarrhea, while macromolecular conjugates with nanomolar Kd and very slow dissociation, such as the MalH-lectin conjugates, might be effective. Therefore, though surface-targeted glycine and malonic acid hydrazides originally appeared to be attractive candidates for antisecretory therapy, the absorbable CFTR inhibitors, the thiazolidinones and PPQ/BPO compounds, are better development candidates for CFTR inhibitor therapy of enterotoxin-mediated secretory diarrheas.
Polycystic Kidney Disease
PKD is one of the most common human genetic diseases. PKD is a major cause of chronic renal failure in which massive enlargement of fluid-filled cysts leads to progressive destruction of normal kidney tissue. Human autosomal dominant PKD is caused by mutations in one of two genes, PKD1 and PKD2, which encode the interacting proteins polycystin-1 and polycystin-2, respectively [64]. Cyst growth in PKD involves fluid secretion into the cyst lumen coupled with hyperplasia of epithelial cells lining cyst walls, which are of tubular origin and express CFTR [65]. As in the intestine, transepithelial fluid secretion into the cyst lumen in PKD involves Cl− transport through luminal CFTR (Fig. 9A). There is strong evidence for the involvement of CFTR in cyst growth in PKD. CFTR-deficient mice are resistant to cyst formation, and CFTR inhibitors or mutations block cyst formation in cell/organ culture and in vivo models [66-69]. In rare families afflicated with CF and PKD, individuals with both genetic disorders have less severe renal disease than those with PKD alone [70-72]. Cyst expansion in PKD also requires epithelial cell proliferation involving mTor signaling [73], which is the basis of anti-proliferative therapies under development [74,75]. Antisecretory therapy using CFTR inhibitors is predicted to inhibit cyst expansion and complement antiproliferative therapy [76-78].
Fig. (9).
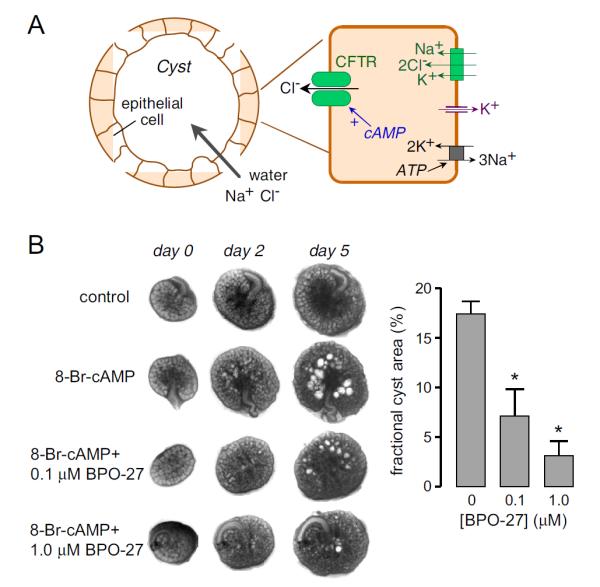
CFTR inhibitor application in polycystic kidney disease. A. Schematic showing involvement of CFTR in cyst fluid secretion in PKD. B. BPO-27 reduces renal cyst growth. (left) Transmission light micrographs of E13.5 embryonic kidneys cultured for indicated days without or with 100 μM 8-Br-cAMP, and with 0, 0.1 or 1 μM BPO-27. (right) Summary of percentage cyst area at 5 days in culture (* P < 0.001). Adapted from ref. 46.
CFTR inhibition therapy for PKD requires inhibitor accumulation at therapeutic levels in kidney. Life-long therapy would likely be required, and hence a candidate drug should have a suitable low toxicity profile. Because potential side-effects of chronic CFTR inhibition therapy include CF lung and pancreatic pathology, as seen in CF, and perhaps constipation, attention to drug pharmacodynamics and tissue accumulation is important. Based on the CF literature, CF pathology would likely require sustained, near-complete CFTR inhibition in target tissues, and, even if inhibition were complete, induction of CF-like pathology would develop over many years. The kidney is an attractive organ for chronic drug therapy because drugs can accumulate preferentially in kidney with minimal extrarenal exposure.
