

Abstract
Vici syndrome is a recessively inherited multisystem disorder characterized by callosal agenesis, cataracts, cardiomyopathy, combined immunodeficiency and hypopigmentation. To investigate the molecular basis of Vici syndrome, we carried out exome and Sanger sequence analysis in a cohort of 18 patients. We identified recessive mutations in EPG5 (previously KIAA1632), indicating a causative role in Vici syndrome. EPG5 is the human homologue of the metazoan-specific autophagy gene epg-5, encoding a key autophagy regulator (ectopic P-granules autophagy protein 5) implicated in the formation of autolysosomes. Further studies demonstrated a severe block of autophagosomal clearance in muscle and fibroblasts from EPG5 mutant patients, resulting in autophagic cargo accumulation in autophagosomes. These findings indicate Vici syndrome as a paradigm of a human multisystem disorder associated with defective autophagy, and suggest a fundamental role of the autophagy pathway in the anatomical and functional formation of organs such as the brain, the heart and the immune system.
Vici syndrome [OMIM 242840]1 is a rare multisystem disorder characterized by callosal agenesis, cataracts, cardiomyopathy, combined immunodeficiency and hypopigmentation. Occurrence in consanguineous families and siblings suggests recessive inheritance1-6.
We identified 18 patients with Vici syndrome of Caucasian (n=11), Arab (n=3), Turkish (n=2), Japanese (n=1) and British-Asian origin (n=1), nine of them previously reported1,4-9. Clinical findings from these patients are summarized in Supplementary Table 1 and illustrated in Supplementary Figure 1.
Muscle biopsies available from 8 patients showed consistent myopathic features5,6, comprising fibre type disproportion with type 1 atrophy, increased internal nucleation and abnormal glycogen accumulation (data not shown). On electron microscopy (Figure 1), there was redundancy of basal lamina with material between layers suggesting exocytosis of debris. There were numerous vacuole-like areas and dense bodies possibly of lysosomal origin. Myofibrils were lacking in many fibres. Mitochondria were of variable size, abnormal distribution and morphology.
Figure 1. Ultrastructural abnormalities in Vici syndrome.
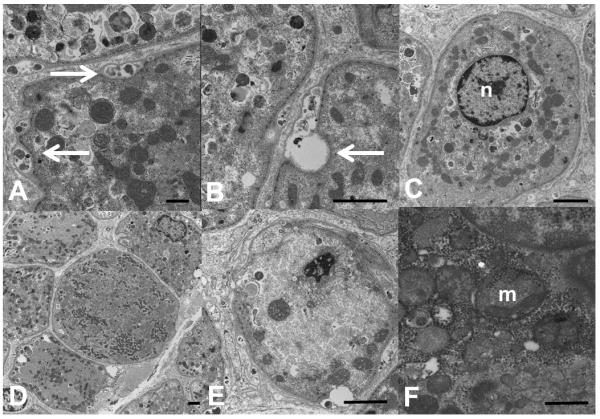
Muscle biopsy from Patient 4.1, electron microscopy, transverse sections. In many fibres, there is material between layers of basal lamina (A, arrows, bar = 500 nm), or overt exocytic vacuoles (B, arrow, bar = 2 microns). Some fibres show a single centralized nucleus (C, bar = 2 microns). Mitochondria are of variable size and distribution. In some fibres, they form a loop around the nucleus in the periphery of the fibre resembling a necklace (C), in others they form clusters (D, bar = 2 microns). Appearance of cristae is often abnormal (E, bar = 2 microns; F, bar = 500 nm). n = nucleus, m = mitochondrion.
We sequenced the exomes of 4 individuals with Vici syndrome from 3 families, one multiplex consanguineous family and two non-consanguineous families with one affected child each, and identified only one gene, EPG5 (previously KIAA1632) on chromosome 18q12.3, in which we found mutations in all affected individuals. Analysis of all EPG5 coding exons in a total of 15 Vici syndrome families with 18 affected individuals showed homozygosity or compound heterozygosity for truncating mutations (including mutations affecting the invariant donor and acceptor splicing recognition sites) in 10 and compound heterozygosity for a truncating and missense mutations in 2 families. Patient 7.1 was homozygous for a mutation affecting the penultimate base of exon 2, predicted to cause aberrant splicing (Table 1). Parental studies suggested recessive inheritance with no carrier manifestations.
Table 1. Genetic findings in patients with Vici syndrome.
Mutation nomenclature follows the recommended guidelines (www.hgvs.orf/mutnomen). Nucleotide numbering for KIAA1632 is based on GenBank Reference Sequence Number NM_020964.2 and denotes the adenosine of the annotated translation start codon as nucleotide position +1. The genotype in the original probands reported by Dionisi-Vici and colleagues1 (Family 1), is an implied genotype based on heterozygous EPG5 variants identified in the parents. Based on sequencing of cDNA derived from patient fibroblast cultures, the homozygous intronic variants identified in Patient 4.1 were predicted to result in a frameshift and premature stop codon insertion, p.Phe1604Glyfs*20. Further quantitative PCR (qPCR) and Western blot studies on tissue from this patient indicated the presence of an unstable or quickly-degraded polypeptide (data not shown). The heterozygous missense variants identified in Patient 6.1 in trans were absent from in-house exome data and from 372 control chromosomes.
