

Abstract
Recent studies have shown that the concentrations of proteins expressed from orthologous genes are often conserved across organisms and to a greater extent than the abundances of the corresponding mRNAs. However, such studies have not distinguished between evolutionary (e.g., sequence divergence) and environmental (e.g., growth condition) effects on the regulation of steady-state protein and mRNA abundances. Here, we systematically investigated the transcriptome and proteome of two closely related Pseudomonas aeruginosa strains, PAO1 and PA14, under identical experimental conditions, thus controlling for environmental effects. For 703 genes observed by both shotgun proteomics and microarray experiments, we found that the protein-to-mRNA ratios are highly correlated between orthologous genes in the two strains to an extent comparable to protein and mRNA abundances. In spite of this high molecular similarity between PAO1 and PA14, we found that several metabolic, virulence, and antibiotic resistance genes are differentially expressed between the two strains, mostly at the protein but not at the mRNA level. Our data demonstrate that the magnitude and direction of the effect of protein abundance regulation occurring after the setting of mRNA levels is conserved between bacterial strains and is important for explaining the discordance between mRNA and protein abundances.
Keywords: Transcriptomics, proteomics, Pseudomonas aeruginosa
Introduction
Recent systematic studies have shown that mRNA and protein abundances within an organism are less correlated than expected both in eukaryotes1−5 and prokaryotes.2,5−8 Surprisingly, the abundances of orthologous proteins in Caenorhabditis elegans and Drosophila melanogaster were shown to be highly conserved and correlated better with each other (Rs = 0.79) than with the corresponding mRNA concentrations within each organism (Rs = 0.44 in C. elegans and Rs = 0.36 in D. melanogaster).3 A later analysis of seven different organisms (two prokaryotes and five eukaryotes) confirmed that orthologous protein abundances were generally more correlated between organisms than the abundances of mRNA and protein within organisms.2 On the basis of these comparisons, we hypothesized that each protein may exhibit an evolutionarily conserved preference for certain steady-state abundances but that the precise mechanisms employed to set these levels (i.e., the relative contributions played by transcriptional, post-transcriptional, translational, and/or degradative processes) may differ between organisms.9 Thus, the degree to which mRNA-level and post-mRNA-level processes contribute to the setting of a given protein’s steady-state abundance may differ between organisms provided that the final target levels of the protein are properly set.
Although it is technically difficult to measure the contributions of transcriptional and post-transcriptional processes to establishing mRNA and protein abundances inside cells,4 by measuring steady-state levels under defined conditions it is possible to identify differences in protein and mRNA abundances. Such differences indicate potential cases of post-transcriptional regulation. (Note that we will generally use the term post-transcriptional to indicate the combined effect of all forms of protein abundance regulation acting after the setting of mRNA levels, including translational and degradative mechanisms.) In particular, protein-to-mRNA ratios form a simple summary statistic that is useful both for detecting specific genes likely to be regulated post-transcriptionally and for measuring the evolutionary conservation of all post-transcriptional regulatory processes.
Several previous studies have measured protein and mRNA abundances in bacteria.2,6−8,10−12 However, these have never been compared across multiple bacterial species or strains. Protein and mRNA abundances in Desulfovibrio vulgaris were analyzed under multiple growth conditions;6,7 however, because of low mass spectrometry resolution, the number of proteins consistently observed under multiple conditions allowed only for the investigation of global trends in post-transcriptional regulation. Similarly, protein abundances from Mycoplasma pneumoniae were measured under different growth conditions8 and integrated with previously published transcriptome data.10 The protein and mRNA abundances were not correlated within M. pneumoniae, and the authors concluded that post-transcriptional regulation plays a large role in this bacterium. However, it is difficult to compare these results to those of other species because of the small number of M. pneumoniae genes. More advanced techniques, such as single-cell imaging combined with in situ hybridization11 and transcriptome profiling with short-read sequencing (RNA–seq),12 have been used to measure protein and mRNA abundances in bacteria. Such studies have confirmed that mRNA abundances are insufficient to predict protein abundances and that some key regulatory genes, including virulence factors, are post-transcriptionally regulated. However, published studies have used different growth conditions for each bacterium, so it is difficult to determine whether the divergences between protein and mRNA abundances are conserved for orthologous genes across organisms.
To investigate the divergence of protein and mRNA expression when controlling for sequence divergence, we measured the protein and mRNA concentrations of 703 genes from two strains of the bacterium Pseudomonas aeruginosa, strain PAO113 and strain UCBPP-PA1414 (hereafter referred to as PAO1 and PA14). Although these two strains have highly similar genomes (96.7% of total PAO1 genes and 90.7% of total PA14 genes are orthologous as one-to-one relations), previous studies have reported that they are physiologically quite different, including virulence in various model organisms.15,16 Both strains were grown under identical conditions, and mRNA and protein abundances were measured using the identical microarray and LC–MS/MS platforms. Because the majority of genes are highly conserved at the nucleotide sequence level between these strains, any measurement bias derived from divergence between orthologous genes is therefore minimized. We further controlled for such bias by limiting our analyses to microarray probes with perfect matches to the corresponding genomes.
We confirmed previous observations that protein and mRNA abundances between the two strains are more correlated than the protein and mRNA abundances within each strain.2 We further showed that the protein-to-mRNA ratios between PAO1 and PA14 are well-correlated, suggesting that mechanisms regulating protein abundances downstream of transcription are conserved. Despite this high correlation, there were important differences between the two strains, and we showed that in these cases protein and mRNA measurements can be used to identify post-transcriptional regulation.
Materials and Methods
Strains and Growth Conditions
P. aeruginosa PAO113 (a subline from Barbara Iglewski) and UCBPP-PA1414 were grown in 25 mL of synthetic cystic fibrosis sputum medium (SCFM) in 250 mL flasks, which mimics the nutritional environment of the cystic fibrosis lung,17 at 37 °C with shaking at 250 rpm. Cells were harvested at an OD600 from 0.4 to 0.5. We performed two biological replicates for each experiment. Cells for transcriptome and proteome analyses were not collected simultaneously, but they were grown under identical conditions. We also performed growth curve assays by diluting overnight cultures grown in SCFM to OD600 0.01 in 25 mL of SCFM (using 250 mL flasks). Cells were grown at 37 °C with shaking at 250 rpm. The OD600 was measured every 30 min to generate a growth curve, from which the doubling time (30–40 min for both strains) was determined (data not shown).
DNA Microarrays
The detailed microarray protocol has been described elsewhere.2,18 Briefly, cultures were mixed 1:1 with RNAlater (Ambion), an RNA-stabilizing agent. RNA was isolated using the RNeasy mini kit (Qiagen), and cDNA was prepared for hybridization to Affymetrix GeneChip microarrays (array identifier Pae_G1a). GeneChips were washed, stained, and scanned using an Affymetrix fluidics station at the University of Iowa DNA core facility. These data are available at the NCBI GEO database (accession number GSM546278–GSM546281) as part of a previous study.18
Transcriptome Analyses
We preprocessed microarray CEL files with the RMA method using the affy package (version 1.32.1)19 in R (version 2.14.1) with default options. To assign microarray probesets to genes, we downloaded probe sequences from the Affymetrix Web site (http://www.affymatrix.com) and mapped them to both the PAO1 genome (GenBank accession number NC_002516.2)20 and the PA14 genome (GenBank accession number NC_008463.1)21 using Exonerate (version 2.20).22 Probes that mapped uniquely were then remapped to P. aeruginosa PAO1 and PA14 cDNA sequences (downloaded from PseudoCAP,23 version 2009-Nov-23). We assigned probe sets to genes if 12–14 probes in a probeset were mapped to a single gene. Differential expression analysis between the two strains was conducted using an empirical Bayesian method implemented in the limma package (version 3.10.3).24 Genes with greater than 2-fold changes and less than 0.05 false discovery rate (FDR) cutoffs were considered differentially expressed.
