

Abstract
Virus infections have been implicated in both initiation of and protection from autoimmune diseases, such as type 1 diabetes (T1D). In this review we intend to reflect on recent evidence how viruses might on the one hand be involved in the pathogenesis of T1D and on the other hand induce a state of protection from autoimmune-mediated damage. It is important to acknowledge that human individuals encounter more than just one virus infection in their lifetime. Therefore, it is important to integrate more than just one possible environmental triggering factor for autoimmune diseases to occur.
Keywords: animal model, enterovirus, hygiene hypothesis, molecular mimicry, pathogens
Introduction
Looking at recent reviews on the role of viruses in type 1 diabetes (T1D) one thing becomes clear. Besides genetic predisposition, environmental factors, such as virus infections, do indeed appear to play a role in the etiology of the disease, at least for some of the T1D cases. It is, however, controversially discussed what kind of role they are playing. Many original articles reporting studies in animal models demonstrate a role as inducers or accelerators of disease, whereas others provide evidence for a protective role supporting the so-called ‘hygiene hypothesis'. In this review we will summarize evidence from epidemiological studies and from animal models that support either a beneficial or detrimental role of viruses in T1D. The emerging concept is that viruses can have a dual role in T1D and that the sensitive balance of the immune system is continuously affected by environmental factors. The overarching challenge is to determine what effect the sum of all environmental factors that we encounter in our lifetime has on the etiology of autoimmune diseases, such as T1D.
Epidemiology
Genetic predisposition is an important factor in the etiology of T1D. The predominant genetic loci are the HLA class I and II genes that provide either susceptibility (HLA DRB1*03, HLA DRB1*04 and HLA DQB1*0302) to or protection (HLA DQB1*0602) from disease.1, 2 In addition, several other diabetes loci such as ptpn22 and the insulin gene itself for example, have been reported and the polymorphism in the expression of the corresponding genes has been demonstrated to influence the etiology and/or pathogenesis of T1D.1 However, several observations lead to the conclusion that other factors have to contribute to the clinical manifestation and the etiology of T1D. First, not all individuals that carry a predominance of susceptibility genes develop T1D.2 Second, even individuals with protective genetic loci develop disease.2 Finally, there is no strict concordance between homozygous twins.3 Even under circumstances where the risk for T1D is extreme, such as for children with the genotype HLA DRB1*03, HLA DRB1*04 and DQB1*0302 in families with two or more affected family members, the disease penetration is not absolute.4 Thus, additional environmental factors are necessary to initiate and/or propagate the disease. Viruses are prime candidate for such environmental factors, since they activate the innate as well as the adaptive immune system and thereby cause acute and often chronic inflammation. In addition, more recently, evidence has been accumulating that viruses and other infections can operate as mitigating factors to reduce the incidence of T1D in animal models,5, 6 thus supporting some of the aspects of the hygiene hypothesis (see below).7, 8, 9
Epidemiological evidence for viruses as promoters of T1D
Epidemiologically, virus infections have been associated with T1D since quite a while. For example, human enteroviruses, such as coxsackievirus B (CVB), have been associated with T1D since the late 1960s10 and have been found in pancreatic isolates of T1D patients.11, 12, 13 Further, infections with viruses such as rotavirus,14 mumps virus,15 rubella virus 16 and cytomegalovirus 17 have been associated with the development of T1D. Analysis of pancreases of children with fatal infections revealed that destruction of β-cells was associated with CVB, cytomegalovirus and varicella-zoster virus infections.18 Further, inflammatory infiltrates have been predominantly found after CVB infection.18 Due to large-scale vaccination programs many viruses, such as mumps and rubella viruses, have been eradicated in many countries with high T1D incidences. At the same time the incidences of autoimmune diseases and allergies increased in these countries. Therefore, it is quite unlikely that these viruses are involved in the etiology of T1D and that rather the lack of such infections might contribute to the increase in T1D incidence over the last decades (see a more detailed discussion of the hygiene theory below). The strongest evidence for an involvement of viruses in the pathogenesis if T1D derives from enteroviruses (see Ref. 19 for a recent review on that topic). For example, CVB3 and CVB4 RNA have been detected in the blood of recent onset T1D patients.20, 21, 22 It has been shown that the presence of enterovirus RNA in the serum is indeed a risk factor for β-cell autoimmunity and T1D.23 A recent study embedded in the ‘Diabetes and Autoimmunity Study in the Young' program investigated the frequency of antibodies to islet antigens and the presence of enterovirus RNA in serum or rectal swabs of genetically predisposed children.24 The children have been examined periodically and it was found that the progression to T1D was significantly increased in seroconverted children in intervals following enterovirus infection.24 These findings indicate that in genetically predisposed children carrying antibodies to islet antigens enterovirus infection might push an autoimmune condition to overt disease. In other studies, enterovirus proteins have been detected by immunohistochemistry in the pancreas and even within the islets of Langerhans of recent onset T1D patients.25, 26, 27 CVB4 has been successfully isolated from pancreata of T1D patients.12 However, there is some controversy about those data since the isolated CVB4 strain was homologous to a strain that was used in the Dotta lab at that time.12 Interestingly, such pancreatic enterovirus isolates have been demonstrated to induce T1D in mice.28 Furthermore, it has been shown that such virus isolates are indeed able to infect and destroy human islet cells in vitro.29 However, according to a recent review by Tracy et al., there is no convincing evidence indicating that the CVB4 strain is more diabetogenic than the other serotypes (CVB1–6).19