| Family | Patient | Consanguinity | Variant 1 | Variant 2 | ||
|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | Nucleotide | Amino acid | |||
| 1 | 1.1 | No | c.4588C>T | p.Gln1530* | c.5704dupT | p.Tyr1902Leufs*2 |
| 1.2 | No | c.4588C>T | p.Gln1530* | c.5704dupT | p.Tyr1902Leufs*2 | |
| 2 | 2.1 | No | c.2413-2A>G | - | c.6724delA | p.Met2242Cysfs*5 |
| 3 | 3.1 | No | c.1253-1G>T | - | c.5110-1G>C | - |
| 4 | 4.1 | Yes | c.4952+1G>A | p.Phe1604Glyfs*20 | c.4952+1G>A | p.Phe1604Glyfs*20 |
| 5 | 5.1 | Yes | c.3481C>T | p.Arg1161* | c.3481C>T | p.Arg1161* |
| 5.2 | Yes | c.3481C>T | p.Arg1161* | c.3481C>T | p.Arg1161* | |
| 6 | 6.1 | No | c.5835T>A | p.Cys1945* | c.1370T>C c.2351A>C |
p.Leu457Pro p.Gln784Pro |
| 7 | 7.1 | Yes | c.1007A>G | p.Gln336Arg | c.1007A>G | p.Gln336Arg |
| 8 | 8.1 | No | c.2575G>T | p.Glu859* | c.6232C>T | p.Arg2078* |
| 8.2 | No | c.2575G>T | p.Glu859* | c.6232C>T | p.Arg2078* | |
| 9 | 9.1 | Yes | c.4751T>A | p.Leu1584* | c.4751T>A | p.Leu1584* |
| 10 | 10.1 | No | c.2719-1G>A | - | c.6295dupA | p.Ser2099Lysfs*5 |
| 11 | 11.1 | Yes | c.6724delA | p.Met2242Cysfs*5 | c.6724delA | p.Met2242Cysfs*5 |
| 12 | 12.1 | No | c.6005_6006dupAG | p.Leu2003Serfs*30 | c.6112T>C | p.Cys2038Arg |
| 13 | 13.1 | Yes | c.4783C>T | p.Gln1595* | c.4783C>T | p.Gln1595* |
| 14 | 14.1 | Yes | - | - | - | - |
| 15 | 15.1 | No | - | - | - | - |
Patients 14.1 and 15.1 did not have any pathogenic sequence variants; quantitative fluorescent PCR (QF-PCR) did not identify any copy number variations in patient 14.1. Although the absence of EPG5 mutations in these individuals indicates the possibility of locus heterogeneity (supported by the presence of heterozygous SNPs in EPG5 in Patient 14.1, who comes from a consanguineous partnership), we cannot exclude the possibility of splice or promoter variants, or mutations affecting other regulatory elements. Sequencing of the human homologues of the epg genes identified by Tian et al10, EI24 and VMP1, did not reveal any mutations.
EPG5 (or KIAA1632) consists of 44 exons, encodes a protein of 2579 amino acids11 and is predominantly expressed in the CNS, skeletal and cardiac muscle, thymus, immune cells, lung and kidneys. EPG5 encodes the human homologue of epg-5, a protein with a key role in the autophagy pathway of multicellular organisms10. EPG5 (or KIAA1632) has previously been found to be mutated in cancer tissue12, a finding shared with other genes implicated in the autophagy machinery13.
Autophagy is an evolutionarily highly conserved lysosomal degradation pathway with fundamental roles in cellular homeostasis, including nutrient provision during fasting or increased metabolic demands, removal of defective proteins or organelles, and defence against infections14-19. A role in normal embryonic development has been suggested20 and is supported by the recent observation of a link between autophagy and early differentiation events in human embryonic stem cells21. Autophagy has also emerged as a key regulator of cardiac and skeletal muscle homeostasis and functional remodelling22-24. The process of autophagy involves several tightly regulated steps, 1) formation of an isolation membrane (the phagophore), 2) fusion of the phagophore to form a double-membranated autophagosome and, finally, 3) fusion of the autophagosome with a lysosome to form the autolysosome, where both captured membrane and contents are degraded. Defects within the autophagy pathway can be broadly divided into those with impaired autophagosome formation, and those with decreased autolysosome clearance.
Membrane dynamics during autophagy are highly conserved from yeast to mammals, but along the evolutionary trajectory involve an increasingly complex molecular machinery: In yeast, a set of more than 30 ATG (autophagy-related) genes encode proteins involved in the autophagic cascade18; this conserved group is modulated in higher organisms by mammalian-specific factors, mainly with a role in autophagosome-lysosome fusion. Epg-5 is one of four higher eukaryote-specific autophagy genes (in addition to epg-2, epg-3 and epg-4) which regulate specific autophagy steps in multicellular organisms and are ubiquitously expressed in early development10. Epg-3, -4 and -5 play an important role in starvation-induced autophagy, as suggested by the reduced survival of mutants in the absence of food. Epg-5 appears specifically involved in a late step of autophagy, the autophagic degradation of protein aggregates, as evidenced by defects of phagolysosome formation in the absence of epg-525, accumulation of non-degradative autolysosomes following knockdown of mammalian EPG-5 in HeLa cells and accelerated autophagic degradation following epg-5 overexpression10.