LC–MS/MS Proteomics Experiments
The detailed proteomics protocol was described in a previous study.2 Briefly, cells were lysed three times with a French press, and cellular lysate was collected from the supernatant after centrifugation for 20 min at 10 000 rpm. Lysis buffer consisted of 25 mM Tris-HCl (pH 7.5), 5 mM DTT, 1.0 mM EDTA, and 1× CPIOPS (Calbiochem protease inhibitor cocktail). Fifty microliters of diluted cell lysate (2 mg/mL; diluted with 50 mM Tris-HCl buffer) was incubated at 55 °C for 45 min with 50 μL of trifluoroethanol (TFE) and 15 mM dithiothreitol (DTT) and was then incubated with 55 mM iodoacetamide (IAM) in the dark for 30 min. After diluting the sample to 1 mL with buffer (50 mM TrisHCl, pH 8.0), 1:50 w/w trypsin was added for a 4.5 h digestion, which was then halted by adding 20 μL of formic acid, resulting in 2% v/v. The sample was lyophilized, resuspended with buffer C (95% H2O, 5% acetonitrile, and 0.01% formic acid), and contaminants removed with C18 tips (Thermo Fisher). The eluted sample was again lyophilized, resuspended with 120 μL of buffer C, and filtered through a Microcon-10 filter (for 45 min at 14 000g at 4 °C). Each sample was injected five times into an LTQ-Orbitrap Classic mass spectrometer (Thermo Electron; mass resolution 60 000; top12 ms2 selection strategy), and data were collected in a 0–90% acetonitrile gradient over 5 h with a Agilent Zorbax C18 column. The raw files from the MS/MS experiments are available at http://www.marcottelab.org/index.php/PSEAE_ref.2013, and the data were also deposited to the ProteomeXchange under identifier PXD000684.
Proteomics Analyses
RAW files were searched independently using the P. aeruginosa PAO1 and PA14 protein sequence database (downloaded from the PseudoCAP database, version 2009-Nov-23).23 The database for each strain contained the same number of randomly shuffled protein sequences as the decoy database. We used Bioworks/SEQUEST (Thermo Electron; version 3.3.1 SP1),25 X!Tandem with k-score (version 2009.10.01.1 LabKey and ISB, included in TPP 4.3.1 package),26,27 InsPecT (version 20100331),28 and MS-GFDB (version 06/16/2011)29 for the database search. We used the same search parameters as described previously30 except that MS-GFDB was newly added for the current study (with the settings –t 300 ppm –c13 1 –nnet 0 –n 2). Then, we combined these results with MSblender30 and considered peptide−spectrum matches with an estimated FDR less than 0.01. Subsequently, we calculated APEX scores5,31 with weighted spectral counts per protein (using a FDR < 0.01 estimated by MSblender). Because MSblender only provides peptide-level probabilities, we set each protein probability as 1.0 using the original APEX formula
To estimate Oi values for each protein, we analyzed both SEQUEST and X!Tandem results of each biological replicate with PeptideProphet26 and ProteinProphet,32 and this output was then used with the APEX GUI program.33 APEX Oi values were trained on proteins with ProteinProphet probabilities greater than 0.99 using a FDR < 0.01. We used 25 000 (arbitrary unit of protein concentration) as the APEX normalization constant. We confirmed that the Oi values calculated with SEQUEST results and those from the X!Tandem results were well-correlated, and we used the mean of these values as the representative Oi values for individual proteins. Oi values for both strains and all proteomics analysis data are available at http://www.marcottelab.org/index.php/PSEAE_ref.2013.
Differentially expressed proteins were identified using QSPEC (version 2; 5000 burn-ins and 20 000 iterations; a 2-fold change and FDR < 0.05 were required to determine differentially expressed proteins)34 on normalized APEX scores. Genes were omitted from the differential expression analysis if the sum of two strains’ APEX scores was less than 1.0. For MS1-intensity-based quantification, we analyzed same data with MaxQuant35 (version 1.4.1.2) using the default option.
Orthology and 5′ Sequence Analysis
To define orthologous genes between PAO1 and PA14, we used InParanoid (version 4.1)36 with protein sequences of each strain downloaded from PseudoCAP (version 2009-Nov-23). To analyze the 5′ sequences of cDNAs containing the Shine–Dalgarno motif, we extracted 50 bp DNA fragments around the translational start site of each gene (from −25 to +25 bp) and calculated the Gibbs free energy of hybridization to the 3′ end of 16S rRNA (33 bp for PAO1 and 31 bp for PA14; PAO1 differs in having an extra AA at the end). This analysis used the modified melt.pl wrapper script in UNAfold (version 3.8)37 and the associated hybrid-min program.
Other Statistical Analyses
Translationally repressed genes (those for which no protein was observed in the shotgun proteomics analysis in spite of reasonably high mRNA abundance) were identified on the basis of calculating two mRNA abundance distributions for genes with or without accompanying protein observations. We identified the mRNA abundance value for which a protein had a ≥80% chance of being observed by proteomics. To increase the stringency further, we sorted all genes with mRNA signals greater than the 80% protein observation threshold, and we removed the genes with the lowest 20% of all mRNA abundance (PAO1 cutoff is 691.0 and PA14 cutoff is 758.0). To identify genes with high or low protein-to-mRNA ratios (Supporting Information Table S4), we calculated protein-to-mRNA ratios of all 703 gene pairs identified in both strains using the mean of the two replicates and then selected the top 50 and bottom 50 genes of the list. For KEGG pathway enrichment analyses, we considered proteins observed in at least one strain or mRNAs observed in both PAO1 and PA14 as a background for testing pathway enrichment. p values of enrichment were estimated by measuring random chances that equal to or greater than the number of reported genes in each pathway were selected among 10 000 trials.
Chloramphenicol Disk Diffusion Assay
PAO1 and PA14 were diluted to OD600 ∼0.01 in 25 mL of SCFM in 250 mL flasks. Cells were grown at 37 °C with shaking at 250 rpm until they reached the exponential phase (OD600 0.4–0.6), at which point they were spread on half of an SCFM agar plate using a sterile cotton swab. Sterile discs containing 0, 1.5, and 2.5 mg/mL chloramphenicol were placed on the plates, which were then incubated overnight at 37 °C. Three biological replicates were performed.
Results
Strong Correlation and Evolutionary Conservation of Protein and mRNA Abundances
To investigate the relationship between protein and mRNA abundance in two strains of the same species, we first analyzed the correlation between protein and mRNA abundances in P. aeruginosa strains PAO1 and PA14. We were able to detect 5345 mRNAs and 1652 proteins in both strains when considering only one-to-one orthologues. Although all detectable mRNAs and proteins were used to determine differential expression, to focus on the highest accuracy measurements, we analyzed the 703 gene pairs with consistent protein abundances in both strains for all correlation analyses (summarized in Figure 6; all values before filtering are available in Supporting Information Table S7). As shown in Figure 1, both the protein and mRNA abundances were highly correlated between PAO1 and PA14 (Rs = 0.95 for mRNA and Rs = 0.89 for protein), which is better than the correlation observed between protein and mRNA abundance within each strain (Rs = 0.64 for PAO1 and Rs = 0.65 for PA14). In contrast to previous studies showing that the correlation in protein abundance is higher than that of mRNA abundance,2,3,38 we observed a better correlation between mRNA abundances than for protein abundances. This may reflect the high degree of relatedness between these strains. Alternatively, previous studies measured mRNA abundance in heterogeneous cell types and in organisms grown under different conditions,2,3 so it is possible that mechanisms setting transcript abundance are more sensitive than mechanisms setting protein abundance.
