

Abstract
MicroRNAs (miRNAs) are an abundant class of evolutionarily conserved, small, non-coding RNAs that post-transcriptionally regulate expression of their target genes. Emerging evidence indicates that miRNAs are important regulators that control the development, differentiation and function of different immune cells. Both CD4+CD25+Foxp3+ regulatory T (Treg) cells and invariant natural killer T (iNKT) cells are critical for immune homeostasis and play a pivotal role in the maintenance of self-tolerance and immunity. Here, we review the important roles of miRNAs in the development and function of iNKT and Treg cells.
Keywords: invariant natural killer T cells, microRNA, regulatory T cells
Introduction
MicroRNAs (miRNAs) were first discovered in Caenorhabditis elegans in 1993.1 They are evolutionarily conserved, small, single-stranded, non-coding RNAs that repress target genes at the post-transcriptional level by antisense binding to their target 3′-UTRs. Approximately one thousand miRNAs have been discovered in humans and mice, which constitutes about 3% of each genome.2, 3 It is estimated that about 60% of human protein-coding genes are under the regulation of miRNAs, which clearly indicates the powerful roles of miRNAs in gene regulation.4, 5 Indeed, accumulated studies suggest that miRNA-mediated RNA interference is emerging as an important regulatory pathway for various biological processes. In the past few years, through extensive use of microarray technology and high-throughput sequencing, investigators have assessed global miRNA expression in the hematopoietic system and found cell lineage-specific changes in miRNA expression during the differentiation of hematopoietic stem cells.6, 7, 8 More recent functional studies using gain- and loss-of-function approaches have uncovered important roles for miRNAs in the development, homeostasis and function of a variety of immune cell types. Both CD4+CD25+Foxp3+ regulatory T (Treg) cells and invariant natural killer T (iNKT) cells are critical for immune homeostasis and play a pivotal role in the maintenance of self-tolerance and immunity. This review covers the recent findings of miRNA-mediated regulation in Treg and iNKT cell development and function.
Biogenesis of miRNAs
Most miRNAs reside in intergenic regions and use their own gene promoter for expression.9, 10 Approximately 40% of miRNAs lie in introns of protein and non-protein coding regions.11 miRNAs are transcribed by RNA polymerase II, resulting in a primary miRNA (pri-miRNA).10 The double-stranded RNA hairpin structure in a pri-miRNA is bound by DiGeorge Syndrome Critical Region 8 (DGCR8). Forming a complex with the RNase III enzyme Drosha, DGCR8 orients the catalytic domain of Drosha that releases hairpins from pri-miRNAs, resulting in pre-miRNAs.12 Pre-miRNAs are then actively exported out of the nucleus by Exportin-5, which recognizes the 3′ end of the pre-miRNA hairpin processed by Drosha.13 As described in Figure 1, the exported pre-miRNA hairpin is further processed by RNase III enzyme Dicer into a ∼21-nucleotide duplex, one strand of which incorporates into the RNA-induced silencing complex (RISC).14 Once incorporated into a RISC, the miRNA is situated to regulate the target genes by degradation of mRNA through direct cleavage or by inhibition of protein translation. During miRNA maturation in the cytoplasm, the Argonaute protein, the critical component of RISC, is thought to stabilize the guide strand, which is important for miRNA function.15 The direct cleavage of the mRNA will cause a reduction of the target mRNA level, whereas the inhibition of protein translation does not change the mRNA level. DGCR8-deleted embryonic stem cells, which lack all canonical mature miRNAs, proliferate profoundly slower than DGCR8-competent embryonic stem cells and accumulate in the G1 phase of the cell cycle.16, 17 The loss of Dicer leads to embryonic lethality in mouse development.18, 19 These studies suggest the critical role of miRNAs in development. The discovery of miRNAs as important regulators of development in model organisms suggested the potential role of miRNAs in the immune system.
Figure 1.

MicroRNA (miRNA) biogenesis. RNA polymerase II transcribes miRNA genes, generating long primary transcripts (pri-miRNAs). In the nucleus, the RNase III-type enzyme Drosha processes the long primary transcripts (pri-miRNA), yielding hairpin precursors (pre-miRNA). Exportin 5 transports the pre-miRNAs from the nucleus into the cytoplasm. The pre-miRNA hairpins are further processed by Dicer into mature miRNAs, which are incorporated into the Argonaute (Ago) protein and form the RNA-induced silencing complex (RISC) together with Dicer. Once incorporated into the RISC, the miRNAs then guide the RISC to the target genes and repress target gene expression by destabilizing the target mRNAs or suppressing protein translation.