To further investigate the autophagy pathway in Vici syndrome, we performed additional immunofluorescence studies on skeletal muscle tissue from 2 patients (2.1, 3.1): We observed marked fibre atrophy and fibre size inhomogeneity with upregulation of the sarcomere-associated autophagy proteins p62/SQSTM1 and Nbr126-28 (Figure 2), with numerous positive puncta (autophagosomes) in many fibres compared to normal controls that were also positive for LC3 but not for sarcomeric proteins. The same fibres also showed strong upregulation of MURF2, a p62/SQSTM1-linked muscle ubiquitin E3-ligase29. Immunofluorescence microscopy and analysis of EM pictures indicated no obvious myofibrillar abnormalities, in contrast to secondary autophagy defects due to mutations in BAG330 or the kinase domain of titin26,31. This suggests that EPG5 may not be involved in the autophagic degradation of myofibrillar components.
Figure 2. Accumulation of Nbr1-positive puncta in skeletal muscle of Vici patient.

Transverse sections from muscle biopsies from a normal control (bottom panel) and Patient 3.1 (top panel) were stained with monoclonal antibody against Nbr1 (green channel), polyclonal MURF2 antibody (red channel), and counterstained with the anti-titin M-band antibody T-M8ra (blue channel). Accumulation of Nbr1 in puncta (autophagosomes, arrowheads in top panel), and of MURF2, as well as fibre inhomogeneity with marked fibre atrophy characterise Vici muscle. Numerous fibres of very small cross-sectional area (arrows in top panel) with high content of MURF2 and Nbr1 puncta are frequently seen. Scale bar: 10 μm.
We hypothesized that the increase in p62/SQSTM1 and Nbr1 in atrophic muscle fibres reflects a block in the autophagy pathway, in keeping with the accumulation of autolysosomes observed after epg-5 knockdown in multicellular organisms10, and the observation of severe muscle atrophy and cardiac failure following autophagy inhibition22,32,33. To further investigate if the increased p62/SQSTM1 and Nbr1 levels are reflective of increased upstream induction or downstream blockade of the autophagy pathway, we exposed patient-derived and healthy control fibroblasts for 12 hours to the autophagy inducer rapamycin (an inhibitor of the mTORC1 complex) and the autophagy inhibitor bafilomycin (an inhibitor of the autolysosomal H+ ATPase required for acidification and hence degradation of lysosomal contents34). Untreated patient-derived cells showed increased levels of p62/SQSTM1, Nbr1 and LC3, particularly of processed, lipidated LC3-II. Induction of autophagy by rapamycin, or block of autophagosomal clearance by bafilomycin, led to strong accumulation of Nbr1 and p62/SQSTM1 in control cells at 12 hours, while the combined upstream activation by rapamycin and the block of clearance by bafilomycin led to further accumulation of these proteins, as well as of LC3-II at 12 hours (Figure 3), as expected. In contrast, the elevated levels of Nbr1 and p62/SQSTM1 increased no further in Vici syndrome patient cells following rapamycin treatment, or combined treatment. This suggests that the induction of early steps in autophagy, including the processing of LC3-I to LC3-II, is unimpaired in Vici syndrome, while the clearance of autophagosomal cargo is nearly saturated. These observations are supported by immunofluorescence microscopy in Vici patient fibroblasts: we observed a high level of baseline Nbr1 and p62/SQSTM1 positive puncta (Supplementary Figure 2) as well as LC3- and p62/SQSTM1 positive puncta (Supplementary Figure 3) compared to control cells, indicative of the accumulation of autophagosomes in the epg-5 deficient cells. In contrast, we found that the fusion of LC3-positive puncta with lysosomes, as indicated by the colocalisation of LC3 with lysosome-associated membrane proteins (LAMP1), is reduced in Vici patient fibroblasts (Figure 4). Similarly, the fusion of Nbr1-positive puncta with LAMP1 positive lysosomal vesicles was strongly reduced (Supplementary Figure 4). Lastly, baseline levels of lysine-63 polyubiquitylated proteins (a measure of ubiquitylation products destined for autolysosomal degradation) were elevated in Vici cells (Supplementary Figure 5). Together, these results indicate a severe deficit in autophagosomal clearance associated with mutations in EPG5, resulting in the accumulation of autophagic cargo in Nbr1 and SQSTM1-positive autophagosomes and impaired fusion with lysosomes.
Figure 3. Autophagy is blocked at a late stage in Vici syndrome.