Epidemiological evidence for viruses as protectors from T1D
Based on the epidemiological evidence mentioned above, one might be tempted to condemn viruses solely as promoters of autoimmune diseases, such as T1D. However, there is also epidemiological evidence that viruses or other pathogens might have a protective effect. Overall an inverse relation between the incidence of prototypical infectious diseases and the incidence of immune disorders has been observed for the years from 1950 to 2000 in the United States.30 Due to vaccinations and increased sanitary standards, infections with pathogens such as measles or mumps virus have been reduced to very low levels. Along these lines the ‘hygiene hypothesis' states that the human immune system that evolved in conjunction with parasites and other pathogens actually requires repeated encounters with various infectious threats in order to remain properly balanced or ‘tuned'. The absence of the majority of these pathogens in regions with high sanitary standards and broad availability and distribution of antibiotics might be one reason why the human immune system sometimes turns against its host (see reviews by Dunne and Cooke31 and Schubert32). Several migration studies for autoimmune diseases, such as T1D and multiple sclerosis (MS), support the hygiene hypothesis. For example, the risk for MS is much lower in equatorial countries with lower sanitary conditions and interestingly, the risk changes for individuals of similar ethic background who migrate from a low-risk to a high-risk region before the age of 15 years and vice versa.33 In contrast, individuals migrating from a high-risk to a low-risk region after the age of 15 years, maintain their high risk for MS. Thus, the surrounding environment seems to predetermine, whether an individual develops MS or is protected from disease later in life.33, 34 Similar to MS, the prevalence of T1D is much lower in equatorial countries compared to regions in northern Europe or northern America that have high sanitary standards but also high frequencies of T1D.35 Many epidemiological studies have also been conducted in Finland, which has one of the highest incidence rates of T1D in the world. The incidence of T1D has been steadily increasing over the last decades, whereas at the same time the number of enterovirus infections has been dropping.36 The EPIVIR project investigated the association of enterovirus infections and the incidence of T1D.37 It was found that in countries with the highest incidences of T1D, such as Finland or Sweden, the levels of antibodies to enterovirus antigens in the serum of infants were rather low. In contrast, T1D was less frequent in countries with relatively high enterovirus exposures (i.e., Estonia, Germany, Hungary, Lithuania and Russia).37 Further, a very interesting comparison between Finland and Russian Karelia, which both have a similar genetic background but different rates of enterovirus infection, revealed an inverse correlation between the presence of antibodies to enterovirus antigens and T1D incidence.38 Similar to the situation with MS, migration studies suggest that individuals do not retain their low risk odds to develop T1D after migrating to a high-risk location. Migrant Asian children aged 0–16 years moving from a low-risk to a high-risk location (in this case from Pakistan to the United Kingdom) showed a rising incidence of childhood diabetes, which was approaching that of the indigenous population.39 Thus, from an epidemiological point of view, viruses might have a dual role in the development of T1D, which would explain contradictory reports.
Besides the genetic susceptibility and sanitary standards resulting in divergence in exposure to infections, the exposure to sunlight might influence the prevalence of certain autoimmune diseases in different geographical regions. It is intriguing that the severity of MS and T1D are both fluctuating seasonally. Exacerbations occur more frequently in spring after the long winter period with low daily hours of sunlight exposure (for review, see Cantorna and Mahon40). Similarly, vitamin D levels are fluctuating seasonally, since adequate levels of vitamin D are generated via sunlight exposure of the skin. The vitamin D metabolite 1alpha,25-dihydroxy vitamin D3 promotes the differentiation of dendritic cells and the generation of regulatory T cells.40 Thus, high vitamin D levels might protect from autoimmunity by establishing a more regulatory milieu. Indeed, vitamin D3 treatment protects non-obese diabetic (NOD) mice from T1D41 most likely by activation of dendritic cell-induced apoptosis of autoaggressive T cells.41, 42 Alternatively or in addition, certain viral infections such as enteroviruses also exhibit seasonality, which might add to this phenomenon.43
How can viruses enhance or block T1D?