Each of the three chemical classes of CFTR has been tested in PKD models. In an initial study, a series of thiazolidinone and glycine hydrazide analogs were screened using an established MDCK cell cyst model [79]. The best thiazolidionone, tetrazolo-CFTRinh-172, and the best glycine hydrazide, an absorbable phenyl-derivative, Ph-GlyH-101, completely inhibited the growth of MDCK cell cysts without effect on cell proliferation. These compounds also inhibited cyst number and growth by > 80 % in an established embryonic kidney cyst model involving 4-day organ culture of E13.5 mouse kidneys in 8-Br-cAMP-containing medium. Subcutaneous delivery of each compound to neonatal, kidney-specific pkd1 knockout mice, which normally develop large renal cysts in their first week of life, produced stable and therapeutic inhibitor concentrations of >3 μM in urine and kidney. Seven-day treatment resulted in reduced kidney size and cyst volume and preserved renal function compared to untreated mice. However, inhibitor efficacy was incomplete, likely because the kidney-specific pkd1 gene knockout mouse model is an imperfect model for testing of antisecretory therapy, as many developing cysts communicate with tubular fluid. Another concern was the low micromolar EC50 for tetrazolo-CFTRinh-172 and Ph-GlyH-101, respectively, as nanomolar EC50 is desirable.
The PPQ-class CFTR inhibitors PPQ-102 and BPO-27 showed greater potency than the thiazolidiones and glycine hydrazides in the embryonic kidney cyst model [45,46]. (Fig. 9B) shows progressive renal cyst formation and growth in 8-Br-cAMP agonist-treated cultures, as seen by transmitted light microscopy. Kidney growth without cysts was seen in the absence of 8-Br-cAMP. Cyst growth in 8-Br-cAMP-treated kidneys was greatly reduced by inclusion of BPO-27 the culture medium, with IC50 ~ 100 nM. Similar studies done with PPQ-102 gave IC50 ~ 500 nM. Notably, the PPQ-class compounds in this model were also effective in reducing the size of pre-formed cysts, indicating cyst fluid accumulation is an interplay between fluid absorptive and secretory processes; however, the extent is unclear to which cyst fluid accumulation is reversible at various stages in human PKD. We conclude that BPO-27 is a potential candidate for further preclinical development for antisecretory therapy of PKD, based on its efficacy, favorable pharmacokinetic profile and renal accumulation in mice [46].
CONCLUDING REMARKS
Small molecule screening has yeilded several classes of nanomolar-potency CFTR inhibitors that target different regions of the CFTR protein, including its extracellular pore. CFTR inhibitors, particularly the original compounds CFTRinh-172 and GlyH-101, have been used extensively in CF research to verify the molecular identity and physiological role of Cl− currents in various cells and tissues. The good potency and pharmacological properties of the CFTR inhibitors, and their demonstrated in efficacy in various preclinical models, support their further development for therapy of PKD and enterotoxin-mediated secretory diarrheas.
ACKNOWLEDGEMENTS
We wish to thank former contributors in our lab to CFTR inhibitor research, including Dr. Tonghui Ma for small-molecule screening, Dr Sujatha Jayaraman for assay development, Dr. Nitin Sonawane for synthetic chemistry, and Drs. Baoxue Yang, Dan Zhao, Byung-Ju Jin, Chenyuan Yao and Wan Namkung for biological studies. Our CFTR inhibitor work has been funded by the NIH and Cystic Fibrosis Foundation.