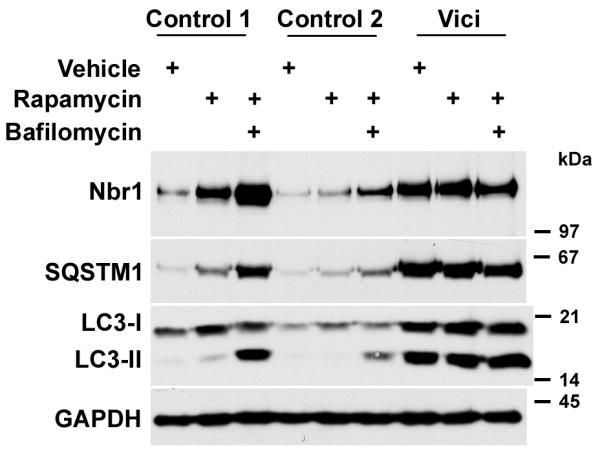
Accumulation of autophagy adaptors Nbr1, p62/SQSTM1 and the phagophore membrane component LC3- is induced in control and Vici-patient fibroblasts (Patient 4.1) by 12 h treatment with rapamycin, or dual treatment with rapamycin and bafilomycin. In control cells, rapamycin induces slight accumulation of unprocessed LC3-I and processed LC3-II, Nbr1 and p62/SQSTM1; dual induction of autophagy by rapamycin and block of autophagosomal degradation by bafilomycin leads to marked accumulation of all autophagosome components. In Vici patient fibroblasts, strong baseline accumulation of all components is visible that is largely unresponsive to both drugs.
Figure 4. Fusion of LC3-positive puncta with lysosomes in Vici syndrome.

In control fibroblasts subjected to 6h bafilomycin treatment, lysosomal structures were detected by staining with monoclonal anti-LAMP1. Numerous LC3-positive autophagosomes are found engulfed by the LAMP1-positive vesicular structures (arrowheads). In contrast, Vici patients fibroblasts consistently show smaller LC3-positive puncta that only sporadically colocalise with LAMP1, with many isolated LC3-positive puncta (arrows). Note that LC3 signal in Vici cells occurs mostly at the rim of LAMP1-positive structures, not centrally. Scale bar: 5 μm
To see if these changes also involve upstream regulatory mechanisms of autophagy, we analysed the AKT-mTOR protein kinase pathway, a major pathway controlling the expression of autophagy proteins by inhibiting the transcriptional activity of FoxO transcription factors35. We observed drastically reduced phosphorylation of the two activating sites in AKT (Ser308 and 473) and consequentially reduced phosphorylation of the AKT substrate GSK3beta on the inhibitory Ser21 site (Supplementary Figure 6). While total FoxO3 protein levels were elevated about 5-fold, the ratio of phosphorylated FoxO was decreased to about 0.6 (see also Supplementary Figure 7). These observations suggest a profound derailment of autophagy in Vici syndrome on multiple regulatory levels, including its transcriptional regulation via the AKT pathway.
These findings are all in agreement with histopathological features consistent with defective autophagy, such as prominence of autophagic vacuoles, storage of abnormal material and secondary mitochondrial abnormalities, the latter likely to reflect the evolutionarily conserved role of the autophagy pathway in maintaining mitochondrial quality and function36. On the clinical level, skeletal and cardiac muscles involvement also feature prominently in other conditions with primary disturbance of autophagy such as X-linked recessive Danon disease [OMIM 300257], caused by failure of correct autolysosome formation due to mutations in the lysosomal LAMP-2 gene37. Disturbed autophagy is also an important secondary pathogenetic mechanism in other neuromuscular disorders26,38-40. In the heart, autophagy plays an important role in the constant renewal of the post-mitotic cardiomyocyte41 and as a mechanism to meet nutrient requirements at times of increased metabolic demands42,43. Of note, microscopic cardiac changes on post mortem in one patient were much more pronounced in the left compared to the right ventricle9, suggesting a relation to the degree of physiological workload.
Additional defects of CNS development, immune regulation and skin pigmentation in Vici syndrome patients implicate autophagy in a wider range of cellular processes. In the brain, autophagy has been extensively investigated as a key pathogenic mechanism in various neurodegenerative disorders [for review17,44]. The observation of callosal agenesis and disturbed neuromigration in patients with Vici syndrome indicates aberrant autophagy also as a cause of disturbed embryonic CNS development, a notion so far only supported by the observation of increased neuronal proliferation and severe neural tube defects in mice with null mutations in AMBRA1, a positive autophagy regulator with predominant expression in neural tissues45. In the immune system, autophagy is generally involved in the delivery of microorganisms to lysosomes (“xenophagy”) and, more specifically, in trafficking events that activate immunity46. As indicated by observations in the T-cell specific atg5−/− knockout mouse, autophagy is also directly important for T-cell survival and proliferation47, whilst our findings also indicate a role of autophagy in B-cell homeostasis. The finding of generalized hypopigmentation in Vici syndrome supports a functional relationship between melanogenesis and the autophagic machinery as has been suggested recently48.
In conclusion, our findings indicate Vici syndrome as a paradigm of a human multisystem disorder associated with defective autophagy. The wide range of associated clinical manifestations suggests epg-5 as a pivotal protein within the autophagic machinery in different tissues. Other genes within the same pathway are plausible candidates for cases of Vici syndrome unlinked to the EPG5 locus.
METHODS
Subjects
A total of 18 individuals with Vici syndrome were included in this study. Patients were included in the study if four of the five major diagnostic criteria (callosal agenesis, cataracts, cardiomyopathy, hypopigmentation and immunodeficiency) were fulfilled. Clinical features of the patients included in this study are summarized in Supplementary Table 1 and illustrated in Supplementary Figure 1. Research ethics committee approval (Ref No 09/H0807/089 “Identification of genes underlying neurodevelopmental disorders” and Ref No 06/Q0406/33 “Setting up of a rare diseases biological samples Biobank for research to facilitate pharmacological, gene and cell therapy trials in neuromuscular disorders”) was obtained. Informed consent was obtained from all individuals and legal guardians of minors.