Many viruses, including entroviruses, cause a massive response by the immune system with the goal to quickly eliminate the viral threat to the host. On the one hand, viruses activate the innate immune system causing a strong inflammation at the site of infection and activate natural killer cells, macrophages and dendritic cells. On the other hand, the intracellular pathogens induce an adaptive immune response that is mostly dominated by cytotoxic CD8 T cells. Thus, viruses induce a very aggressive immune response that under certain circumstances might damage the host and subsequently cause autoimmunity. Several mechanisms by which the antiviral immune response might be detrimental for the host have been suggested. First, infection might directly infect and damage the target cell resulting in spontaneous (virus-induced) or an immune-mediated cell lysis. Target cell antigen presentation will be massively elevated including presentation of determinants of normally sequestered antigens that have not yet been seen by the immune system. Second, viruses might carry determinants with structural similarity to components of the host. Thereby, an immune response directed against the virus might in addition attack the similar structure in the host as well. This concept has been termed ‘molecular mimicry'.44, 45, 46, 47 Third, the strong inflammatory response that is caused in the infected tissue might generate an environment that allows further attraction and activation of aggressive immune cells that normally would not have migrated in critical number to the site of inflammation. Viruses might therefore generate a ‘fertile field' for subsequent autoimmune damage.48 It is important to note that the presence of cytokines and other inflammatory factors per se might impair β-cells function.49, 50
Experimental evidence for such mechanisms to be involved in the pathogenesis of T1D has derived from a multitude of animal models. The NOD mouse that spontaneously develops T1D, possibly due to an aberrant responsiveness to immune regulation51 is one of the most prominent models, since T1D slowly progresses and reflects several aspect of human T1D. Among enteroviruses the CVB replicates best in the mouse due to the close similarity between the murine and human coxsackie–adenovirus receptor,52 which is expressed Interestingly, different CVB strains have opposite effects on the outcome of T1D in the NOD mouse. Infection with CVB4 promotes T1D most likely by induction of bystander damage rather than molecular mimicry.53 The observed exacerbation of T1D requires both uptake and activation of antigen-presenting cells54 as well as presence of a pre-existing critical mass of autoreactive T cells within the islets of Langerhans.55 This later observation is intriguing since it demonstrates that CVB4 might rather accelerate an ongoing autodestructive process than de novo induce T1D. It is interesting to note that even infection of regular wild-type mice with CVB4 causes insulitis, low serum insulin levels and moderately elevated blood glucose values.56 In contrast to CVB4, the strain CVB3 seems to have a dual effect on T1D.19 Infection of young (4–6 weeks old) NOD mice with CVB3 failed to accelerate T1D, but provided a long-term (for at least 10 months of age) protection from disease.57 Interestingly, the replication properties and the dose of administration of CVB3 substrains critically influenced the outcome of T1D in the NOD mouse. However, administration of a low dose of the poorly virulent and slowly replicating strain CVB3/GA delayed T1D in prediabetic NOD mice, a higher dose accelerated T1D.58 Infection with a high dose of the rapid replicating strain CVB3/28 even induced disease within one week of infection. Besides the viral dose and the substrain, the viral tropism within the pancreas might also play a role. Whereas CVB4, which can directly infect β-cells, predominantly accelerates T1D, CVB3 infects the exocrine acinar cells of the pancreas and seems to have a dual effect on the pathogenesis of disease.54 Therefore, more in-depth analysis of the precise viral strain and the precise nature and magnitude of infectious events will need to be recorded in epidemiological studies in the future wherever possible.
Although most mechanistic insight derives from a large variety of animal models for T1D, several observation have been made in human individuals or by using isolated human islets of Langerhans. In particular the attention has been focused on the mechanism by which enteroviruses might be able to induce T1D. For example, it has been recently demonstrated that infection of human islets by CVB3 induces a strong inflammatory response resulting in the activation of dendritic cells (DCs). Interestingly, upon phagozytosis of infected islets the DCs induce the expression of interferon-stimulated genes, including the RIG-I-like helicases RIG-I and Mda5, and thus induce an antiviral state that protected the DCs from further infection.59 It is, however, not clear if such an antiviral state of DCs might induce T1D or rather protect from disease.