Figure 6.
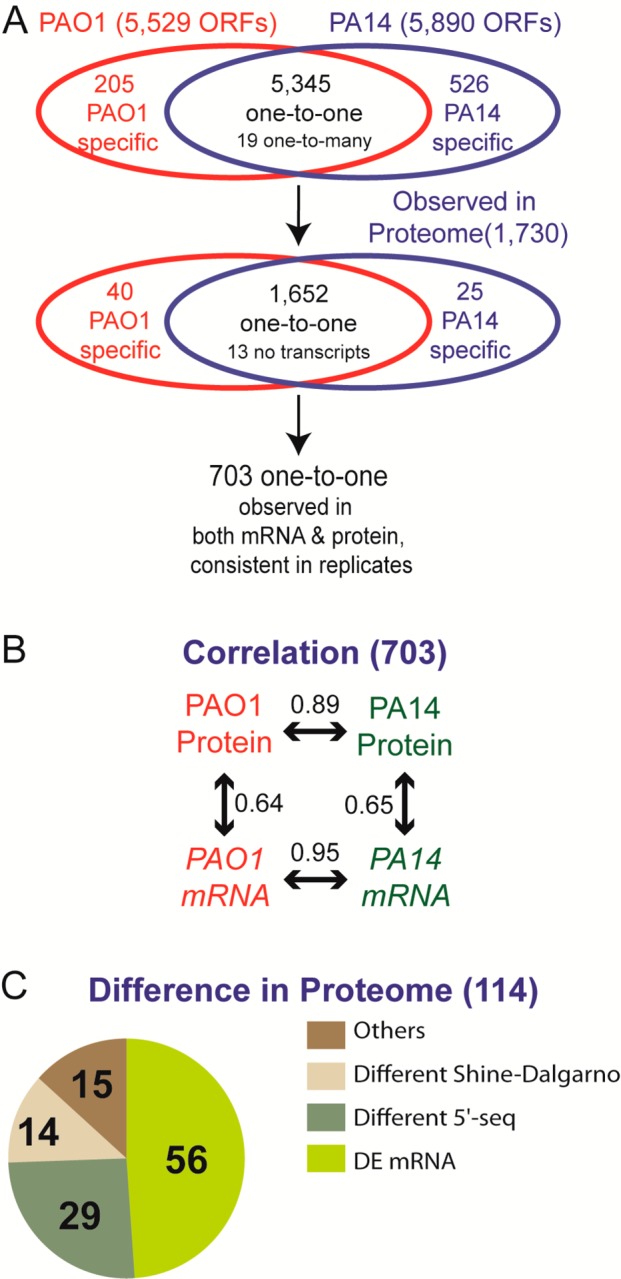
Overview of the proteomic measurements in this study. (A) Out of 5345 genes with one-to-one orthology between PAO1 and PA14, we measured proteome and transcriptome abundance of 703 genes with highly stringent criteria of reproducibility between biological replicates. (B) We observed that protein (Spearman rank correlation 0.89) and mRNA (Spearman rank correlation 0.95) abundances are highly conserved, much more than those abundances within each strain. (C) Out of 114 genes showing significantly different protein levels between PAO1 and PA14, about half of them (56 genes) showed different mRNA abundances and 43 genes showed different 5′ sequences (assuming that different 5′ sequence may affect translation repression efficiency). However, fewer than half of 5′ sequence differences were relevant to differences in the Shine–Dalgarno sequence. Fifteen differentially expressed proteins with identical 5′ sequences and similar mRNA abundances may be regulated by strand-specific post-transcriptional mechanisms (Table 3).
Figure 1.
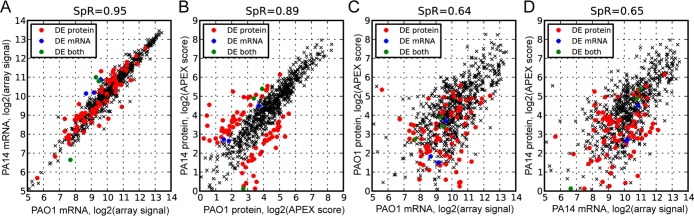
mRNA and protein concentrations are highly correlated between P. aeruginosa strains. (A) Correlation between mRNA abundances from P. aeruginosa PAO1 and PA14 strains. (B) Correlation between protein abundances from PAO1 and PA14. (C) Correlation between protein and mRNA abundances in PAO1. (D) Correlation between protein and mRNA abundances in PA14. Genes were considered to be differentially expressed (DE) if they exhibited a greater than 2-fold expression change and an FDR < 0.05 for both protein and mRNA. Genes without protein observations in either PAO1 or PA14 and genes with high variation between biological replicates are omitted (see Supporting Information Figures S1 and S2 for details). A total of 703 genes are presented (3 DE mRNA genes, 72 DE protein genes, and 3 DE both genes; see the text for details). DE mRNA: differentially expressed genes at the mRNA level but not at the protein level between two strains. DE protein: differentially expressed genes at the protein level but not at the mRNA level. DE both: differentially expressed genes at both the mRNA and protein level. SpR: Spearman rank correlation.
Next, we investigated the differentially expressed genes between PAO1 and PA14. Among the 5377 gene pairs with probesets on the Affymetrix microarray, 150 genes were significantly differentially expressed at the mRNA level (Supporting Information Table S1). Similarly, among the 1730 gene pairs with associated protein abundances, 279 genes were significantly differentially expressed at the protein level (Supporting Information Table S2). Among the 703 genes that we analyzed for correlation between the two strains, 75 gene pairs with differential protein expression and 6 gene pairs with differential mRNA expression (3 gene pairs with differential expression of both protein and mRNA) were identified.
Many of the gene pairs that were significantly differentially expressed between the two strains at the protein level (red circles in Figure 1B) did not show differences at the mRNA level (red circles in Figure 1A), suggesting that these genes are post-transcriptionally regulated. These differentially expressed proteins between the two strains did not exhibit systematic trends in the correlation between protein and mRNA within each strain (red circles in Figure 1C,D), so neither the protein nor the mRNA abundances themselves are the major factors of inconsistency between them. Compared to the genes differentially expressed at the protein level, only a few genes that were differentially expressed at the mRNA level were included in this analysis because most of those (96 out of 150) identified as differentially expressed at the mRNA level between PAO1 and PA14 were not observed in the shotgun proteomics analysis (the sum of the APEX scores for all four biological samples was less than 1.0), likely because of their low abundance.
Protein-to-mRNA Ratios Are Evolutionarily Conserved
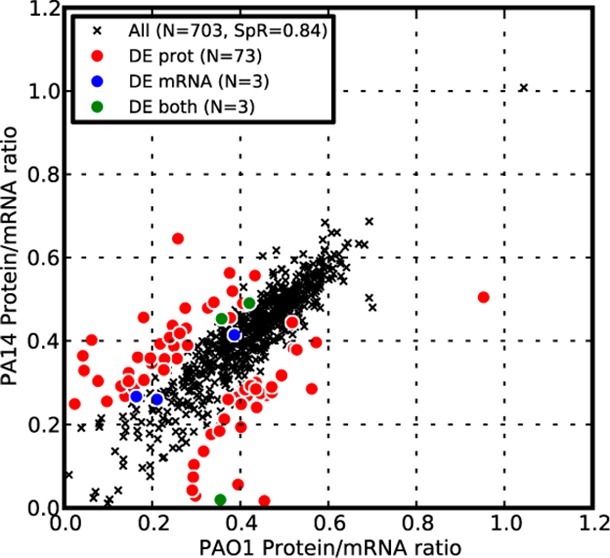
The higher correlation in both protein and mRNA abundances between the two strains, compared to the mRNA and protein abundances within each strain, led us to speculate that the protein-to-mRNA ratios should also be highly correlated between the strains. As shown in Figure 2, the protein-to-mRNA ratios between the strains were indeed highly correlated (Rs = 0.84) and were considerably higher than the correlation of mRNA to protein within each strain. We also observed that most of the differentially expressed genes at the protein level had different protein-to-mRNA ratios (red circles in Figure 2A), supporting the notion that the differences in protein abundance between the two closely related strains were attributable to post-transcriptional regulation.