Exome capture and sequencing
Library construction
Libraries were prepared according to Agilent’s SureSelect Human All-Exon capture (Illumina Paired-End sequencing, Agilent Technologies, Santa Clara, CA, USA) v.1.0.1 protocol, with the following modifications: 3μg of genomic DNA from patients P2, P4, P5.1 and P5.2 was sheared using adaptive focused acoustics (Covaris S2; Covaris, Woburn, MA, USA) using the following conditions: 20% duty cycle, 5 intensity, 200 cycles per burst, frequency sweeping mode for 90 seconds at 4°C to give an average fragment size of 200bp. The fragmented DNA was then subjected to end-repair, phosphorylation, ‘A’ base addition and adapter ligation followed by 6 cycles of PCR amplification.
Solution capture
Solution capture was carried out according to Agilent’s v.1.0.1 protocol with the following modifications: 500ng of prepped library was mixed with 7.5μg of human Cot-1 DNA and dried down to a pellet using a Savant SpeedVac 120 (Thermo Fisher Scientific, Waltham, MA, USA). This was resuspended in 3.4μl water to give a final library concentration of 147ng/μl. Following hybridisation and selection, the libraries were subjected to 10 cycles of PCR amplification.
Sequencing
Target enriched sequencing libraries were quantified by Qubit (Invitrogen, Carlsbad, CA, USA). Denatured libraries were loaded onto an Illumina Genome Analyser IIx GAIIx (IIIumina, Inc., San Diego, CA, USA) flowcell at a concentration of 10pM. Sequencing of the flowcell was carried out as described in the manufacturer’s instructions using one lane of 76 base pair paired-end sequencing per sample.
Read conversion and alignment
Read conversion alignment and variant calling were conducted in parallel using both the NextGENe (SoftGenetics, State College, PA, USA) and Linux-based tools on the King’s College London cluster as described below. Sequencing metrics are described in Supplementary Table 2.
NextGENe Read conversion and alignment
Reads were obtained in qseq format from the GAIIx instrument. NextGENe software was used to quality filter and convert the reads into fasta format. The following filtering settings were used: median phred score of 15, maximum number of uncalled bases of 10, called base number greater than or equal to 25, and trim or reject reads where 10 or more bases have a phred score of 15 or less. Reads not fulfilling these quality criteria were omitted from the alignment.
Quality filtered fasta converted reads were aligned to the whole human genome reference sequence (GRCh37) using the pre-indexed reference in NextGene.
KCL Linux alignment
Sequence reads were aligned to the reference genome (hg18) with the Novoalign aligner (http://novocraft.com), duplicate reads, resulting from pcr clonality or optical duplicates, and reads mapping to multiple locations were excluded from downstream analysis. Single nucleotide substitutions and small insertion deletions were identified and quality filtered within the SamTools software package 49 and in-house software tools. Variants were annotated with respect to genes and transcripts with the SNPClassifier tool 50. Filtering of variants for novelty was performed by comparison to dbSNP131, 1000 Genomes pilot SNP calls (March 2010).
Gene identification strategy and results
Analogous gene identification strategies were employed using output from both NextGENe and KCL/Linux pipelines. Removal of previously identified variants (NextGENe: dbSNP; KCL pipeline: dbSNP, 1000Genomes, in-house exomes) and synonymous changes identified nine genes and eleven genes, from the NextGENe and Linux datasets, respectively, with homozygous sequence variants in both individuals (Patient 5.1, Patient 5.2) from the consanguineous kindred (Supplementary Table 3). Of these genes, five were shared and ten were identified by a single analysis pipeline. Differences between the two datasets can be attributed to alternative variant calling settings and algorithms and to the subtraction of in-house exome data in the KCL pipeline. The sequencing data for candidate genes identified in both analyses were then interrogated in the remaining two exomes (Patient 2.1, Patient 4.1). For both datasets a single gene, EPG5 (or KIAA1632), was identified in which all patients had two truncating mutations in keeping with expected inheritance patterns (Table 1). The homozygous sequence variants in patients 5.1, 5.2 and 4.1 were present at read depths of 48, 52 and 113, respectively. The heterozygous variants in patient 2.1, c.2413-2A>G and c.6724delA, were present in reference:variant read depth ratios of 35:22 and 68:50, respectively.
Sanger sequencing
All EPG5 (or KIAA1632) mutations identified by exome sequencing were confirmed by bi-directional Sanger sequencing. Sequencing primers were designed to EPG5 (NM_020964.2), EI24 (NM_004879.3) and VMP1 (NM_030938.3) using Primer3 (http://frodo.wi.mit.edu/primer3/) to cover all coding exons and intron-exon boundaries. PCR and sequencing reactions were performed as described previously5. Patients without truncating mutations in EPG5 were subject to sequencing of the human homologues of the EPG genes identified by Tian et al 10, EI24 and VMP1. All sequencing primers used in this study are listed in Supplementary Table 4.