Further mechanistic insight has derived from the rat insulin promotor (RIP)-lymphocytic choriomeningitis virus (LCMV) mouse model for T1D. RIP-LCMV mice express the glycoprotein (GP) or the nucleoprotein (NP) of the LCMV under control of the RIP specifically in the β-cells.60 The model uses the concept of molecular mimicry to the extreme using a virus for infection that is identical to the target antigen in islets of Langerhans. Indeed, infection of RIP-LCMV mice with LCMV generates an immune response that results in the destruction of the β-cells bearing the identical (viral) target antigen, thus ensuing T1D within 10–14 days (RIP-LCMV-GP, fast-onset line) or several weeks to months (RIP-LCMV-NP, slow onset).61 LCMV is a natural occurring rodent virus that interestingly has been demonstrated to block the development of T1D in the NOD mouse.62 Similarly, secondary infection of RIP-LCMV-NP mice at a time when the autoimmune destruction of the β-cells was already ongoing (i.e., 4 weeks post-LCMV infection) with a LCMV strain that predominantly replicates outside of the pancreas abrogated the destructive process.5 It was found that the increased inflammation at the auxiliary site attracts aggressive CD8 T cells along a CXCL10 gradient and causes activation-induced cell death.5 Thus, virus infection at an auxiliary site might act as a filter for activated aggressive T cells.63 Another possible mechanism of how viruses might abrogate an ongoing autodestructive process is the induction of a regulatory immune response. Indeed, infection of prediabetic NOD mice with either LCMV or CVB3 reduced the frequency of T1D and delayed the onset of disease by increasing the number of CD4+CD25+ regulatory T cells that produced TGF-β and maintained long-term protection.6 In addition, a transient upregulation of the programmed cell death-1 ligand 1 on lymphoid cells prevented the expansion of diabetogenic CD8 T cells expressing programmed cell death-1.6
Judging from the few examples described above, the complexity of the system becomes already clear. Factors, such as viral strain, time of exposure, tropism and magnitude, influence the pathogenesis of T1D in the mouse model and possibly in human individuals as well. Considering that we all encounter several challenges through our lifetime adds to the complexity. Experiments with multiple sequential infections by heterologous viruses have been demonstrated to modulate the immune repertoire significantly.64 The history of immunological challenges might dramatically influence how the immune system is handling subsequent pathogen infections. Infection of naive RIP-LCMV-NP mouse Pichinde virus (PV), which shares a subdominant epitope of its nucleoprotein (PV-NP) with LCMV-NP, elicits only a marginal anti-NP CD8 T-cell response and does not cause T1D.65 In contrast, when RIP-LCMV-NP mice are infected with LCMV followed by PV T1D is massively accelerated.65 The mechanistic reason behind the observed acceleration was the expansion of aggressive CD8 T cells with reactivity to the subdominant PV/LCMV-NP epitope that confers molecular mimicry. The findings suggest that an experienced immune repertoire reacts differently on a subsequent viral challenge than a naive repertoire. In addition, the data suggest that molecular mimicry of a subdominant epitope might not be sufficient to induce autoimmunity, but might rather accelerate a preexisting autoimmune condition.
How can we tell the difference?
Considering the collected evidence from epidemiological studies and experimental animal models, it becomes more and more clear that not a single environmental factor is responsible for the development of T1D. It appears that multiple factors are involved in the detrimental activation of the immune system in a way that leads to the autoimmune destruction of the pancreatic β-cells and the subsequent clinical T1D. In Figure 1, we propose possible scenarios for the etiology of T1D integrating the genetic predisposition and protective and detrimental environmental factors. We postulate that the odds for T1D might be the highest when genetic predisposition is paired with several detrimental environmental triggering factors including viruses and direct β-cell toxins. In addition, an impaired immune regulation might exacerbate the disease. The Environmental Determinants of Diabetes in the Young (TEDDY) study66 is designed to collect as much information about such multiple triggering and/or protective factors. The TEDDY study keeps track of the diet, infections, allergies and other life experiences of neonates for up to an age of 15 years. After an initial screening for genetic risk factors more than 7000 children will be periodically screened for islet cell-specific autoantibodies. First data from the TEDDY study are expected to be released in a few years and will hopefully bring further insight in the etiology of T1D and/or help to identify protective factors. Unfortunately, the TEDDY study does not foresee sampling of stool and blood during infectious events, which might be a huge drawback when trying to precisely identify the infection agents.
Figure 1.