Footnotes
CONFLICT OF INTEREST The authors confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Riordan JR. CFTR function and prospects for therapy. Ann Rev Biochem. 2008;77:701–26. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- [2].Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J. 2004;23:146–58. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- [3].Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 2012;18:81–91. doi: 10.1016/j.molmed.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sosnay PR, Castellani C, Corey M, et al. Evaluation of the disease liability of CFTR variants. Meth Mol Biol. 2011;742:355–72. doi: 10.1007/978-1-61779-120-8_21. [DOI] [PubMed] [Google Scholar]
- [5].Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–71. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zolnerciks JK, Andress EJ, Nicolaou M, Linton KJ. Structure of ABC transporters. Essays Biochem. 2011;50:43–61. doi: 10.1042/bse0500043. [DOI] [PubMed] [Google Scholar]
- [7].Rosenberg MF, Kamis AB, Aleksandrov LA, Ford RC, Riordan JR. Purification and crystallization of the cystic fibrosis transmembrane conductance regulator (CFTR) J Biol Chem. 2004;279:39051–7. doi: 10.1074/jbc.M407434200. [DOI] [PubMed] [Google Scholar]
- [8].Zhang L, Aleksandrov LA, Zhao Z, Birtley JR, Riordan JR, Ford RC. Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J Struct Bio. 2009;167:242–51. doi: 10.1016/j.jsb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- [9].Rosenberg MF, O’Ryan LP, Hughes G, Zhao Z, Aleksandrov LA, Riordan JR. Three-dimensional structure and localization of a channel gate. J Biol Chem. 2011;286:42647–54. doi: 10.1074/jbc.M111.292268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–5. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- [11].Atwell S, Brouillette CG, Conners K, et al. Protein Eng Des Sel. 2010;23:375–84. doi: 10.1093/protein/gzq004. [DOI] [PubMed] [Google Scholar]
- [12].Mornon JP, Lehn P, Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci. 2009;66:3469–86. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mornon JP, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65:2594–612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Routaboul C, Norez C, Melin P, et al. Discovery of α-aminoazaheterocycle-methylglyoxal adducts as a new class of high-affinity inhibitors of cystic fibrosis transmembrane conductance regulator chloride channels. J Pharmacol Exp Ther. 2007;322:1023–35. doi: 10.1124/jpet.107.123307. [DOI] [PubMed] [Google Scholar]
- [15].Sonawane N, Zegarra-Mora O, Galietta LJ, Verkman AS. α-Aminoazaheterocyclic-methylglyoxyl adducts do not inhibit cystic fibrosis transmembrane conductance chloride channel activity. J Pharmacol Exp Ther. 2008;325:529–35. doi: 10.1124/jpet.107.132357. [DOI] [PubMed] [Google Scholar]
- [16].Verkman AS. Chemical and GFP-based fluorescent chloride indicators. In: Alvarez-Leefmans FJ, Delpire E, editors. Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Disease. Elsevier; 2009. pp. 111–124. [Google Scholar]
- [17].Jayaraman S, Teitler SL, Skalski B, Verkman AS. Long-wavelength iodide-sensitive fluorescent indicators for measurement of functional CFTR expression in cells. Am J Physiol. 1999;277:C1008–18. doi: 10.1152/ajpcell.1999.277.5.C1008. [DOI] [PubMed] [Google Scholar]
- [18].Wachter RM, Yarbough D, Kallio K, Remington SJ. Crystallographic and energetic analysis of binding of selected anions to the yellow variants of green fluorescent protein. J Mol Biol. 2000;301:157–71. doi: 10.1006/jmbi.2000.3905. [DOI] [PubMed] [Google Scholar]
- [19].Jayaraman S, Haggie P, Wachter R, Remington SJ, Verkman AS. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J Biol Chem. 2000;275:6047–50. doi: 10.1074/jbc.275.9.6047. [DOI] [PubMed] [Google Scholar]
- [20].Galietta LJ, Haggie PM, Verkman AS. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001;499:220–24. doi: 10.1016/s0014-5793(01)02561-3. [DOI] [PubMed] [Google Scholar]
- [21].Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–59. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- [22].Ma T, Vetrivel L, Yang H, et al. High-affinity activators of CFTR chloride conductance identified by high-throughput screening. J Biol Chem. 2002;277:37235–41. doi: 10.1074/jbc.M205932200. [DOI] [PubMed] [Google Scholar]
- [23].Ma T, Thiagarajah JR, Yang H, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–58. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pedemonte N, Lukacs GL, Du K, et al. Small molecule correctors of defective ΔF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115:2564–71. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pedemonte N, Sonawane ND, Taddei A, et al. Phenylglycine and sulfonamide correctors of defective ΔF508- and G551D-CFTR chloride channel gating. Mol Pharmacol. 2005;67:1797–1807. doi: 10.1124/mol.105.010959. [DOI] [PubMed] [Google Scholar]