Figure 2.
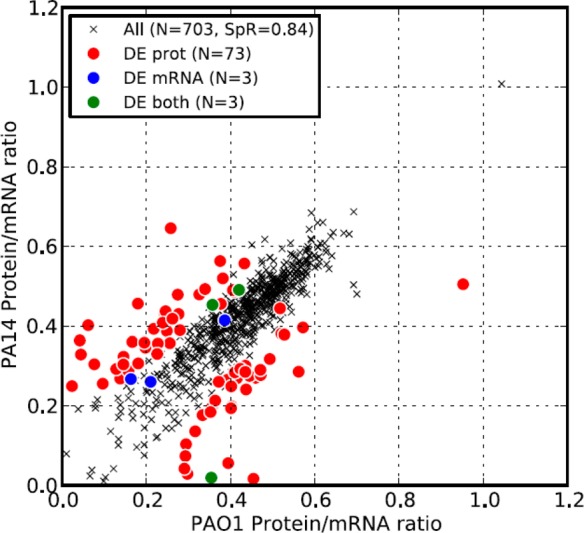
Protein-to-mRNA ratios are well-conserved between P. aeruginosa strains. Correlation of protein-to-mRNA ratios between PAO1 and PA14. Protein-to-mRNA ratios were calculated as the ratio of the log2-transformed APEX score to the log2-transformed microarray signal (ratio is not log transformed). Although the two strains showed strong correlation in their protein-to-mRNA ratios (Spearman rank correlation 0.84, p value < 10–9), most genes with statistically different expression at the protein level also showed a statistically significant difference between the protein-to-mRNA ratios from the two strains (red circles). Only genes for which we detected proteins in both PAO1 and PA14 are presented. DE mRNA: differentially expressed genes at the mRNA level but not at the protein level between two strains. DE protein: differentially expressed genes at the protein level but not at the mRNA level. DE both: differentially expressed genes at both the mRNA and protein level. SpR: Spearman rank correlation.
Differential Expression between the Strains Explains Phenotypic Differences
Although many studies have used both PAO1 and PA14 as reference strains, to our knowledge there has not been a systematic comparison of their molecular characteristics at the transcriptome and proteome levels. Because the genetic differences between PAO1 and PA14 are minor, we expected that phenotypic differences between the two strains might be predominantly explained by underlying differences in mRNA and protein abundances. Among the 114 conserved proteins that had significantly different protein abundances between PAO1 and PA14, 14 genes also showed significantly different mRNA expression levels (Table 1).
Table 1. Fourteen Genes with Significantly Different mRNA and Protein Abundances between PAO1 and PA14a.
| PAO1 identifier | PAO1 protein abundanceb | PA14 protein abundanceb | PAO1 mRNA abundancec | PA14 mRNA abundancec | annotation |
|---|---|---|---|---|---|
| PA0997|pqsB | 0.0 | 38.9 | 36.0 | 1934.0 | homologous to beta-keto-acyl-acyl-carrier protein synthase |
| PA0998|pqsC | 0.0 | 36.6 | 27.5 | 1003.0 | homologous to beta-keto-acyl-acyl-carrier protein synthase |
| PA2235|pslE | 4.2 | 0.0 | 844.5 | 32.0 | hypothetical protein |
| PA2493|mexE | 43.3 | 0.3 | 5211.0 | 307.0 | resistance-nodulation-cell division (RND) multidrug efflux membrane fusion protein MexE precursor |
| PA2494|mexF | 35.3 | 0.0 | 4251.5 | 246.0 | resistance-nodulation-cell division (RND) multidrug efflux transporter MexF |
| PA2495|oprN | 38.7 | 0.0 | 3620.0 | 186.0 | multidrug efflux outer membrane protein OprN precursor |
| PA2667 | 15.0 | 42.0 | 627.0 | 2043.0 | conserved hypothetical protein |
| PA2813 | 4.7 | 0.0 | 392.5 | 141.0 | probable glutathione S-transferase |
| PA4502 | 0.0 | 7.8 | 117.5 | 833.0 | probable binding protein component of ABC transporter |
| PA4771|lldD | 10.5 | 29.6 | 709.5 | 1772.5 | l-lactate dehydrogenase |
| PA4772 | 0.7 | 10.8 | 933.5 | 2008.0 | probable ferredoxin |
| PA4778 | 0.0 | 9.7 | 266.5 | 1392.5 | probable transcriptional regulator |
| PA5261|algR | 6.6 | 1.1 | 207.0 | 100.0 | alginate biosynthesis regulatory protein AlgR |
| PA5289 | 5.0 | 0.0 | 655.0 | 190.5 | hypothetical protein |
A comprehensive list of differentially expressed genes is available in Supporting Information Tables S1 and S2. A >2-fold-change and FDR<0.05 were applied to detect significant differences both in mRNA and protein.
The protein abundance value represents the average APEX score of two biological replicates.
The mRNA abundance value represents the average normalized microarray signals of two biological replicates.
Differentially expressed genes at both the protein and mRNA levels included well-known virulence and antibiotic resistance genes, such as algR and the pqs operon, and the mexEF–oprN operon. It has been shown that overexpression of mexEF–oprN increases resistance to chloramphenicol.39 We therefore hypothesized that PAO1, which shows higher expression of mexEF–oprN, may exhibit higher resistance to chloramphenicol compared to PA14. To test this hypothesis, we used a disk diffusion assay to measure growth inhibition by chloramphenicol. As expected, the zones of inhibition were larger for PA14 with increasing chloramphenicol concentrations, whereas PAO1 growth was minimally inhibited by chloramphenicol (Figure 3). Additionally, genes involved in the metabolism of several amino acids were differentially expressed between PAO1 and PA14 (Table 2), likely highlighting different metabolic characteristics of the two strains.
Figure 3.