microRNAs in Treg cell development and function
The Dicer/Drosha knockout indicates roles for miRNAs in Treg cells
CD4+CD25+Foxp3+ Treg cells arise naturally in the thymus during T-cell differentiation, and the mechanisms that govern Treg cell differentiation and function are still not completely understood. To gain insight into the role of miRNAs in the biology of Treg cells, Cobb et al.20 first analyzed miRNA expression profiles in Treg cells using miRNA microarray analyses. They found that natural Treg cells have an miRNA expression profile similar to that of activated conventional CD4 T cells, which is distinct from that of naive CD4 T cells, suggesting that Treg cells represent a state of partial activation.20 Further studies showed that the ectopic expression of the Treg cell signature transcription factor Foxp3 can confer a partial Treg cell miRNA profile, indicating the direct or indirect role of Foxp3 in controlling the miRNA signature in Treg cells. Given that Dicer is required for functional miRNA processing, several laboratories, including our own, developed mouse models with a conditional deletion of Dicer in the bone marrow and thymus to analyze the role of miRNAs in the development and function of Treg cells. Using a CD4-Cre-mediated Dicer deletion mouse model, Cobb et al.20 first reported that depleting miRNAs in the thymus reduced Treg cell numbers in thymus, spleen and lymph nodes, with normal conventional CD4 and CD8 T-cell development in the thymus. Our laboratory21, 22 further confirmed these findings in mouse models with Dicer deletion in the bone marrow or thymus (Figure 2). Using thymus organ culture and mixed thymus chimeras, subsequent studies20 further indicated the cell-intrinsic fashion of Dicer-dependent Treg cell development in the thymus.
Figure 2.
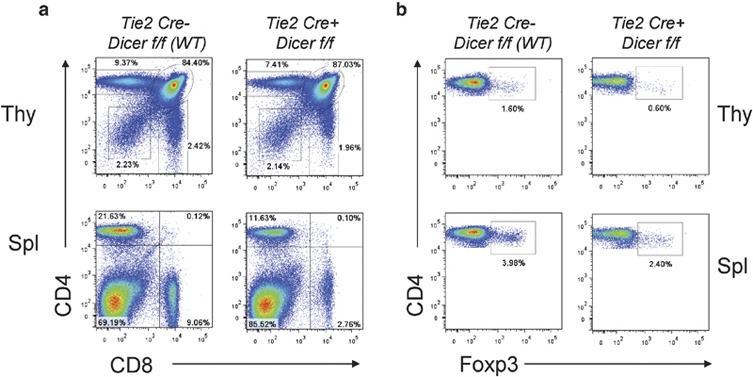
Reduced number of regulatory T (Treg) cells in the absence of microRNAs (miRNAs) in the bone marrow. (a) Flow cytometric analysis of T-cell subsets in the thymus and spleen. (b) Foxp3+ Treg cells were analyzed on gated CD4+ T cells in 6- to 8-week-old mice. The percentage of Foxp3+ Treg cells was significantly reduced in both the thymus and spleen of the Tie2-Cre+.Dicerf/f mouse compared to the thymus and spleen of the Tie2-Cre−.Dicerf/f wild-type (WT) control.
To further identify the specific role of miRNAs in Treg cell development and function, three individual groups identified, almost simultaneously, the critical role of miRNAs in Treg function using mouse models with a Foxp3-Cre-mediated Dicer or Drosha deletion in the Treg cell lineage.23, 24, 25 All three Treg-specific miRNA deletion models show fatal early-onset lymphoproliferative syndrome, phenocopying the mice lacking a functional Foxp3 gene. Rudensky's group23 found that Dicer-deficient Treg cells show impaired peripheral homoeostasis, and Dicer-deficient Treg cells from healthy mice are anergic and functional, although markedly less suppression efficient on a per cell basis compared to the Treg cells from wild-type mice. However, the Dicer-deficient Foxp3+ T cells purified from littermates with inflammation were completely devoid of suppressor activity and show a robust in vitro proliferative response. Interestingly, Dicer-deficient Foxp3+ Treg cells show comparable Foxp3 expression compared to Treg cells from wild-type mice, suggesting that the loss of suppressive capacity was not caused by changes in Foxp3 itself, but rather by low expression of suppressor effector molecules, including cytotoxic T-lymphocyte antigen 4, inducible T-cell costimulator, IL-10, EBV- induced gene3 (Ebi-3) and granzyme B in Dicer-deficient Foxp3+ Treg cells. Bluestone's group24 reported that mice with a Dicer deletion at the time of Foxp3 expression have interrupted Treg cell lineage stability based on their downregulated Foxp3 expression, which positively correlates with mouse age, even though these mice show normal thymic Treg cell development at 2 weeks of age; most importantly, Dicer-deficient Treg cells lose suppressive activity in vivo. Moreover, their microarray studies showed that the majority of the altered genes and proteins in Treg cells from Foxp3-Cre-Dicer knockout mice are consistent with the Rudensky group's report. Consistent with the above findings, Littman's group25 found that the early lymphoproliferative phenotype observed in Treg-specific Drosha-deletion mice was not caused by a loss of Treg cells, but rather by a loss of their suppression functions because Foxp3 Treg cells could still be detected in these mice. The fact that Treg-specific miRNA depletion leads to a much more aggressive autoimmune phenotype than in bone marrow (Tie2)26- or thymus (CD4-Cre)20-specific miRNA deletion indicates roles for miRNAs in regulating conventional T-cell activation, migration and function, which have been evidenced by other studies.27, 28 Overall, the findings from different developmental stage-specific Dicer/Drosha deletion mouse models confirm the critical roles of miRNA-dependent regulation in Treg cell development and function, and in preventing spontaneous inflammation and autoimmunity.