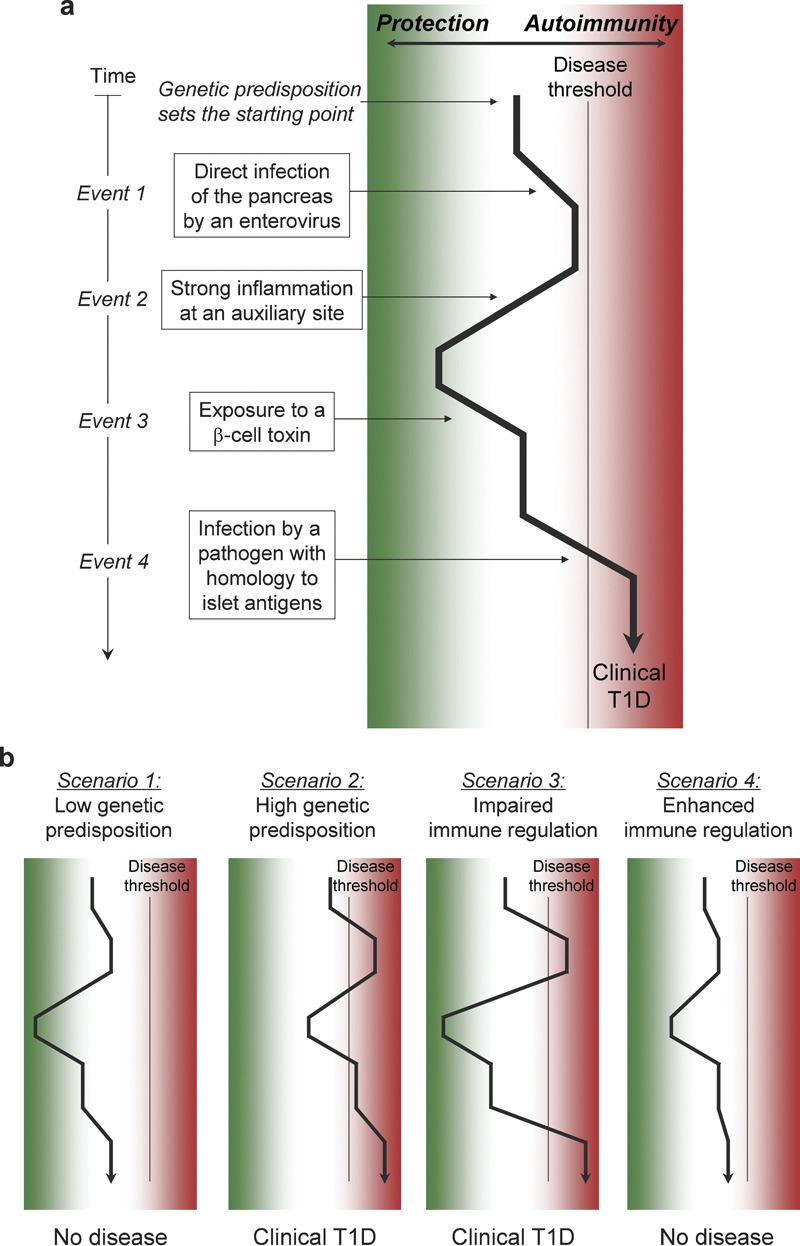
Sequential challenge by environmental factors. (a) Several events occurring throughout an individual's lifetime might influence the balance of the immune system towards autoimmune disease or protection. The genetic predisposition for disease might on the one hand determine a certain starting point and on the other hand influence the magnitude of the different immune reactions against environmental challenges. Several events including detrimental and protective virus infections, exposure to xenobiotica and molecular mimicry might be involved in the modulation of the immune repertoire and its balance. The accumulation of detrimental events combined with a sufficient genetic susceptibility might push the immune balance over a certain threshold for the development of clinical autoimmune disease. (b) Several scenarios might be feasible. First, a low genetic predisposition might be a reason for which the disease threshold is never reached. In contrast, a high predisposition might accelerate the autoimmune process. Further, an impaired immune regulation might enhance all events that involve an activation of the immune system, but might have no influence on the consequences of a β-cell toxin. Last, enhanced immune regulation by viral activation of regulatory T cells or by enhanced exposure to sunlight and elevated vitamin D levels might cause a general dampening of the immune response and the disease threshold might not be reached. T1D, type 1 diabetes.
An interesting, simple and unifying concept for how to distinguish T1D enhancing from protective enteroviral infections was put forward based on investigations by Tracy et al. (as discussed here previously), who observed that more severe infections enhanced diabetes in the NOD, whereas lower-replicating strains were protective.19 This implies that antiviral vaccines should transform deleterious more severe infections into protective ones and there is indeed an initiative in Finland to develop such a vaccine and test this concept. It should also be possible to test this concept in prospective studies such as TEDDY, but to achieve this it might have been necessary to also recover samples from children precisely at the time of febrile infections, which would likely encompass the more deleterious enterovirus encounters.