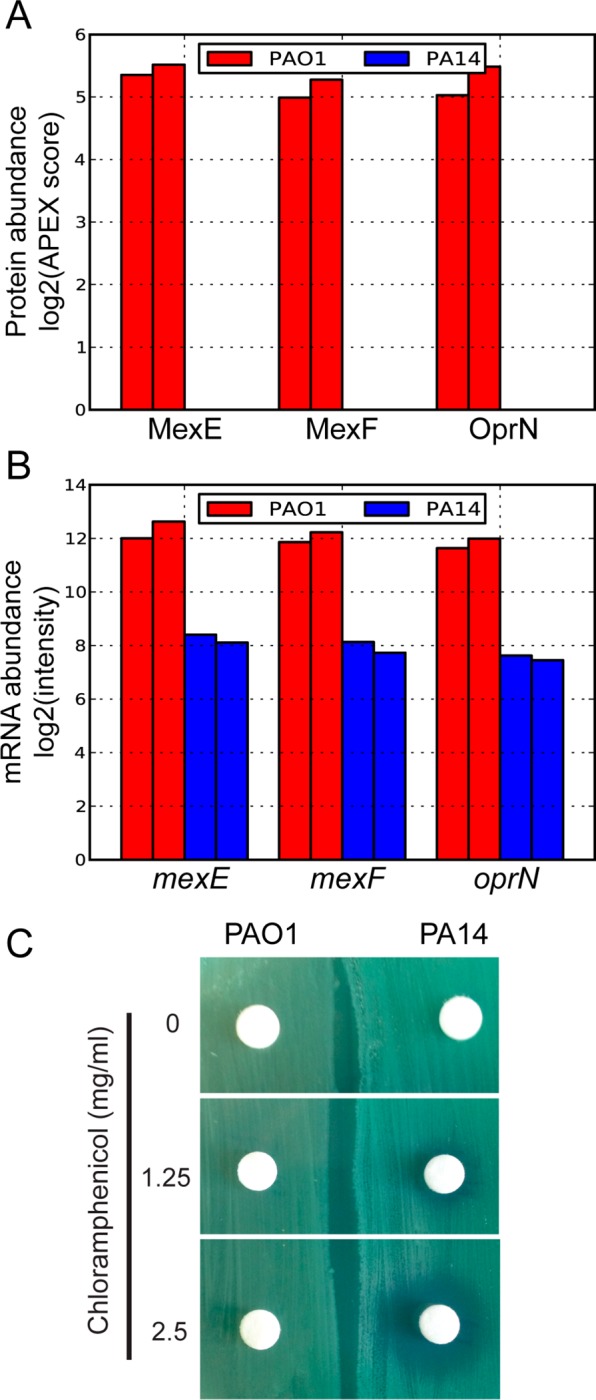
. Differential expression of the mexEF–oprN operon explains differential chloramphanicol resistance in P. aeruginosa PAO1 and PA14 strains. (A) Protein expression levels of MexEF–OprN in PAO1 and PA14. Two biological replicates are plotted for each strain, as shown by the same color bars. In PA14, we could not detect MexF or OprN, and the protein abundance score for MexE was low (∼0.2). (B) mRNA levels of mexEF–oprN in PAO1 and PA14. Two biological replicates are plotted for each strain, as shown by the same color bars. (C) Chloramphenicol disk diffusion assay. Exponentially growing PAO1 and PA14 were swabbed on an agar plate and exposed to increasing levels of chloramphenicol. Three biological replicates were performed, and a representative is shown.
Table 2. KEGG Pathways Enriched in Differentially Expressed Proteins between PAO1 and PA14a.
| KEGG pathway name | p value | genes |
|---|---|---|
| mismatch repair | 0.015 | PA1529, PA1532, PA1816 |
| nucleotide excision repair | 0.001 | PA1529, PA3002, PA4234 |
| DNA replication | 0.000 | PA1529, PA1532, PA1816, PA4931 |
| nicotinate and nicotinamide metabolism | 0.004 | PA0143, PA3625, PA4920 |
| selenocompound metabolism | 0.035 | PA0849, PA1642 |
| fatty acid biosynthesis | 0.014 | PA1609, PA1610, PA2965, PA3645 |
| phenylalanine, tyrosine, and tryptophan biosynthesis | 0.040 | PA0650, PA0870, PA0872 |
| phenylalanine metabolism | 0.004 | PA0865, PA0870,PA0872, PA5304 |
| pyrimidine metabolism | 0.020 | PA0849, PA1532, PA1816, PA3625, PA3654 |
A hypergeometric test was used to evaluate significance of enrichment with 10 000-fold bootstrapping. Two pathways marked in bold (nucleotide excision repair and pyrimidine metabolism) were enriched in genes showing different protein abundances but not differences in mRNA abundances or Shine–Dalgarno-binding free energies.
Intrinsic Factors to Control Post-Transcriptional Regulation
We identified 114 differentially expressed proteins between PAO1 and PA14 (Supporting Information Table S2). Fifty six of these proteins (49%) also had concordant mRNA levels to proteins, although only 14 were significant according to our statistical cutoffs. Higher mRNA levels most likely explain the differences in protein levels, suggesting that the remaining 58 genes have a different post-transcriptional regulation mechanism. In addition to differences in the gene repertoires between the two strains, these genes may prove to be a useful resource for choosing reference strains for P. aeruginosa experiments.
To determine whether differential protein-to-mRNA ratios for the remaining 58 genes can be explained by ribosomal binding energy, we analyzed the Shine–Dalgarno motif under the assumption that high-affinity ribosome binding may correspond to increased translation efficiency40 (Supporting Information Table S5). To normalize for the effect of differential mRNA abundance, we compared protein-to-mRNA ratios instead of protein abundance. Differences in protein-to-mRNA ratios for 14 gene pairs (12%) could be accounted for by differences in ribosome binding energy.
We could not identify the cause of the remaining 39% of gene pairs with differential protein abundances (Supporting Information Table S6). In light of the high sequence similarity of orthologous genes between PAO1 and PA14, we suspect that intrinsic sequence features, such as sequence length, are unlikely, in general, to explain these differences. Also, our group reported that various sequence signatures and mRNA concentrations can only explain 66% of protein abundances in a human cell line;1 thus, these sequences may be the target of extrinsic post-transcriptional regulation such as small noncoding RNAs or RNA-binding proteins.41
Evidence for Translational Repression
Finally, the systematic measurement of protein and RNA abundances allowed us to select specific candidate genes for translational regulation. In particular, if protein abundance is solely proportional to mRNA abundance and the protein detection is mainly governed by abundance, then genes with high mRNA signal but undetected protein should be candidates for translational repression. To search for such cases, we identified those genes that were not observed in the shotgun proteomics experiment but had reasonably high mRNA expression levels. By comparing mRNA levels of genes for which we detected protein to those of which we did not, we identified an mRNA abundance threshold corresponding to an 80% chance of protein detection (Figure 4). To enrich for true cases of translational repression further, we additionally filtered out the 20% of genes with the lowest mRNA abundances (Supporting Information Table S4). Of the 181 genes identified as translationally repressed in this analysis, 97 genes showed significant repression at the protein level in both PAO1 and PA14.
Figure 4.
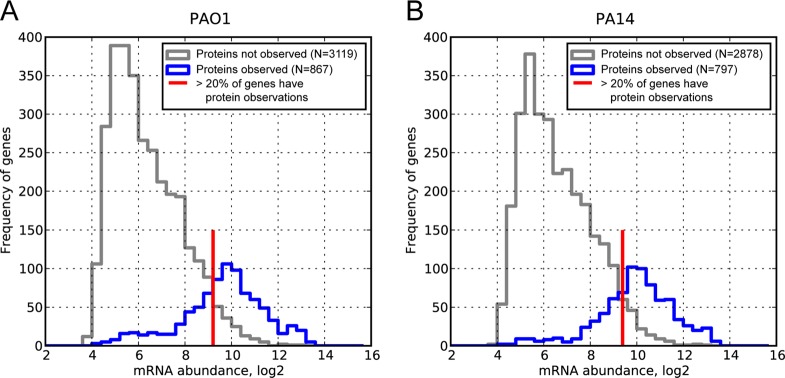
Candidates for translation repression can be selected from histograms of mRNA concentrations with and without corresponding protein observations: (A) PAO1 and (B) PA14. The gray line represents the frequencies of genes with a microarray signal (log2-transformed) for which proteins were not observed in shotgun proteomics. The blue line represents the frequencies of genes with a microarray signal for which we also measured the corresponding protein. Some genes with high microarray signals are still not observed at the protein level (gray line to the right of the red mark) and may be translationally repressed or targeted for degradation. To identify candidates for such regulation, we considered mRNAs for which protein was not observed with abundances above the red line (corresponding to an 80% chance of detecting protein). To increase the stringency further, we sorted all genes with microarray signals greater than the cutoff and discarded the bottom 20% of the list. In total, 181 genes were identified as putatively translationally repressed, and 97 showed in both PAO1 and PA14 (Supporting Information Table S4). Because it is possible that low detectability in shotgun proteomics can produce this bias, we corrected for mass spectrometry detectability using the APEX method.