miR-155 regulates Treg cell development
The defect in Treg cell homeostasis and function in mice lacking all miRNAs and the differential expressions of miRNAs in the Treg versus effector T cells raised the question as to how individual miRNA plays distinct roles in Treg cell homeostasis and function. Foxp3 is an X-chromosome-encoded transcription factor that is essential for the development, homeostasis and function of Treg cells.29, 30 Genome-wide analysis of Foxp3-binding genes revealed that Foxp3 directly or indirectly regulates several thousands of genes in Treg cells.30, 31 As the first miRNA candidate for investigation, miR-155 expression is directly regulated by Foxp3,31, 32, 33 indicating the potential involvement of miR-155 in regulating Treg cell differentiation, maintenance or function. However, conventional T cells lacking Foxp3 and B cells also transiently upregulate miR-155 following activation in humans and mice,20, 34 suggesting that Foxp3 is not the sole factor that regulates and maintains the expression of miR-155. By using miR-155 knockout (KO) mice, Lu et al. reported that a loss of miR-155 did not markedly impair Treg cell suppressor function. However, the deficiency of miR-155 in Treg cells reduced Treg cell numbers and negatively impacted Treg cell proliferative potential in a cell-autonomous fashion, with increased expression of suppression of cytokine signaling 1 (SOCS1), decreased activation of signal transduction and activation of transcription 5 (STAT5).33 This finding is consistent with the result of defective IL-2 production or IL-2 receptor expression showing interrupted Treg cell peripheral survival and the loss of self-tolerance.30 These studies indicated that Foxp3-driven miR-155 expression plays a critical role in the maintenance of Treg cells by targeting SOCS1, leading to increased sensitivity of IL-2R.33 Kohlhaas et al. further reported that miR-155-deficient mice show a reduced number of Treg cells, both in the thymus and periphery, due to the defect in development. However, miR-155-deficient Treg cells show normal in vitro suppression activity and can prevent colitis induced by the adoptive transfer of CD4+CD45RBhigh T cells into lymphopenic hosts.35 Thus, the current data indicate that miR-155 contributes to Treg cell development and homeostasis, but Treg cell function could be controlled by additional miRNAs.
miR-146a regulates Treg cell function
Similar to the expression pattern of miR-155, Treg cells and activated conventional T cells exhibit augmented miR-146a expression.20 miR-146a KO mice develop severe lympho- and myeloproliferative syndrome at 6 months of age.36 In contrast to the reduced number of Treg cells in mice lacking miR-155,33 miR-146a-deficient mice contained a significantly increased number of Foxp3+ Treg cells in the periphery, combined with heightened proliferative activity and a modest increase in activation markers.37 As miR-146a is an important negative regulator of myeloid cell responses to cytokines and Toll-like receptor ligands, the increased miR-146a-deficient Treg cell number could potentially come from the heightened activation status of dendritic cells.37 However, mixed bone marrow chimera experiments confirmed the cell intrinsic property of the augmented Treg cell numbers in miR-146a-deficient mice and demonstrated a dramatic functional defect of miR-146-deficient Treg cells similar to that observed in Dicer-deficient Treg cells, suggesting an indispensable role for miR-146 in Treg cell-mediated immunological tolerance. The loss of miR-146a results in increased production of the proinflammatory Th1 cytokine Interferon-gamma (IFN-γ) by both miR-146a-deficient conventional T cells and Foxp3+ Treg cells, and purified miR-146a-deficient Treg cells adoptively transferred together with Foxp3 KO CD4+ effector T cells into lymphopenic recipients failed to restrain Th1 responses.37 Furthermore, it is known that an IFN-γ blockage prevents autoimmune disease in mice with miR-146a-deficient Treg cells.37 Thus, in addition to Foxp3, miR-146a plays a role in preventing the acquisition of Th1-like properties and restraining the production of the proinflammatory cytokine IFN-γ by Treg cells.
miR-146a was identified as a negative feedback regulator of myeloid cell responses to cytokines and Toll-like receptor ligands in Toll-like receptor signaling by targeting IL-1R-associated kinase 1, IL-1R-associated kinase 2 and TNF receptor-associated factor 6 during inflammation.38, 39 Given that signal transducer and activator transcription 1 (STAT1) is a key transcription factor downstream of IFN-γ receptor signaling, it is possible that the increased Stat1 expression in miR-146a-deficient Treg cells could account for the IFN-γ-/Th1-dependent autoimmunity in miR-146a KO mice, as suggested by recent human-related studies, even though computational algorithms relying on miRNA seed sequence analysis failed to identify mouse STAT1 as an miR-146a putative target.37, 40 Luciferase reporter assays and high-throughput sequencing of RNA isolated by a crosslinking immunoprecipitation approach confirmed the partial complementary miR-146 binding site in the mouse STAT1 gene.37 The target role of STAT1 in miR-146-mediated Treg cell regulation further supported by two independent confirmations using different animal models in which STAT1 hemizygosity significantly reduce the severity of the disease observed in mice harboring miR-146-deficient Treg cells, while Treg-specific SOCS1 KO/Foxp3 KO chimeras develop IFN-γ-mediated Th1 immunopathology similar to that observed in the presence of miR-146 KO Treg cells.37 These results suggest that IFN-γ-dependent Th1-mediated pathogenesis is under the control of miR-146a through the promotion of Treg cell-mediated Th1 regulation and by limiting Th1 responses (Figure 3). Overall, miR-146a expression in Treg cells is crucial for Treg-mediated control of Th1 responses, in a significant part, through targeting STAT1.