- [26].Yang H, Shelat AA, Guy RK, et al. Nanomolar-affinity small-molecular activators of ΔF508-CFTR chloride channel gating. J Biol Chem. 2003;278:35079–35085. doi: 10.1074/jbc.M303098200. [DOI] [PubMed] [Google Scholar]
- [27].De La Fuente R, Namkung W, Mills S, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008;73:758–68. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- [28].Namkung W, Phuan P, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of CaCC conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–74. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. Faseb J. 2011;25:4048–62. doi: 10.1096/fj.11-191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis of health benefits of green tea and red wine. Faseb J. 2010;24:4178–86. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Galietta LJ, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol. 2001;281:C1734–42. doi: 10.1152/ajpcell.2001.281.5.C1734. [DOI] [PubMed] [Google Scholar]
- [32].Thiagarajah JR, Song Y, Haggie PM, Verkman AS. A small molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 2004;18:875–877. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- [33].Salinas D, Haggie PM, Thiagarajah JR, et al. Submucosal gland dysfunction as a primary defect in cystic fibrosis. FASEB J. 2005;19:431–33. doi: 10.1096/fj.04-2879fje. [DOI] [PubMed] [Google Scholar]
- [34].Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 2004;558:52–6. doi: 10.1016/S0014-5793(04)00011-0. [DOI] [PubMed] [Google Scholar]
- [35].Kopeikin Z, Sohma Y, Li M, Hwang TC. On the mechanism of CFTR inhibition by a thiazolidinone derivative. J Gen Physiol. 2010;136:659–71. doi: 10.1085/jgp.201010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Caci E, Caputo A, Hinzpeter A, et al. Evidence for direct CFTR inhibition by CFTRinh-172 based on arginine 347 mutagenesis. Biochem J. 2008;413:135–42. doi: 10.1042/BJ20080029. [DOI] [PubMed] [Google Scholar]
- [37].Stahl M, Stahl K, Brubacher MB, Forrest JN., Jr Divergent CFTR orthologs respond differently to the channel inhibitors CFTRinh-172, glibenclamide, and GlyH-101. Am J Physiol Cell Physiol. 2012;302:C67–76. doi: 10.1152/ajpcell.00225.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sonawane ND, Muanprasat C, Nagatani R, Song Y, Verkman AS. In vivo pharmacology and antidiarrheal efficacy of a thiazolidinone CFTR inhibitor in rodents. J Pharm Sci. 2005;94:134–43. doi: 10.1002/jps.20228. [DOI] [PubMed] [Google Scholar]
- [39].Sonawane ND, Verkman AS. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure-activity analysis. Bioorg Med Chem. 2008;16:8187–8195. doi: 10.1016/j.bmc.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–37. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Norimatsu Y, Ivetac A, Alexander C, et al. Locating a plausible binding site for an open channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator. Mol Pharmacol. 2012 doi: 10.1124/mol.112.080267. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sonawane ND, Hu J, Muanprasat C, Verkman AS. Luminally active, nonabsorbable CFTR inhibitors as potential therapy to reduce intestinal fluid loss in cholera. FASEB J. 2006;20:130–32. doi: 10.1096/fj.05-4818fje. [DOI] [PubMed] [Google Scholar]
- [43].Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem Biol. 2008;15:718–28. doi: 10.1016/j.chembiol.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology. 2007;132:1234–44. doi: 10.1053/j.gastro.2007.02.018. [DOI] [PubMed] [Google Scholar]
- [45].Tradtrantip L, Sonawane ND, Namkung W, Verkman AS. Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J Med Chem. 2009;52:6447–55. doi: 10.1021/jm9009873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem. 2011;54:5468–77. doi: 10.1021/jm200505e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Petri WA, Jr, Miller M, Binder HJ, Levine MM, Dillingham R, Guerrant RL. Enteric infections, diarrhea, and their impact on function and development. J Clin Invest. 2008;118:1277–90. doi: 10.1172/JCI34005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Thiagarajah JR, Verkman AS. CFTR inhibitors for treating diarrheal disease. Clin Pharm Therapeut. 2012;92:287–90. doi: 10.1038/clpt.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ball JM, Tian P, Zeng CQ, Morris AP, Estes MK. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science. 1996;272:101–4. doi: 10.1126/science.272.5258.101. [DOI] [PubMed] [Google Scholar]
- [50].Morris AP, Scott JK, Ball JM, Zeng CQ, O’Neal WK, Estes MK. NSP4 elicits age-dependent diarrhea and Ca2+ mediated I− influx into intestinal crypts of CF mice. Am J Physiol. 1999;277:G431–44. doi: 10.1152/ajpgi.1999.277.2.G431. [DOI] [PubMed] [Google Scholar]
- [51].Venkatasubramanian J, Ao M, Rao MC. Ion transport in the small intestine. Curr Opin Gastroenterol. 2010;26:123–28. doi: 10.1097/MOG.0b013e3283358a45. [DOI] [PubMed] [Google Scholar]