Using KEGG pathway enrichment analysis, we identified ribosomal proteins (PA3745, PA4432, and PA5049) with significantly high protein-to-mRNA ratios. We also found that genes involved in terpenoid backbone biosynthesis (PA3627 and PA4557), nucleotide excision repair (PA1529 and PA4234), and one carbon (folate) metabolism (PA0944 and PA1843) showed low protein-to-mRNA ratios. Genes involved in oxidative phosphorylation (PA1582|sdhD, PA2643|nuoH, PA2645|nuoJ, PA2646|nuoK, PA2648|nuiM, and PA4430) had reasonably high mRNA levels, but we did not detect protein for them, suggesting that these genes may be translationally repressed.
Modeling Post-Transcriptional Regulation
If most gene regulation occurred at the level of transcription and RNA degradation, then we might expect protein abundance to be directly proportional to mRNA abundance, as a constant level of protein is translated from mRNA. However, recent studies show that variation in mRNA concentrations can only explain a fraction (one-third to one-half) of the resulting variation in final protein concentrations.1,4,9 On the basis of the observed conservation of protein-to-mRNA ratios across the two closely related P. aeruginosa strains, we argue that these ratios can predict post-transcriptional regulation. To model this, first we assumed a linear relationship between log-transformed protein and mRNA concentrations within each organism (Figures 1 and 3). With the additional assumption of steady state for both protein and mRNA abundances (degradation and synthesis are not considered separately, and they are assumed to be constant across the cell population over time), their linear relationship can be described as
where P, M, and ε represent the protein abundance, the mRNA abundance, and the random error term, respectively. Here, αspeciesX is the global translational efficiency in species X, representing how many proteins can be produced from a given mRNA amount. It should be noted that αspeciesX does not account for dynamic features in translation, such as protein and mRNA degradation and time delay in translation. If gene-specific post-transcriptional regulation is negligible, then global translation efficiency should be dominant, resulting in a constant protein-to-mRNA ratio for all genes in a given species (αspeciesX). However, as shown in Figure 2, the protein-to-mRNA ratios of PAO1 and PA14 were not a constant, and those of orthologous genes in two strains were highly correlated to each other. On the basis of these observations, we revised the equation as follows
Here, βspeciesX,geneY is the gene Y-specific post-transcriptional regulation factor in species X, representing the adjustment of translation for individual genes to their mRNA levels. We concluded that gene-specific post-transcriptional regulation factors, βspeciesX,geneY, were conserved between PAO1 and PA14. Indeed, as shown in Figure 5, we can predict protein abundance more accurately when we incorporate these gene-specific post-transcriptional regulation factors with mRNA abundance. It should be noted that these post-transcriptional regulation factors will vary depending on environmental conditions and related post-transcriptional regulators such as noncoding RNA and RNA-binding proteins. Although our model does not distinguish translation and degradation separately, the specific parameter αspeciesX and gene-specific factor βspeciesX,geneY incorporate these processes implicitly.
Figure 5.

Protein abundance is well-predicted from mRNA abundance and conserved protein-to-mRNA ratio. (A) PAO1 predicted protein abundance was calculated with PAO1 mRNA abundance multiplied by PA14 protein-to-mRNA ratio. (B) Similarly, PA14 predicted protein abundance was calculated with PA14 mRNA abundance multiplied by PAO1 protein-to-mRNA ratio. Compared to Figure 1, it is clear that gene-specific protein-to-mRNA ratios help to predict protein abundance from mRNA abundance more accurately. SpR: Spearman rank correlation.
Discussion
In this study, we measured the correlation between mRNA and protein abundances of 703 orthologous gene pairs in two P. aeruginosa reference strains, PAO1 and PA14 (summarized in Figure 6). (Fifteen such genes with significantly different protein abundance between PAO1 and PA14 without accompanying differences in mRNA abundance or 5′ sequence are given in Table 3.) Similar to previous studies, we observed that mRNA and protein abundances of orthologous genes are less well correlated within each strain (Rs = 0.64–0.65) than the protein and mRNA abundances between the two strains (Rs = 0.89 and 0.95, respectively). Because we examined mRNA and protein levels in two P. aeruginosa strains grown under identical conditions, we were able to focus the analysis on evolutionary differences (e.g., sequence divergence) while controlling for the influence of different environments.
Table 3. Fifteen Genes with Significantly Different Protein Abundance between PAO1 and PA14 but without Accompanying Differences in mRNA Abundance or 5′ Sequence.
| PAO1 identifier | PAO1 protein abundancea | PA14 protein abundancea | PAO1 mRNA abundanceb | PA14 mRNA abundanceb | products |
|---|---|---|---|---|---|
| PA0022 | 13.8 | 3.9 | 106.5 | 113.0 | conserved hypothetical protein |
| PA0331|ilvA1 | 7.3 | 2.4 | 535.0 | 605.5 | threonine dehydratase, biosynthetic |
| PA0508 | 6.7 | 0.0 | 39.0 | 45.0 | probable acyl-CoA dehydrogenase |
| PA4315|mvaT | 32.6 | 70.7 | 5368.0 | 5937.5 | transcriptional regulator MvaT, P16 subunit |
| PA4420 | 2.6 | 7.2 | 614.0 | 662.0 | conserved hypothetical protein |
| PA4438 | 31.1 | 12.6 | 731.5 | 780.0 | conserved hypothetical protein |
| PA4461 | 18.0 | 7.8 | 2341.0 | 2603.0 | probable ATP-binding component of ABC transporter |
| PA4557|lytB | 2.2 | 5.8 | 382.0 | 420.0 | LytB protein |
| PA4837 | 0.0 | 3.9 | 63.0 | 65.0 | probable outer membrane protein precursor |
| PA5013|ilvE | 47.7 | 16.9 | 1504.5 | 1760.0 | branched-chain amino acid transferase |
| PA5018|msrA | 5.1 | 17.7 | 367.0 | 406.0 | peptide methionine sulfoxide reductase |
| PA5201 | 28.5 | 7.3 | 1453.0 | 1523.5 | conserved hypothetical protein |
| PA5286 | 9.4 | 3.6 | 818.0 | 1366.5 | conserved hypothetical protein |
| PA5343 | 8.3 | 3.0 | 193.5 | 246.5 | hypothetical protein |
| PA5535 | 0.0 | 17.1 | 42.0 | 42.5 | conserved hypothetical protein |
The protein abundance value represents the average APEX score of two biological replicates.
The mRNA abundance value represents the average normalized microarray signals of two biological replicates.
In contrast to previous studies,2,3,38 we observed that for the two P. aeruginosa strains mRNA abundances are more conserved than protein abundances (0.95 and 0.89 in comparison to roughly −0.01 and 0.57 from other studies2). One possible explanation for this is that transcriptional regulation is more sensitive to environmental conditions than post-transcriptional regulation. A high correlation of mRNA abundances would be observed only when the data are collected under very similar conditions, as was done in this study. Alternatively, in previous studies, differences in microarray platforms and sequence hybridization may be larger than expected, accounting for the observed lower correlation of mRNA concentrations. Of course, the higher correlation in mRNA abundances that we observed compared to protein abundances may also be explained by lower variation between biological replicates in the mRNA measurements as compared to the protein measurements. However, after filtering out inconsistently observed genes between biological replicates (see Supporting Information Figures S1 and S2 for details), we found a very high correlation (Spearman rank correlation > 0.95) between replicates for both mRNA and protein abundances, so it is unlikely that the experimental variance between mRNA and protein abundances significantly affected our results. Thus, the apparent post-transcriptional buffering of divergence of mRNA concentrations across organisms does not seem to hold true when accounting for any differences in environmental conditions and focusing simply on two very closely related strains: both transcriptional and post-transcriptional regulation appears to diverge between the two P. aeruginosa strains and has additive effects on the final protein concentrations.