Figure 3.
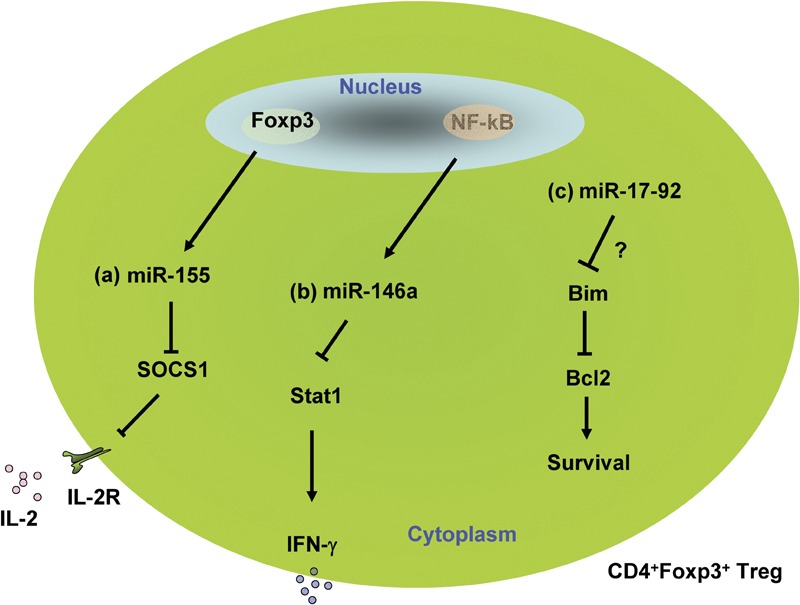
Regulation of regulatory T (Treg) cells by microRNAs (miRNAs). (a) Foxp3 directly regulates the expression of miR-155, which negatively regulates expression of suppressor of cytokine signaling 1 (SOCS1); SOCS1 renders Treg cells resistant to IL-2 responsiveness, controlling Treg cell survival. (b) miR-146a expression in Treg cells is crucial for preventing the acquisition of Th1-like properties and restraining the production of the proinflammatory cytokine interferon-gamma(IFN-γ) through the targeting of Stat1. (c) miR-17-92 may regulate Treg cells through targeting Bim.
miR-17-92 cluster regulate Treg cell function
The miR-17-92 transcript encoded by mouse chromosome 14 (and human chromosome 13) is the precursor for 7 mature miRNAs (miR-17-5p, miR-17-3p, miR-18a, miR-19a, miR-20a, miR-19b and miR-92). miR-17-92 is expressed by many human hematopoietic cell neoplasms. T or B cell-specific overexpression of miR-17-92 leads to increased proliferation and survival of B cells and T cells, thereby causing lymphoproliferative disease and autoimmunity in mice. B and T cells in miR-17-92 transgenic mice exhibit enhanced proliferation and reduced cell death associated with down-regulation of Bim and PTEN.41 However, the role of Treg cells in the miR-17-92 overexpression-induced lymphoproliferative phenotype is still unknown. More recently, preliminary studies reported at the Immunology 2011 Annual Meeting by Bluestone's group42 suggest that the miR-17-92 cluster was dispensable for Treg cell numbers and proper functioning under homeostatic conditions, even in ageing mice. However, mice with a Treg-specific miR-17-92 deletion develop much more severe experimental autoimmune encephalitis than control mice, and miR-17-92-deficient Treg numbers are significantly reduced in the spinal cord, while antigen-specific effector T cells are present in normal numbers, indicating that the miR-17-92 cluster may be essential for Treg cell survival and function under inflammatory conditions.42 The related mechanisms underlying miR-17-92-mediated Treg cell functional regulation are currently under investigation. Mice lacking IL-2 have more than a twofold reduction in the percentage of Foxp3+ CD4 T cells in the peripheral lymphoid organs. To address whether ablating the Bim-related death pathway would rescue Foxp3+ cells, Barron et al.43 crossed IL-2−/− mice with Bim−/− mice. In double-knockout mice lacking IL-2 and Bim, there was a significant increase in the proportion of Foxp3+ Treg cells. Thus, it could be expected that miR-17-92 may regulate Treg cell numbers through targeting Bim.