Acknowledgments
UC is supported by grants of the German Research Foundation. MGvH is supported by a scholar award of the Juvenile Diabetes Research Foundation and a Program Project Grant of the National Institute of Health to the La Jolla Institute for Allergy and Immunology.
References
- Todd JA. Etiology of type 1 diabetes. Immunity. 2010;32:457–467. doi: 10.1016/j.immuni.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity. 2010;32:468–478. doi: 10.1016/j.immuni.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo MJ, Rewers M, Yu L, Garg S, Pilcher CC, Elliott RB, et al. Genetic determination of islet cell autoimmunity in monozygotic twin, dizygotic twin, and non-twin siblings of patients with type 1 diabetes: prospective twin study. BMJ. 1999;318:698–702. doi: 10.1136/bmj.318.7185.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio E, Hummel M, Walter M, Schmid S, Ziegler AG. IDDM1 and multiple family history of type 1 diabetes combine to identify neonates at high risk for type 1 diabetes. Diabetes Care. 2004;27:2695–700. doi: 10.2337/diacare.27.11.2695. [DOI] [PubMed] [Google Scholar]
- Christen U, Benke D, Wolfe T, Rodrigo E, Rhode A, Hughes AC, et al. Cure of prediabetic mice by viral infections involves lymphocyte recruitment along an IP-10 gradient. J Clin Invest. 2004;113:74–84. doi: 10.1172/JCI200417005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi CM, Estes EA, Oldham JE, von Herrath MG. Immunoregulatory mechanisms triggered by viral infections protect from type 1 diabetes in mice. J Clin Invest. 2009;119:1515–1523. doi: 10.1172/JCI38503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers S, Kaufmann SH. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: lifestyle changes affecting the host-environment interface. Clin Exp Immunol. 2010;160:10–14. doi: 10.1111/j.1365-2249.2010.04120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi CM, von Herrath MG. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: viruses, autoimmunity and immunoregulation. Clin Exp Immunol. 2010;160:113–119. doi: 10.1111/j.1365-2249.2010.04128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatenoud L, You S, Okada H, Kuhn C, Michaud B, Bach JF. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: immune therapies of type 1 diabetes: new opportunities based on the hygiene hypothesis. Clin Exp Immunol. 2010;160:106–112. doi: 10.1111/j.1365-2249.2010.04125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble DR, Kinsley ML, FitzGerald MG, Bolton R, Taylor KW. Viral antibodies in diabetes mellitus. Br Med J. 1969;3:627–630. doi: 10.1136/bmj.3.5671.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hello H, Paananen A, Eskelinen M, Ylipaasto P, Hovi T, Salmela K, et al. An enterovirus strain isolated from diabetic child belongs to a genetic subcluster of echovirus 11, but is also neutralised with monotypic antisera to coxsackievirus A9. J Gen Virol. 2008;89:1949–1959. doi: 10.1099/vir.0.83474-0. [DOI] [PubMed] [Google Scholar]
- Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–5120. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JW, Austin M, Onodera T, Notkins AL. Virus-induced diabetes mellitus: isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- Honeyman MC, Coulson BS, Stone NL, Gellert SA, Goldwater PN, Steele CE, et al. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes. 2000;49:1319–1324. doi: 10.2337/diabetes.49.8.1319. [DOI] [PubMed] [Google Scholar]
- Hyoty H, Leinikki P, Reunanen A, Ilonen J, Surcel HM, Rilva A, et al. Mumps infections in the etiology of type 1 (insulin-dependent) diabetes. Diabetes Res. 1988;9:111–116. [PubMed] [Google Scholar]
- Gale EA. Congenital rubella: citation virus or viral cause of type 1 diabetes. Diabetologia. 2008;51:1559–1566. doi: 10.1007/s00125-008-1099-4. [DOI] [PubMed] [Google Scholar]
- Pak CY, Eun HM, McArthur RG, Yoon JW. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet. 1988;2:1–4. doi: 10.1016/s0140-6736(88)92941-8. [DOI] [PubMed] [Google Scholar]
- Jenson AB, Rosenberg HS, Notkins AL. Pancreatic islet-cell damage in children with fatal viral infections. Lancet. 1980;2:354–358. [PubMed] [Google Scholar]