Inheritance studies and mutation frequency in the general population
ABI traces for mutant alleles identified in Families 1-13 are shown in Supplementary Figure 8. Parental samples were available in 11 of the 13 families in which sequence variants were detected. In all cases the sequence variants exhibited segregation in accordance with autosomal recessive inheritance. This figure includes segregation of the missense variants identified in patients 6.1 and 12.1 on the opposite allele to the truncating mutations identified in these families. Additionally, unaffected sibs were available for segregation analysis in families 5 and 9 and were shown to have genotypes consistent with the expected inheritance pattern. Using Merlin (http://www.sph.umich.edu/csg/abecasis/Merlin/index.html) we calculated a LOD score of 8.304 across all families (assuming a recessive model with full penetrance and no phenocopies, a marker allele frequency of 0.000444444 and a disease frequency of 0.00001).
Sequence variant in silico analysis and control chromosome studies
The missense variants, p.Leu457Pro and p.Gln784Pro, identified in patient 6.1 were subject to in silico analysis using the Alamut software suite (http://www.interactive-biosoftware.com/). The affected amino acids show moderate and low levels of conservation, respectively, with Grantham distances of 98 and 76. The SIFT (http://sift.jcvi.org/) and Align GVGD (http://agvgd.iarc.fr/agvgd_input.php) prediction tools indicated neither change likely to be disease causing, but PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) predicted both changes to be probably damaging. Neither variant is predicted to cause aberrant splicing. Both variants were absent from 374 in-house control chromosomes and are absent from greater than 9000 chromosomes on the exome variant server (http://evs.gs.washington.edu/EVS/). It is therefore unclear whether either or both of these variants are responsible for the presumed loss of function of this allele, found in trans with the p.Cys1945X mutation identified in this patient. Undetected mutations, such as large deletions, duplications, inversions, deep intronic splice mutations or promoter mutations, in cis with these missense variants cannot be eliminated as the true causative change on this allele. The p.Cys2038Arg missense variant identified in patient 12.1 was subject to in silico analysis in the same manner. Cys2038 is highly conserved (to fruit fly). The Cysteine to Arginine Grantham distance is 180. PolyPhen-2 and SIFT predict the change to affect protein function, although conversely Align GVGD predicts the change unlikely to be pathogenic. The amino acid change is absent from exome variant server, although an amino acid change at the same location (p.Cys2038Gly) has been observed with a frequency of 1 in 9990 alleles. Other mutations on this allele undetectable by sequencing of coding regions cannot be eliminated, but the high degree of conservation and the disruption of a Cysteine residue, often involved in protein tertiary structure, indicate that this variant may be the second disease-causing mutation in the patient.
Sequence variants affecting the canonical donor and acceptor splice sites were observed in four families (2, 3, 4, 10) and are predicted to abolish splicing according to the prediction tools in Alamut, namely SpliceSiteFinder-like, MaxEntScan, NNSPLICE and Human Splicing Finder. The homozygous variant c.1007A>G (p.Gln336Arg) detected in patient 7 affects the penultimate base of exon 2 and is predicted by SpliceSiteFinder-like and NNSPLICE to abolish the donor splice site; scores for MaxEntScan and Human Splicing Finder are also reduced. Additionally this variant is absent from the exome variant server dataset. This variant seems therefore highly likely to be the pathogenic allele in this patient.
Quantitative fluorescent PCR
Quantitative fluorescent PCR (QF-PCR) was used to interrogate exon copy number in patient 14.1. Quantitative fluorescent PCR (QF-PCR) performed using a two-stage PCR with fluorescently-labelled primers complementary to a tag sequence incorporated into the exon-specific primers in a multiplex reaction (full details available upon request) 51.
cDNA sequencing
Nested PCR primers flanking exon 28 of EPG5 were designed using Primer3. RNA was extracted from fibroblast culture from Patient 4.1 using the RNeasy kit (QIAGEN NV, Venlo, The Netherlands) and reverse transcribed using SuperScript® III First-Strand Synthesis SuperMix (Life Technologies) using an oligo dT primer. The resulting cDNA was subject to a two-stage nested PCR reaction and sequenced. PCR and sequencing reactions were performed as described previously5.
Quantitative real-time PCR (qPCR)
qPCR was used to interrogate relative transcript abundance in patient 4.1. cDNA was generated as above. Dual-labelled probes and primers were designed for EPG5 and ACTB using the IDT real time PCR assay design tool (www.idtdna.com). Quantitative real-time PCR reactions were carried out using TaqMan® Gene Expression Master Mix in a 25μl reaction on an ABI 7900 (Life Technologies). Analysis was conducted using the ΔΔCt method (according to Applied Biosystems ‘Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR’).