- [52].Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82:245–89. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- [53].Murek M, Kopic S, Geibel J. Evidence for intestinal chloride secretion. Exp Physiol. 2010;95:471–78. doi: 10.1113/expphysiol.2009.049445. [DOI] [PubMed] [Google Scholar]
- [54].Field M. Mechanisms of action of cholera and Escherichia coli enterotoxins. Am J Clin Nutr. 1979;32:189–96. doi: 10.1093/ajcn/32.1.189. [DOI] [PubMed] [Google Scholar]
- [55].Thiagarajah J, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 2004;126:511–19. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- [56].Grubb BR. Ion transport across the jejunum in normal and cystic fibrosis mice. Am J Physiol. 1995;268:G505–13. doi: 10.1152/ajpgi.1995.268.3.G505. [DOI] [PubMed] [Google Scholar]
- [57].Berschneider HM, Knowles MR, Azizkhan RG, et al. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988;2:2625–9. doi: 10.1096/fasebj.2.10.2838365. [DOI] [PubMed] [Google Scholar]
- [58].O’Loughlin EV, Hunt DM, Gaskin KJ, et al. Abnormal epithelial transport in cystic fibrosis jejunum. Am J Physiol. 1991;260:G758–63. doi: 10.1152/ajpgi.1991.260.5.G758. [DOI] [PubMed] [Google Scholar]
- [59].Morris AP, Scott JK, Ball JM, Zeng CQ, O’Neal WK, Estes MK. NSP4 elicits age-dependent diarrhea and Ca2+ mediated I− influx into intestinal crypts of CF mice. Am J Physiol. 1999;277:G431–44. doi: 10.1152/ajpgi.1999.277.2.G431. [DOI] [PubMed] [Google Scholar]
- [60].Hoque KM, Woodward OM, van Rossum DB, et al. Epac1 mediates protein kinase A-independent mechanism of forskolin-activated intestinal chloride secretion. J Gen Physiol. 2010;135:43–58. doi: 10.1085/jgp.200910339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Namkung W, Finkbeiner WE, Verkman AS. CFTR-adenylyl cyclase I association responsible for UTP activation of CFTR in well-differentiated primary human bronchial cell cultures. Mol Biol Cell. 2010;21:2639–48. doi: 10.1091/mbc.E09-12-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dechecchi MC, Tamanini A, Berton G, Cabrini G. Protein kinase C activates chloride conductance in C127 cells stably expressing the cystic fibrosis gene. J Biol Chem. 1993;268:11321–5. [PubMed] [Google Scholar]
- [63].Jin BJ, Thiagarajah JR, Verkman AS. Convective washout prevents the antidiarrheal efficacy of enterocyte surface-targeted antisecretory drugs. J Gen Physiol. 2013;141:261–72. doi: 10.1085/jgp.201210885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- [65].Brill SR, Ross KE, Davidow CJ, Ye M, Granthan JJ, Caplan MJ. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc Natl Acad Sci U S A. 1996;93:10206–11. doi: 10.1073/pnas.93.19.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996;50:208–18. doi: 10.1038/ki.1996.304. [DOI] [PubMed] [Google Scholar]
- [67].Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148–57. doi: 10.2215/CJN.00330705. [DOI] [PubMed] [Google Scholar]
- [68].Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–87. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- [69].Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl− secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–38. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- [70].Cotton CU, Avner ED. PKD and CF: an interesting family provides insight into the molecular pathophysiology of polycystic kidney disease. Am J Kidney Dis. 1998;32:1081–3. doi: 10.1016/s0272-6386(98)70088-5. [DOI] [PubMed] [Google Scholar]
- [71].O’Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1998;32:976–83. doi: 10.1016/s0272-6386(98)70072-1. [DOI] [PubMed] [Google Scholar]
- [72].Xu N, Glockner JF, Rossetti S, Babovich-Vuksanovic D, Harris PC, Torres VE. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J Nephrol. 2006;19:529–34. [PubMed] [Google Scholar]
- [73].Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493–02. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- [74].Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi: 10.1097/MNH.0b013e3283262ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Torres VE, Boletta A, Chapman A, et al. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin J Am Soc Nephrol. 2010;5:1312–29. doi: 10.2215/CJN.01360210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs. 2010;19:315–28. doi: 10.1517/13543781003588491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Masoumi A, Reed-Gitomer B, Kelleher C, Schrier RW. Potential pharmacological interventions in polycystic kidney disease. Drugs. 2007;67:2495–510. doi: 10.2165/00003495-200767170-00004. [DOI] [PubMed] [Google Scholar]
- [78].Torres VE. Treatment strategies and clinical trial design in ADPKD. Adv Chronic Kidney Dis. 2010;17:190–204. doi: 10.1053/j.ackd.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1300–10. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]