- Tracy S, Drescher KM, Jackson JD, Kim K, Kono K. Enteroviruses, type 1 diabetes and hygiene: a complex relationship. Rev Med Virol. 2010;20:106–116. doi: 10.1002/rmv.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoletti L, Hober D, Hober-Vandenberghe C, Belaich S, Vantyghem MC, Lefebvre J, et al. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J Med Virol. 1997;52:121–127. doi: 10.1002/(sici)1096-9071(199706)52:2<121::aid-jmv1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Schulte BM, Bakkers J, Lanke KH, Melchers WJ, Westerlaken C, Allebes W, et al. Detection of enterovirus RNA in peripheral blood mononuclear cells of type 1 diabetic patients beyond the stage of acute infection. Viral Immunol. 2010;23:99–104. doi: 10.1089/vim.2009.0072. [DOI] [PubMed] [Google Scholar]
- Clements GB, Galbraith DN, Taylor KW. Coxsackie B virus infection and onset of childhood diabetes. Lancet. 1995;346:221–223. doi: 10.1016/s0140-6736(95)91270-3. [DOI] [PubMed] [Google Scholar]
- Lonnrot M, Salminen K, Knip M, Savola K, Kulmala P, Leinikki P, et al. Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: a prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J Med Virol. 2000;61:214–220. [PubMed] [Google Scholar]
- Stene LC, Oikarinen S, Hyöty H, Barriga KJ, Norris JM, Klingensmith G, et al. Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: The Diabetes and Autoimmunity Study in the Young (DAISY) Diabetes. 2010;59:3174–3180. doi: 10.2337/db10-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JW, Austin M, Onodera T, Notkins AL. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179. doi: 10.1056/NEJM197905243002102. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Hilton DA, Tauriainen S, Hyoty H, Bone AJ, et al. Use of antisera directed against dsRNA to detect viral infections in formalin-fixed paraffin-embedded tissue. J Clin Virol. 2010;49:180–185. doi: 10.1016/j.jcv.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Chatterjee NK, Nejman C, Gerling I. Purification and characterization of a strain of coxsackievirus B4 of human origin that induces diabetes in mice. J Med Virol. 1988;26:57–69. doi: 10.1002/jmv.1890260109. [DOI] [PubMed] [Google Scholar]
- Elshebani A, Olsson A, Westman J, Tuvemo T, Korsgren O, Frisk G. Effects on isolated human pancreatic islet cells after infection with strains of enterovirus isolated at clinical presentation of type 1 diabetes. Virus Res. 2007;124:193–203. doi: 10.1016/j.virusres.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- Dunne DW, Cooke A. A worm's eye view of the immune system: consequences for evolution of human autoimmune disease. Nat Rev Immunol. 2005;5:420–426. doi: 10.1038/nri1601. [DOI] [PubMed] [Google Scholar]
- Schubert C. News feature: the worm has turned. Nat Med. 2004;10:1271–1272. doi: 10.1038/nm1204-1271. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccone P, Fehervari Z, Phillips JM, Dunne DW, Cooke A. Parasitic worms and inflammatory diseases. Parasite Immunol. 2006;28:515–523. doi: 10.1111/j.1365-3024.2006.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, et al. Relationship between the incidence of type 1 diabetes and maternal enterovirus antibodies: time trends and geographical variation. Diabetologia. 2005;48:1280–1287. doi: 10.1007/s00125-005-1780-9. [DOI] [PubMed] [Google Scholar]
- Viskari H, Ludvigsson J, Uibo R, Salur L, Marciulionyte D, Hermann R, et al. Relationship between the incidence of type 1 diabetes and enterovirus infections in different European populations: results from the EPIVIR project. J Med Virol. 2004;72:610–617. doi: 10.1002/jmv.20033. [DOI] [PubMed] [Google Scholar]
- Seiskari T, Kondrashova A, Viskari H, Kaila M, Haapala AM, Aittoniemi J, et al. Allergic sensitization and microbial load—a comparison between Finland and Russian Karelia. Clin Exp Immunol. 2007;148:47–52. doi: 10.1111/j.1365-2249.2007.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodansky HJ, Staines A, Stephenson C, Haigh D, Cartwright R. Evidence for an environmental effect in the aetiology of insulin dependent diabetes in a transmigratory population. BMJ. 1992;304:1020–1022. doi: 10.1136/bmj.304.6833.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantorna MT, Mahon BD. Mounting evidence for vitamin D as an environmental factor affecting autoimmune disease prevalence. Exp Biol Med (Maywood) 2004;229:1136–1142. doi: 10.1177/153537020422901108. [DOI] [PubMed] [Google Scholar]
- Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. A 1alpha,25-dihydroxyvitamin D3 analog enhances regulatory T-cells and arrests autoimmune diabetes in NOD mice. Diabetes. 2002;51:1367–1374. doi: 10.2337/diabetes.51.5.1367. [DOI] [PubMed] [Google Scholar]