It is also possible that a small group of highly abundant genes, such as housekeeping genes, may dominate the interspecies correlation patterns we observed in this study. Unlike studies between more distant organisms, such as fly and worm,3 where the proportion of housekeeping genes may be higher among the conserved genes, here we analyzed two bacterial strains of the same species and do not expect housekeeping genes to dominate the set of orthologues. Also, as shown in Figure 1, the correlation patterns are quite consistent over a wide range of abundance both in protein and mRNA. Thus, the correlation is unlikely to derive primarily from highly abundant housekeeping genes. We also identified genes with significantly high or low protein-to-mRNA ratios (Supporting Information Table S3) and genes with high mRNA abundances that we did not detect in our proteomics experiment (Supporting Information Table S4). Low protein-to-mRNA ratios or translational repression may be alternatively explained by low detectability. Shotgun proteomics can introduce certain biases because of inefficient peptide ionization and unavailability of informative tryptic peptides, which could potentially account for some of these proteins. One possible scenario is that post-translational protein modifications could be missed when searching the mass spectrometry data: proteins might appear to be at lower concentration when, in reality, they are post-translationally modified, which hinders their identification. However, most of the proteins we observed in this study were identified by two or more peptides (653 PAO1 proteins and 664 PA14 proteins, respectively, out of 703 one-to-one protein pairs used in the correlation analysis), making it unlikely that low protein-to-mRNA ratios were caused by systematic bias because of unidentified modified peptides. Another possibility to explain translational repression is a systematic bias of mass spectrometry against certain types of proteins, such as membrane proteins. Although we identified more proteins localized in the cytoplasm than those localized in other cellular compartments, we found no significant bias in protein-to-mRNA ratios according to localization (Supporting Information Figure 3), confirming that the translational repression we observed here was not due to technical bias.
To validate our findings in the correlation analysis further, we reanalyzed all proteomics data using an alternative label-free quantification method based on MS1 intensities and confirmed the same trends for protein-to-mRNA ratios to be more conserved across species than for protein abundances to be correlated with mRNA abundances within species (Supporting Information Figure 5 and Supporting Information Table 8). Recently developed techniques improving detectability and precision of both the proteome and transcriptome, such as selected reaction monitoring (SRM) and RNA–seq, would be helpful to minimize the potential for systematic bias further, as has been already shown in several studies of single organisms.12,42 Also, techniques discriminating newly synthesized mRNAs and proteins by labeling4 should allow the contributions of different post-transcriptional regulatory mechanisms to the observed divergence of mRNA and protein abundances to be determined.
Interestingly, several virulence genes in P. aeruginosa were differentially expressed at the protein level under our growth conditions. The mexEF–oprN operon was more highly expressed in PAO1 at both the mRNA and protein levels. We confirmed that PAO1 is more resistant to chloramphenicol, a substrate of mexEF–oprN efflux pump, than PA14. Although MexT is a known regulator of the mexEF–oprN operon,39,43 both its mRNA and protein expression was low under our growth conditions. Thus, the high expression of this operon in PAO1 may be independent of mexT expression.44
P. aeruginosa produces 4-quinolones, a structurally diverse group of small molecules that act as cell–cell signals and antibiotics. The products required for 4-quinolone biosynthesis and regulation are encoded by the pqs operons (pqsA–E, pqsH, and pqsL). Notably, PA14 expressed all of them at higher levels than PAO1 (Supporting Information Figure S4), confirming that they are differentially regulated between PAO1 and PA14 in SCFM. By testing for overrepresented KEGG pathways among the differentially expressed proteins (p value < 0.05 estimated by resampling), we also observed significant differences in several key metabolic pathways including amino acid metabolism, which can impact 4-quinolone levels because of the shared metabolic precursors between these pathways. Given the low mRNA abundances for genes in this pathway in PAO1, translational repression appears to be unlikely to explain these differences; rather, PA14 appears to have upregulated this pathway relative to PAO1 at the transcript level. Our approach to measure protein and mRNA abundances between closely related organisms complements previous studies on protein and mRNA concentrations in a single organism under different conditions.6−8,10,45,46 Although these studies report similarly discordant tendencies of protein and mRNA abundances and the importance of post-transcriptional regulation, the detailed mechanisms of post-transcriptional regulation are still unclear. Mechanisms of translational suppression can include altering the mRNA structure of translation initiation sites47 and the presence of cis-encoded antisense RNAs.48 Recently, several studies have reported the genome-wide effect of trans-acting post-transcriptional regulation in bacteria, such as small RNAs41,49−51 and RNA-binding proteins,52,53 which can impact RNA synthesis, stability, sequestration, and degradation. In the future, it will be interesting to investigate the contribution of these regulatory mechanisms to the observed discordance between protein and mRNA levels in various organisms.
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Science Foundation, Cancer Prevention Research Institute of Texas, U.S. Army Research (58343-MA), ARO award (W911NF-12-1-0390), and the Welch Foundation (F1515) to E.M.M. and from the NIH and Cystic Fibrosis Foundation to M.W. C.V. acknowledges funding by the NYU Whitehead Fellowship and the NYU University Research Challenge Fund. M.W. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease.