Immunostaining and confocal microscopy
Skeletal muscle sections were lightly fixed in 4% paraformaldehyde, followed by permeabilisation in 0.05% Tween-20 in phosphate-buffered saline (PBS) and blocking by 10% goat serum in PBS. Sections were immunostained using antibodies against p62/SQSTM1 (Abcam, ab56416), Nbr1 (Abcam, ab55474), and MURF2 (Abcam, ab4387) and counterstained with an affinity-purified rabbit polyclonal antibody against M-band titin (T-M8ra52) using standard procedures53. Fibroblasts were cultured for the times indicated, washed briefly in phosphate-buffered saline (PBS) and fixed in methanol for 10 mins at −20°C. Cells were immunostained using antibodies against SQSTM/p62 (as above), nbr1 (Novus Biologicals, NBP1-71703), LAMP 1 (monoclonal H4A3, Abcam, ab25630) and LC3B (Cell Signaling Technology, 2775S). Fluorescent secondary antibodies were from Jackson Immunoresearch and Molecular Probes. The specimens were analyzed by confocal microscopy using a Zeiss LSM-510 Meta microscope under 63x magnification and 2-4 times zoom, with identical gain settings for image recording for all related samples.
Cell culture studies and Western blotting
Fibroblast cultures were maintained in Dulbecco’s modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin (100U/ml), streptomycin (100μg/ml) and 50μg/ml uridine in 3 or 10 cm culture dishes in a 5% CO2 atmosphere. Cells were treated with 100nM rapamycin and/or 200nM bafilomycin for 5-12 hours before lysis, compared to equal concentrations of vehicle (DMSO). Cells were harvested after washing twice with PBS directly into Laemmli sample buffer, and lysates were run on 10-15% SDS-PAGE gels. Protein loading was normalised to actin staining and GAPDH-immunoreactivity in Western blot. Western blots were carried out using standing procedures53, and antibodies to LC3B (CellSignalling Technology), nbr1 (Abcam) and p62 (Abcam). All phospho and pan antibodies (AKT, FOXO, GSK3β, mTOR, p70S6K) were obtained from Cell Signaling Technology and antibodies against SQSTM1/p62, nbr1 and LC3B were as above. Densitometric quantification was carried out as previously described 29.
Supplementary Material
Acknowledgements
We are grateful to the individuals with Vici syndrome and their families for their participation in this study. We like to thank our colleagues in the Genomics Facility of the Comprehensive Biomedical Research Centre of Guy’s & St Thomas’ NHS Foundation Trust for their support. We also like to thank the following physicians for their input and productive discussions: Professor Don Creel and Professor Robert O. Hoffman, John A. Moran Eye Center, University of Utah, Salt Lake City, USA; Professor Lihadh Al-Gazali, Department of Paediatrics, Faculty of Medicine & Health Sciences, Al-Ain, UAE. H.J. was supported by a grant from the Guy’s and St Thomas’ Charitable Foundation (Grant number 070404). M.G. and A.L.K. were supported by the Leducq Foundation Transatlantic Network “Proteotoxicity” (11 CVD 04) and the Medical Research Council of Great Britain (MR/J010456/1). M.G. holds the British Heart Foundation Chair of Molecular Cardiology. H.J. would like to dedicate this work to the memory of Rahul Ghosh (10.10.2006 – 04.01.2007), his first patient with Vici syndrome.
Footnotes
Weblinks:
The URLs for the data presented herein are as follows:
(http://frodo.wi.mit.edu/primer3/)
(http://www.sph.umich.edu/csg/abecasis/Merlin/index.html)
(http://www.interactive-biosoftware.com/)
(http://agvgd.iarc.fr/agvgd_input.php)
(http://genetics.bwh.harvard.edu/pph2/)
Author contributions
T.C. designed the experiments, performed whole exome capture, Sanger sequencing, cDNA sequencing, quantitative PCR (qPCR) analysis, analysed data and wrote the manuscript. A.L.K. and B.B. performed immunostaining, confocal microscopy, cell culture studies and Western blotting. Z.U performed qPCR analysis. F.S, M.A.S, S.Y. and S.A. prepared and performed whole exome capture and analysed the exome sequencing data. C.D-V., E.B., V.M.M, M.A-O., S.K., C.K. G.F.H, F.W., A.E.t.H., R.C.R., D.M., R.M., M.H., E.S., D.S., P.M.K., C.W., F.M.F., S.A-K., J.H. and M.D.C. provided clinical data. S.B., I.B., H.H.G. and C.A.S. provided and analysed neuropathological data. S.M. and D.J. provided clinical data and oversaw genetic aspects of the research. M.G. analysed data obtained from immunostaining, confocal microscopy, cell culture studies and Western blotting, and wrote the manuscript. H.J. provided clinical and neuropathological data, analysed exome and Sanger sequencing data, oversaw all aspects of the research and wrote the manuscript. H.J. declares on behalf of all authors that there are no competing financial interests.