REGULATION of iNKT CELLS BY miRNAs
iNKT cells are another subset of regulatory T cells in mice and humans. iNKT cells possess the properties of both T cells and natural killer cells, as they co-express a rearranged T-cell receptor Vα14-Jα18 (in humans, Vα24-Jα18) in combination with a limited set of Vβ chains and several NK cell receptors. Upon antigenic stimulation, iNKT cells rapidly and robustly produce a broad range of cytokines, which allow them to modulate the function of variety of other immune cells, including dendritic cells, Th17 and Th1 T cells, and to regulate both innate and adaptive immune responses.44, 45, 46, 47 In addition, iNKT cells can secrete perforin, granzymes and FasL when activated and mediate protective immune functions, including tumor rejection and protection against infectious microbes.48, 49, 50 Defective iNKT development and/or inappropriate iNKT activation and function are associated with a broad spectrum of diseases, ranging from autoimmunity, cancer, infection and allergies to allograft rejection.51, 52, 53, 54 iNKT cell-based immunotherapy has been applied to clinical trials for the treatment of immunization-associated diseases and cancer.55, 56
iNKT cells develop in the thymus. After being positively selected by CD1d expressed on CD4+CD8+ double-positive thymocytes, iNKT cells undergo a development and maturation process defined by the sequential downregulation of CD24 and upregulation of CD44 and NK1.1.46, 57, 58 The first stage of iNKT cells (stage 0) are defined as CD24+CD44− and NK1.1−, followed by the CD24−CD44− and NK1.1− (stage 1). After stage 1, iNKT cells acquire an activated or memory phenotype marked by increased CD44 expression (stage 2). A final maturation step that occurs either in the thymus or in the peripheral organs is accompanied by the expression of NK1.1 and other NK cell receptors (stage 3) (Figure 4a).46, 59 Both immature and mature iNKT cells release cytokines when stimulated with either glycolipid antigens, such as α-GalCer, or non-specific antigen, phorbol myristate acetate and ionomycin. However, during the process of maturation, the production of IFN-γ by iNKT cells gradually increases, while IL-4 production decreases, demonstrating a cytokine profile switch from Th2 to Th1 (Figure 4b).46 The development of iNKT cells is controlled by multiple transcription factors, cytokines and signaling molecules involved in the SLAM/Fyn/SAP/PKC pathways; deletion of any one of these molecules or factors impairs iNKT cell development and function, indicating that multiple regulatory mechanisms concomitantly control the development of iNKT cells.52, 60 However, the detailed mechanisms and related networks that drive iNKT development and maturation are still not completely understood.
Figure 4.
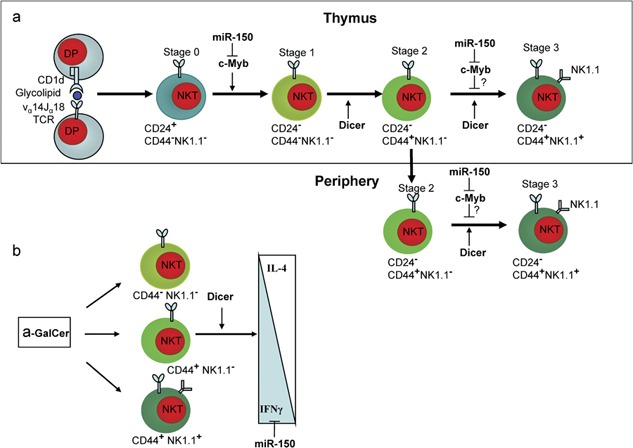
microRNAs (miRNAs) regulate invariant natural killer T (iNKT) cell development and function. (a) Dicer and miR-150 regulate iNKT cell development and maturation. miR-150 may differentially regulate NKT cell early-stage proliferation and late-stage maturation. (b) Dicer and miR-150 regulate iNKT cell activation and cytokine production.