- Decallonne B, van Etten E, Overbergh L, Valckx D, Bouillon R, Mathieu C. 1Alpha,25-dihydroxyvitamin D3 restores thymocyte apoptosis sensitivity in non-obese diabetic (NOD) mice through dendritic cells. J Autoimmun. 2005;24:281–289. doi: 10.1016/j.jaut.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Fisman DN. Seasonality of infectious diseases. Annu Rev Public Health. 2007;28:127–143. doi: 10.1146/annurev.publhealth.28.021406.144128. [DOI] [PubMed] [Google Scholar]
- Christen U, Hintermann E, Holdener M, von Herrath MG. Viral triggers for autoimmunity: is the ‘glass of molecular mimicry' half full or half empty. J Autoimmun. 2010;34:38–44. doi: 10.1016/j.jaut.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen U, von Herrath MG. Induction, acceleration or prevention of autoimmunity by molecular mimicry. Mol Immunol. 2004;40:1113–1120. doi: 10.1016/j.molimm.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Damian RT. Molecular mimicry: antigen sharing by parasite and host and its consequences. Am Nat. 1964;98:129–149. [Google Scholar]
- Oldstone MB. Molecular mimicry as a mechanism for the cause and as a probe uncovering etiologic agent(s) of autoimmune disease. Curr Top Microbiol Immunol. 1989;145:127–136. doi: 10.1007/978-3-642-74594-2_11. [DOI] [PubMed] [Google Scholar]
- von Herrath MG, Fujinami RS, Whitton JL. Microorganisms and autoimmunity: making the barren field fertile. Nat Rev Microbiol. 2003;1:151–157. doi: 10.1038/nrmicro754. [DOI] [PubMed] [Google Scholar]
- Rhode A, Pauza ME, Barral AM, Rodrigo E, Oldstone MB, von Herrath MG, et al. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol. 2005;175:3516–3524. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- D'Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci USA. 2008;105:19857–19862. doi: 10.1073/pnas.0810713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimuth P, Philipson L, Carson SD. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol. 2008;323:67–87. doi: 10.1007/978-3-540-75546-3_4. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–785. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Ilic A, Fine C, Balasa B, Sarvetnick N. Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin Immunol. 2004;110:134–144. doi: 10.1016/j.clim.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Ottendorfer EW, Ellis TM, Gauntt CJ, Atkinson MA. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes. 2000;49:708–711. doi: 10.2337/diabetes.49.5.708. [DOI] [PubMed] [Google Scholar]
- Coleman TJ, Gamble DR, Taylor KW. Diabetes in mice after Coxsackie B4 virus infection. Br Med J. 1973;3:25–27. doi: 10.1136/bmj.3.5870.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracy S, Drescher KM, Chapman NM, Kim KS, Carson SD, Pirruccello S, et al. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J Virol. 2002;76:12097–12111. doi: 10.1128/JVI.76.23.12097-12111.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Kim K, Kono K, Drescher KM, Chapman NM, Tracy S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J Virol. 2006;80:5637–5643. doi: 10.1128/JVI.02361-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte BM, Kramer M, Ansems M, Lanke KH, van Doremalen N, Piganelli JD, et al. Phagocytosis of enterovirus-infected pancreatic beta-cells triggers innate immune responses in human dendritic cells. Diabetes. 2010;59:1182–1191. doi: 10.2337/db09-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: Role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- von Herrath MG, Dockter J, Oldstone MB. How virus induces a rapid or slow onset insulin-dependent diabetes mellitus in a transgenic model. Immunity. 1994;1:231–242. doi: 10.1016/1074-7613(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Oldstone MB. Prevention of type I diabetes in nonobese diabetic mice by virus infection. Science. 1988;239:500–502. doi: 10.1126/science.3277269. [DOI] [PubMed] [Google Scholar]
- Christen U, von Herrath MG. Infections and autoimmunity—good or bad. J Immunol. 2005;174:7481–7486. doi: 10.4049/jimmunol.174.12.7481. [DOI] [PubMed] [Google Scholar]
- Selin LK, Welsh RM. Plasticity of T cell memory responses to viruses. Immunity. 2004;20:5–16. doi: 10.1016/S1074-7613(03)00356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen U, Edelmann KH, McGavern DB, Wolfe T, Coon B, Teague MK, et al. A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest. 2004;114:1290–1298. doi: 10.1172/JCI22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homepage TEDDY Study Available from: http://teddy.epi.usf.edu/