REFERENCES
- 1.Vici CD, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am. J. Med. Genet. 1988;29:1–8. doi: 10.1002/ajmg.1320290102. [DOI] [PubMed] [Google Scholar]
- 2.del Campo M, et al. Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. Am. J. Med. Genet. 1999;85:479–85. doi: 10.1002/(sici)1096-8628(19990827)85:5<479::aid-ajmg9>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- 3.Chiyonobu T, et al. Sister and brother with Vici syndrome: agenesis of the corpus callosum, albinism, and recurrent infections. Am. J. Med. Genet. 2002;109:61–6. doi: 10.1002/ajmg.10298. [DOI] [PubMed] [Google Scholar]
- 4.Miyata R, et al. Sibling cases of Vici syndrome: sleep abnormalities and complications of renal tubular acidosis. Am. J. Med. Genet. A. 2007;143:189–94. doi: 10.1002/ajmg.a.31584. [DOI] [PubMed] [Google Scholar]
- 5.McClelland V, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am. J. Med. Genet. A. 2010;152A:741–7. doi: 10.1002/ajmg.a.33296. [DOI] [PubMed] [Google Scholar]
- 6.Al-Owain M, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am. J. Med. Genet. A. 2010;152A:1849–53. doi: 10.1002/ajmg.a.33421. [DOI] [PubMed] [Google Scholar]
- 7.Said E, Soler D, Sewry C. Vici syndrome--a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am. J. Med. Genet. A. 2012;158A:440–4. doi: 10.1002/ajmg.a.34273. [DOI] [PubMed] [Google Scholar]
- 8.Finocchi A, et al. Immunodeficiency in Vici syndrome: a heterogeneous phenotype. Am. J. Med. Genet. A. 2012;158A:434–9. doi: 10.1002/ajmg.a.34244. [DOI] [PubMed] [Google Scholar]
- 9.Rogers CR, Aufmuth B, Monesson S. Vici Syndrome: A Rare Autosomal Recessive Syndrome with Brain Anomalies, Cardiomyopathy, and Severe Intellectual Disability. Case Reports in Genetics. 2011;2011:4. doi: 10.1155/2011/421582. Article ID 421582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–55. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Halama N, Grauling-Halama SA, Beder A, Jager D. Comparative integromics on the breast cancer-associated gene KIAA1632: clues to a cancer antigen domain. Int. J. Oncol. 2007;31:205–10. [PubMed] [Google Scholar]
- 12.Sjoblom T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 13.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 16.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 17.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007;6:304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 18.Klionsky DJ, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010:6. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 20.Di Bartolomeo S, Nazio F, Cecconi F. The role of autophagy during development in higher eukaryotes. Traffic. 2010;11:1280–9. doi: 10.1111/j.1600-0854.2010.01103.x. [DOI] [PubMed] [Google Scholar]
- 21.Tra T, et al. Autophagy in human embryonic stem cells. PLoS One. 2011;6:e27485. doi: 10.1371/journal.pone.0027485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandri M. Autophagy in skeletal muscle. FEBS letters. 2010;584:1411–6. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 23.Portbury AL, Willis MS, Patterson C. Tearin’ up my heart: proteolysis in the cardiac sarcomere. J. Biol. Chem. 2011;286:9929–34. doi: 10.1074/jbc.R110.170571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Curr. Hypertens. Rep. 2009;11:406–11. doi: 10.1007/s11906-009-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, et al. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J. Cell Biol. 2012;197:27–35. doi: 10.1083/jcb.201111053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lange S, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 27.Waters S, Marchbank K, Solomon E, Whitehouse C, Gautel M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett. 2009;583:1846–52. doi: 10.1016/j.febslet.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 28.Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009;5:732–3. doi: 10.4161/auto.5.5.8566. [DOI] [PubMed] [Google Scholar]
- 29.Perera S, Holt MR, Mankoo BS, Gautel M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev. Biol. 2011;351:46–61. doi: 10.1016/j.ydbio.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selcen D, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of Neurology. 2009;65:83–9. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edstrom L, Thornell LE, Albo J, Landin S, Samuelsson M. Myopathy with respiratory failure and typical myofibrillar lesions. J. Neurol. Sci. 1990;96:211–28. doi: 10.1016/0022-510x(90)90134-9. [DOI] [PubMed] [Google Scholar]
- 32.Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6:307–9. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- 33.Taneike M, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010:6. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 34.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiaffino S, Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, et al. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3:337–46. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- 37.Nishino I, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–10. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- 38.Fukuda T, et al. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006;14:831–9. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lunemann JD, et al. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 2007;61:476–83. doi: 10.1002/ana.21115. [DOI] [PubMed] [Google Scholar]
- 40.Fujita E, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum. Mol. Genet. 2007;16:618–29. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 41.Terman A, Brunk UT. Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc. Res. 2005;68:355–65. doi: 10.1016/j.cardiores.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 42.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature Medicine. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 43.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–8. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 44.Williams A, et al. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr. Top. Dev. Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 45.Fimia GM, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 46.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. The J. Exp. Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ganesan AK, et al. Genome-wide siRNA-based functional genomics of pigmentation identifies novel genes and pathways that impact melanogenesis in human cells. PLoS Genetics. 2008;4:e1000298. doi: 10.1371/journal.pgen.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li K, Stockwell TB. VariantClassifier: A hierarchical variant classifier for annotated genomes. BMC Res. Notes. 2010;3:191. doi: 10.1186/1756-0500-3-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heath KE, Day IN, Humphries SE. Universal primer quantitative fluorescent multiplex (UPQFM) PCR: a method to detect major and minor rearrangements of the low density lipoprotein receptor gene. J. Med. Genet. 2000;37:272–80. doi: 10.1136/jmg.37.4.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obermann WM, et al. The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J. Cell Biol. 1996;134:1441–53. doi: 10.1083/jcb.134.6.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harlow E, Lane D. Antibodies, a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 1988. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.