The discovery of miRNAs and their essential role in immune system development opens a new research avenue for iNKT cell-related studies. By generating hematopoietic-specific Dicer deletion mouse models, we have first uncovered the important role of miRNAs in iNKT cell development and function.26 Tie2 kinase is specifically expressed by hematopoietic progenitors and endothelial cells. The Tie2-Cre-mediated Dicer deletion resulted in significantly reduced miRNA gene expression in the hematopoietic lineages. Consistent with previous reports in CD4-Cre- or Lck-Cre-mediated thymic deletion of Dicer,20 we found that the Dicer deletion did not dramatically affect thymic conventional CD4+ and CD8+ T-cell development, but influenced Treg cells. Interestingly, a substantial reduction in iNKT cell number was observed in both the thymus and peripheral lymphoid organs from Dicer knockout mice, indicating the special critical dependence of iNKT cells on Dicer-dependent miRNAs.26 The maturation of Dicer-deficient iNKT cells was largely blocked at stage 2 of iNKT development, suggesting that miRNAs are required for the later stage transition of iNKT development (Figure 4). Bone marrow transfer experiments further demonstrated that the defective iNKT development was stem cell-intrinsic. Given that Tie2 is also expressed in endothelial cells, we transferred thymic NKT cells from wild-type mice to Dicer KO and wild-type recipients, and we found that the lack of miRNAs in endothelial cells affect iNKT cell homeostasis. In addition to the developmental defects, significantly interrupted iNKT cell activation and elevated IL-4 and IFN-γ production were observed in either the α-GalCer- or phorbol myristate acetate/ionomycin-stimulated iNKT cells from Dicer KO mice suggesting the involvement of miRNAs in iNKT functional regulation (Figure 4b). Therefore, the developmental and functional defect of iNKT cells observed in Tie2-cre-Dicer KO mice may result from both intrinsic (hematopoietic cells) and extrinsic (endothelial cells) defects.26
Soon after our report was published, Fedeli et al. further confirmed the critical role of miRNAs in iNKT cell development using CD4-Cre- and Lck-Cre-derived thymus-specific Dicer KO mice.61 Meanwhile, defective iNKT cell homeostasis was identified in thymus-specific Dicer deletion mice, manifested by increased cell death, especially iNKT cells of developmental stage 2, suggesting a possible link between cell division and death of iNKT cells in the absence of miRNAs.61 In agreement with these results on defective iNKT cell development and homeostasis, our studies using the similar thymus-specific deletion mouse models further confirmed the profound functional defect of Dicer-deleted iNKT cells.22 Moreover, the miRNA expression profile specific for iNKT cells has been identified, and it exhibits features of activated/effector T lymphocytes.61 These original studies demonstrated the critical roles of Dicer-dependent miRNA pathways in iNKT cell development and function, and the necessity of identifying the specific miRNAs contributing to the iNKT cell defects observed in Dicer KO mouse models.
Although a central role for the Dicer-dependent miRNA pathway in various aspects of iNKT cell biology was clearly demonstrated by these genetic studies, an understanding of the role of individual miRNAs in this context is lacking. Using miRNA arrays to analyze the miRNA expression profiles from different developmental stages of iNKT cells, we have found more than 50 miRNAs whose expression are differentially regulated during the development of iNKT cells and after activation. MicroRNA miR-223 was first identified bioinformatically and was found to be expressed in the hematopoietic system, especially in the myeloid compartment.62 Recent studies from Camargo's group showed that miR-223 is highly expressed in neutrophils and macrophages and negatively regulates the proliferation and differentiation of neutrophils by downregulating the transcription factor Mef2c.63 Interestingly, we found that miR-223 is also highly expressed in immature iNKT cells compared to mature iNKT cells, and miR-223 is upregulated after iNKT cell activation.64 Given that several potential miR-223 target genes, including SLAM, IKK2, ETS1 and ROR-γt, are involved in iNKT cell development, the hypothesis was raised that miR-223 might be involved in iNKT cell development. Using a miR-223-deficient mouse model, we evaluated the role of miRNA-223 in normal iNKT cell development, activation and function. Our results show that the lack of miR-223 does not dramatically interrupt iNKT cell development and function, indicating that the expression of miR-223 in the early stage of iNKT cells may not be critical for normal iNKT cell development and their function. Our data may also suggest the potential redundancy of different miRNAs in iNKT cell developmental regulation.64 As miR-223 was also dramatically upregulated in activated iNKT cells, the potential roles of miR-223 in activation-induced iNKT cell population expansion, contraction and anergy induction are under investigation.
Compared to miR-223, miR-150 is rarely expressed in immature iNKT cells, but is upregulated in mature iNKT cells. Lanier's group65 and our group66 both reported at the 2011 AAI meeting that miR-150 regulates NK and NKT cell development. Using conventional miR-150 KO mice, we found that deletion of miR-150 partially blocks both thymic and peripheral iNKT cell maturation. Moreover, unlike Dicer-deficient iNKT cells, miR-150-deficient iNKT cells have increased IFN-γ production after stimulation with α-GalCer, but not with phorbol myristate acetate/ionomycin stimulation, indicating that miR-150 may negatively regulate iNKT function mainly through suppressing targets upstream of the TCR signaling pathway (unpublished data). Notably, Lanier's group reported that overexpression of miR-150 dramatically reduced the iNKT cell number and blocked NKT cell development in the early stage of iNKT cell development, further indicating the importance of the dynamic regulation of miR-150 levels in the development of iNKT cells. The transcription factor c-Myb has been shown to be a direct target of miR-150.67 The c-Myb gene is abundantly expressed in immature immune cells, which is in mirror opposite of miR-150 expression levels in iNKT cells. It has been recently reported that c-Myb is absolutely required for early iNKT cell development, and c-Myb has a central role in priming double-positive thymocytes to enter the iNKT lineage by simultaneously regulating CD1d expression, the half-life of double-positive cells and the expression of SLAMF1, SLAMF6 and SAP.68 Interestingly, previous studies reported that the ectopic expression of c-Myb blocked the induced differentiation of hematopoietic cell lines. Furthermore, discrete threshold levels of c-Myb activity appear to be required for differentiation along individual hematopoietic cell lineages.69 Given the opposite expression levels of miR-150 and c-Myb in iNKT cells during their development and the critical roles of miR-150 and c-Myb in iNKT cell development and maturation, it is likely that the dynamic expression of miR-150 is required for normal iNKT cell development by controlling the levels of c-Myb expression. However, more substantial evidence is needed to confirm the target role of c-Myb in miR-150-mediated iNKT cell regulation.