Supporting Information Available
mRNA abundance correlates highly between biological replicates; protein abundance correlates highly between biological replicates; abundance distributions of proteins localized in different cellular compartments; expression of pqs (4-quinolone) genes; alternate protein quantification methods show similar trends; genes with significant differential mRNA expression; genes with significant differential protein expression; top50/bottom50 genes when sorted by protein-to-mRNA ratio; candidate genes for translational repression or targeted degradation; explanation of differential protein expression by translation efficiency; differential protein expression that cannot be explained by mRNA difference and translation efficiency; raw data for mRNA and protein expression measurements prior to filtering; and protein abundance based on MS1-intensities. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Vogel C.; Abreu R.; de S.; Ko D.; Le S.-Y.; Shapiro B. A.; Burns S. C.; Sandhu D.; Boutz D. R.; Marcotte E. M.; Penalva L. O.; et al. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400-1–400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent J. M.; Vogel C.; Kwon T.; Craig S. A.; Boutz D. R.; Huse H. K.; Nozue K.; Walia H.; Whiteley M.; Ronald P. C.; et al. Protein abundances are more conserved than mRNA abundances across diverse taxa. Proteomics 2010, 10, 4209–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrimpf S. P.; Weiss M.; Reiter L.; Ahrens C. H.; Jovanovic M.; Malmström J.; Brunner E.; Mohanty S.; Lercher M. J.; Hunziker P. E.; et al. Comparative functional analysis of the Caenorhabditis elegans and Drosophila melanogaster proteomes. PLoS Biol. 2009, 7, e1000048-1–e1000048-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [DOI] [PubMed] [Google Scholar]
- Lu P.; Vogel C.; Wang R.; Yao X.; Marcotte E. M. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007, 25, 117–124. [DOI] [PubMed] [Google Scholar]
- Nie L.; Wu G.; Zhang W. Correlation between mRNA and protein abundance in Desulfovibrio vulgaris: A multiple regression to identify sources of variations. Biochem. Biophys. Res. Commun. 2006, 339, 603–610. [DOI] [PubMed] [Google Scholar]
- Nie L.; Wu G.; Zhang W. Correlation of mRNA expression and protein abundance affected by multiple sequence features related to translational efficiency in Desulfovibrio vulgaris: A quantitative analysis. Genetics 2006, 174, 2229–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T.; Schmidt A.; Güell M.; Kühner S.; Gavin A.-C.; Aebersold R.; Serrano L. Quantification of mRNA and protein and integration with protein turnover in a bacterium. Mol. Syst. Biol. 2011, 7, 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C.; Marcotte E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell M.; Van Noort V.; Yus E.; Chen W.-H.; Leigh-Bell J.; Michalodimitrakis K.; Yamada T.; Arumugam M.; Doerks T.; Kühner S.; et al. Transcriptome complexity in a genome-reduced bacterium. Science 2009, 326, 1268–1271. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y.; Choi P. J.; Li G.-W. W.; Chen H.; Babu M.; Hearn J.; Emili A.; Xie X. S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 2010, 329, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omasits U.; Quebatte M.; Stekhoven D. J.; Fortes C.; Roschitzki B.; Robinson M. D.; Dehio C.; Ahrens C. H. Directed shotgun proteomics guided by saturated RNA–seq identifies a complete expressed prokaryotic proteome. Genome Res. 2013, 23, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway B. W.; Krishnapillai V.; Morgan A. F. Chromosomal genetics of Pseudomonas. Microbiol. Rev. 1979, 43, 73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati N. T.; Urbach J. M.; Miyata S.; Lee D. G.; Drenkard E.; Wu G.; Villanueva J.; Wei T.; Ausubel F. M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2833–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carilla-Latorre S.; Calvo-Garrido J.; Bloomfield G.; Skelton J.; Kay R. R.; Ivens A.; Martinez J. L.; Escalante R. Dictyostelium transcriptional responses to Pseudomonas aeruginosa: Common and specific effects from PAO1 and PA14 strains. BMC Microbiol. 2008, 8, 109-1–109-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.-W.; Mahajan-Miklos S.; Ausubel F. M. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K. L.; Aye L. M.; Whiteley M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 8079–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse H. K.; Kwon T.; Zlosnik J. E. A.; Speert D. P.; Marcotte E. M.; Whiteley M. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 2010, 1, e00199-1–e00199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L.; Cope L.; Bolstad B. M.; Irizarry R. A. Affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Pham X. Q.; Erwin A. L.; Mizoguchi S. D.; Warrener P.; Hickey M. J.; Brinkman F. S.; Hufnagle W. O.; Kowalik D. J.; Lagrou M.; et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 2000, 406, 959–964. [DOI] [PubMed] [Google Scholar]
- Lee D. G.; Urbach J. M.; Wu G.; Liberati N. T.; Feinbaum R. L.; Miyata S.; Diggins L. T.; He J.; Saucier M.; Déziel E.; et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006, 7, R90-1–R90-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G. S. C.; Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 2005, 6, 31-1–31-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsor G. L.; Lo R.; Ho Sui S. J.; Ung K. S. E.; Huang S.; Cheng D.; Ching W.-K. H.; Hancock R. E. W.; Brinkman F. S. L.; Sui S. J. H. Pseudomonas aeruginosa Genome Database and PseudoCAP: Facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 2005, 33, D338–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. K.Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article3. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Keller A.; Nesvizhskii A. I.; Kolker E.; Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Keller A.; Eng J.; Zhang N.; Li X.-J.; Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 2005, 1, 2005.0017-1–2005.0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner S.; Shu H.; Frank A.; Wang L.-C.; Zandi E.; Mumby M.; Pevzner P. A.; Bafna V. InsPecT: Identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 2005, 77, 4626–4639. [DOI] [PubMed] [Google Scholar]
- Kim S.; Mischerikow N.; Bandeira N.; Navarro J. D.; Wich L.; Mohammed S.; Heck A. J. R.; Pevzner P. A. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 2010, 9, 2840–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon T.; Choi H.; Vogel C.; Nesvizhskii A. I.; Marcotte E. M. MSblender: A probabilistic approach for integrating peptide identifications from multiple database search engines. J. Proteome Res. 2011, 10, 2949–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C.; Marcotte E. M. Calculating absolute and relative protein abundance from mass spectrometry-based protein expression data. Nat. Protoc. 2008, 3, 1444–1451. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Keller A.; Kolker E.; Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Braisted J. C.; Kuntumalla S.; Vogel C.; Marcotte E. M.; Rodrigues A. R.; Wang R.; Huang S.-T.; Ferlanti E. S.; Saeed A. I.; Fleischmann R. D.; et al. The APEX quantitative proteomics tool: Generating protein quantitation estimates from LC–MS/MS proteomics results. BMC Bioinf. 2008, 9, 529-1–529-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.; Fermin D.; Nesvizhskii A. I. Significance analysis of spectral count data in label-free shotgun proteomics. Mol. Cell. Proteomics 2008, 7, 2373–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Remm M.; Storm C. E.; Sonnhammer E. L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [DOI] [PubMed] [Google Scholar]
- Markham N. R.; Zuker M. UNAFold: Software for nucleic acid folding and hybriziation. Methods Mol. Biol. 2008, 453, 3–31. [DOI] [PubMed] [Google Scholar]
- De Sousa Abreu R.; Penalva L. O.; Marcotte E. M.; Vogel C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetar H.; Gilmour C.; Klinoski R.; Daigle D. M.; Dean C. R.; Poole K. mexEF-oprN multidrug efflux operon of Pseudomonas aeruginosa: Regulation by the MexT activator in response to nitrosative stress and chloramphenicol. Antimicrob. Agents Chemother. 2011, 55, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Campbell A.; Karlin S. Correlation between Shine–Dalgarno sequences and gene features such as predicted expression levels and operon structures. J. Bacteriol. 2002, 184, 5733–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E.; Haas D. Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl. Microbiol. Biotechnol. 2011, 91, 63–79. [DOI] [PubMed] [Google Scholar]
- Marguerat S.; Schmidt A.; Codlin S.; Chen W.; Aebersold R.; Bähler J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 2012, 151, 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler T.; Epp S. F.; Curty L. K.; Pechère J. C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Schweizer H. P. Evidence of MexT-independent overexpression of MexEF-OprN multidrug efflux pump of Pseudomonas aeruginosa in presence of metabolic stress. PLoS One 2011, 6, e26520-1–e26520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. V.; Topper S. E.; Hubler S. L.; Hose J.; Wenger C. D.; Coon J. J.; Gasch A. P. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 2011, 7, 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher J. M.; Liebermeister W.; Jules M.; Uhr M.; Muntel J.; Botella E.; Hessling B.; Kleijn R. J.; Le Chat L.; Lecointe F.; et al. Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science 2012, 335, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [DOI] [PubMed] [Google Scholar]
- Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Opin. Microbiol. 2007, 10, 102–109. [DOI] [PubMed] [Google Scholar]
- Sonnleitner E.; Abdou L.; Haas D. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21866–21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E.; Gonzalez N.; Sorger-Domenigg T.; Heeb S.; Richter A. S.; Backofen R.; Williams P.; Hüttenhofer A.; Haas D.; Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 2011, 80, 868–885. [DOI] [PubMed] [Google Scholar]
- González N.; Heeb S.; Valverde C.; Kay E.; Reimmann C.; Junier T.; Haas D. Genome-wide search reveals a novel GacA-regulated small RNA in Pseudomonas species. BMC Genomics 2008, 9, 167-1–167-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J.; Luisi B. F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011, 9, 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E.; Schuster M.; Sorger-Domenigg T.; Greenberg E. P.; Bläsi U.; Blasi U. Hfq-dependent alterations of the transcriptome profile and effects on quorum sensing in Pseudomonas aeruginosa. Mol. Microbiol. 2006, 59, 1542–1558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.