Concluding remarks
A central role for the Drosha/Dicer-dependent miRNA pathway in various aspects of Treg and NKT cell biology has been clearly demonstrated using genetic studies. Recent progress in understanding the immunological function of miRNAs using mouse models with loss- or gain-of-function of individual miRNAs has revealed that miRNAs act as ‘fine-tuners' for the development and functioning of Treg and iNKT cells. However, many gaps remain in our knowledge about miRNA-mediated gene regulation for Treg and iNKT cells under normal and autoimmune/inflammatory conditions. Extra caution should be taken when using in vitro systems to confirm miRNA phenotypes, including their targets, since function and targets of miRNAs in vitro might be different from what is happening in vivo. Given the many potential target mRNAs for each miRNAs, a single miRNA may not be a key player in most cases; multiple miRNAs with common or coordinated target mRNAs are likely to have major roles in gene regulation and may control the specific pathways for cell development and function. Thus, it will be important to identify a set of miRNAs acting cooperatively for Treg and iNKT cell development and function, which may be particularly relevant for the development of miRNA-based immunotherapy. Although the most important findings in this fascinating field are still to come, it is obvious that the miRNA field will affect our future and has more promise than previously expected.
Acknowledgments
This work was support in part by grants from the Juvenile Diabetes Research Foundation International and the Henry Ford Immunology Program startup.
References
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Engels BM, Hutvagner G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene. 2006;25:6163–6169. doi: 10.1038/sj.onc.1209909. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Lodish HF. MicroRNAs as regulators of mammalian hematopoiesis. Semin Immunol. 2005;17:155–65. doi: 10.1016/j.smim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Garzon R, Pichiorri F, Palumbo T, Iuliano R, Cimmino A, Aqeilan R, et al. MicroRNA fingerprints during human megakaryocytopoiesis. Proc Natl Acad Sci USA. 2006;103:5078–5083. doi: 10.1073/pnas.0600587103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan M, Miller CP, Papayannopoulou T, Stamatoyannopoulos G, Song CZ. MicroRNA expression dynamics during murine and human erythroid differentiation. Exp Hematol. 2007;35:1015–1025. doi: 10.1016/j.exphem.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Chendrimada TP, Shiekhattar R. MicroRNA biogenesis: isolation and characterization of the microprocessor complex. Methods Mol Biol. 2006;342:33–47. doi: 10.1385/1-59745-123-1:33. [DOI] [PubMed] [Google Scholar]
- Murchison EP, Hannon GJ. miRNAs on the move: miRNA biogenesis and the RNAi machinery. Curr Opin Cell Biol. 2004;16:223–229. doi: 10.1016/j.ceb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Lund E, Dahlberg JE. Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb Symp Quant Biol. 2006;71:59–66. doi: 10.1101/sqb.2006.71.050. [DOI] [PubMed] [Google Scholar]
- Kai ZS, Pasquinelli AE. MicroRNA assassins: factors that regulate the disappearance of miRNAs. Nat Struct Mol Biol. 2010;17:5–10. doi: 10.1038/nsmb.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1–S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–5. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BS, Hertweck A, Smith J, O'Connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med. 2006;203:2519–2527. doi: 10.1084/jem.20061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Seo KH, He HZ, Pacholczyk R, Meng DM, Li CG, et al. Tie2cre-induced inactivation of the miRNA-processing enzyme Dicer disrupts invariant NKT cell development. Proc Natl Acad Sci USA. 2009;106:10266–10271. doi: 10.1073/pnas.0811119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo KH, Zhou L, Meng D, Xu J, Dong Z, Mi QS. Loss of microRNAs in thymus perturbs invariant NKT cell development and function. Cell Mol Immunol. 2010;7:447–453. doi: 10.1038/cmi.2010.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston A, Lu LF, O'Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205:1993–2004. doi: 10.1084/jem.20081062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, et al. Selective miRNA disruption in Treg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205:1983–1991. doi: 10.1084/jem.20080707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med. 2008;205:2005–2017. doi: 10.1084/jem.20081219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Seo KH, He HZ, Pacholczyk R, Meng DM, Li CG, et al. Tie2cre-induced inactivation of the miRNA-processing enzyme Dicer disrupts invariant NKT cell development. Proc Natl Acad Sci USA. 2009;106:10266–10271. doi: 10.1073/pnas.0811119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kondo E, Takeuchi M, Harashima A, Otani T, Tsuji-Takayama K, et al. miR-155, a modulator of FOXO3a protein expression, is underexpressed and cannot be upregulated by stimulation of HOZOT, a line of multifunctional Treg. PLoS One. 2011;6:e16841. doi: 10.1371/journal.pone.0016841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150–2158. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–1075. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeker LT, Kouchkovsky JE, Bluestone JA.The miR-17-92 cluster is essential for regulatory T cell function in vivo 2011186168.14. [Google Scholar]
- Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O'Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol. 2010;185:6426–6430. doi: 10.4049/jimmunol.0903940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- Morita CT, Verma S, Aparicio P, Martinez C, Spits H, Brenner MB. Functionally distinct subsets of human gamma/delta T cells. Eur J Immunol. 1991;21:2999–3007. doi: 10.1002/eji.1830211215. [DOI] [PubMed] [Google Scholar]
- Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science. 2002;296:553–555. doi: 10.1126/science.1069017. [DOI] [PubMed] [Google Scholar]
- Wallace KL, Marshall MA, Ramos SI, Lannigan JA, Field JJ, Strieter RM, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van KL. NKT cells: T lymphocytes with innate effector functions. Curr Opin Immunol. 2007;19:354–364. doi: 10.1016/j.coi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Kawamura T, Takeda K, Kaneda H, Matsumoto H, Hayakawa Y, Raulet DH, et al. NKG2A inhibits invariant NKT cell activation in hepatic injury. J Immunol. 2009;182:250–258. doi: 10.4049/jimmunol.182.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Hayakawa Y, van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci USA. 2000;97:5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol. 2011;11:131–142. doi: 10.1038/nri2904. [DOI] [PubMed] [Google Scholar]
- Godfrey DI, Berzins SP. Control points in NKT-cell development. Nat Rev Immunol. 2007;7:505–518. doi: 10.1038/nri2116. [DOI] [PubMed] [Google Scholar]
- Leung B, Harris HW. NKT cells in sepsis. Clin Dev Immunol. 2011;2010.pii:414650. doi: 10.1155/2010/414650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif S, Arreaza GA, Zucker P, Delovitch TL. Regulatory natural killer T cells protect against spontaneous and recurrent type 1 diabetes. Ann NY Acad Sci. 2002;958:77–88. doi: 10.1111/j.1749-6632.2002.tb02949.x. [DOI] [PubMed] [Google Scholar]
- Motohashi S.Clinical application of NKT cell system for lung cancer Nippon Rinsho 200563Suppl.4574–578.Japanese. [PubMed] [Google Scholar]
- Knothe S, Mutschler V, Rochlitzer S, Winkler C, Ebensen T, Guzman CA, et al. The NKT cell ligand alphagalactosylceramide suppresses allergic airway inflammation by induction of a Th1 response. Vaccine. 2011;29:4249–4255. doi: 10.1016/j.vaccine.2011.03.068. [DOI] [PubMed] [Google Scholar]
- Gapin L, Matsuda JL, Surh CD, Kronenberg M. NKT cells derive from double-positive thymocytes that are positively selected by CD1d. Nat Immunol. 2001;2:971–978. doi: 10.1038/ni710. [DOI] [PubMed] [Google Scholar]
- Pellicci DG, Hammond KJ, Uldrich AP, Baxter AG, Smyth MJ, Godfrey DI. A natural killer T (NKT) cell developmental pathway involving a thymus-dependent NK1.1−CD4+ CD1d-dependent precursor stage. J Exp Med. 2002;195:835–844. doi: 10.1084/jem.20011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JL, Zhang Q, Ndonye R, Richardson SK, Howell AR, Gapin L. T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells. Blood. 2006;107:2797–2805. doi: 10.1182/blood-2005-08-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Sant'Angelo DB, Nichols KE. Transcriptional control of invariant NKT cell development. Immunol Rev. 2010;238:195–215. doi: 10.1111/j.1600-065X.2010.00962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedeli M, Napolitano A, Wong MP, Marcais A, de LC, Colucci F, et al. Dicer-dependent microRNA pathway controls invariant NKT cell development. J Immunol. 2009;183:2506–2512. doi: 10.4049/jimmunol.0901361. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- Li K, Seo KH, Gao T, Zheng Q, Qi RQ, Wang H, et al. Invariant NKT cell development and function in microRNA-223 knockout mice. Int Immunopharmacol. 2010;11:561–568. doi: 10.1016/j.intimp.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Bezman NA, Chakraborty T, Pellerin A, Bender TP, Lanier LL.miR-150 differentially regulates the development of NK and NKT cells via targeting the transcript factor c-Myb J Immunol 2011186153.37. [Google Scholar]
- Zhou L, Zheng QH, Mi QS.microRNA miR-150 regulates thymic NKT cell development and function J Immunol 2011186153.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- Hu T, Simmons A, Yuan J, Bender TP, berola-Ila J. The transcription factor c-Myb primes CD4+CD8+ immature thymocytes for selection into the iNKT lineage. Nat Immunol. 2010;11:435–441. doi: 10.1038/ni.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Dai G, Tsujino K, Hashimoto K, Huang X, Fujimoto T, et al. Proper levels of c-Myb are discretely defined at distinct steps of hematopoietic cell development. Blood. 2006;108:896–903. doi: 10.1182/blood-2005-09-3846. [DOI] [PubMed] [Google Scholar]