

Abstract
Here, we report the synthesis and characterization of a novel 4-arm poly(lactic acid urethane)-maleate (4PLAUMA) elastomer and its composites with nano-hydroxyapatite (nHA) as potential weight-bearing composite. The 4PLAUMA/nHA ratios of the composites were 1:3, 2:5, 1:2 and 1:1. FTIR and NMR characterization showed urethane and maleate units integrated into the PLA matrix. Energy dispersion and Auger electron spectroscopy confirmed homogeneous distribution of nHA in the polymer matrix. Maximum moduli and strength of the composites of 4PLAUMA/nHA, respectively, are 1973.31 ± 298.53 MPa and 78.10 ± 3.82 MPa for compression, 3630.46 ± 528.32 MPa and 6.23 ± 1.44 MPa for tension, 1810.42 ± 86.10 MPa and 13.00 ± 0.72 for bending, and 282.46 ± 24.91 MPa and 5.20 ± 0.85 MPa for torsion. The maximum tensile strains of the polymer and composites are in the range of 5% to 93%, and their maximum torsional strains vary from 0.26 to 0.90. The composites exhibited very slow degradation rates in aqueous solution, from approximately 50% mass remaining for the pure polymer to 75% mass remaining for composites with high nHA contents, after a period of 8 weeks. Increase in ceramic content increased mechanical properties, but decreased maximum strain, degradation rate, and swelling of the composites. Human bone marrow stem cells and human endothelial cells adhered and proliferated on 4PLAUMA films and degradation products of the composites showed little-to-no toxicity. These results demonstrate that novel 4-arm poly(lactic acid urethane)-maleate (4PLAUMA) elastomer and its nHA composites may have potential applications in regenerative medicine.
Keywords: Polyester urethanes, ceramic composites, urethane composites, bone void fillers, load-bearing fillers, hydroxyapatite composites
1. Introduction
Repair of large weight-bearing bone defects is still a clinical challenge. To assist in bone healing and repair of such bone defects, a mechanically-competent, biodegradable synthetic material is highly desirable. Regarding mechanical properties, an ideal material for bone tissue engineering devices should be able to promote scaffold-host tissue integration, facilitate tissue ingrowth, and maintain structural and mechanical integrity while withstanding multidirectional loads incurred during normal daily activities. However, most available materials are not suitable for this application, as the balance between properties is not easy to replicate [1]. Elastomers are polymers that have shape-memory abilities and unique mechanical properties that make them well-suited to serve as components of load-bearing composites. Studies describing the synthesis of elastomer materials and their composites for bone tissue engineering have previously been published and numerous applications have been proposed [2–12].
In particular, polyester-urethanes are one kind of elastomers and have been increasingly used for the development of bone void fillers and scaffolds, since they combine a hydrolytically-degradable ester backbone with strong chemical linkers that provide elasticity. Short-chain polyester networks have been synthesized for bone tissue engineering applications, but these have limited mechanical resilience and are subject to swelling, high sol content, and fast degradation rates [13, 14]. High molecular weight versions of polymers such as polylactic acid (PLA), poly(lactide-co-glycolide) (PLGA), and poly(caprolactone) (PCL) have been used for load-bearing applications [15–17], but significant hydrophobicity makes them inconvenient for cell attachment and viability [18]. To further enhance the compatibility of the material without compromising degradation and strength, urethane-based scaffolds have also had a push in recent years in bone tissue engineering [19, 20]. Polyurethanes are tough hydrophobic materials with very high mechanical properties; however, these properties are offset by their reduced biocompatibility, which limits their applicability in vivo and in vitro [21]. The combination of polyurethanes and biocompatible, biodegradable short-chain polyesters represents a promising solution to the limitations of each individual material and encouraging early results have been reported. Previous work on polyester urethanes has shown compressive moduli ranging from cements at 49 MPa [22] to pressure-molded composites at 9000 MPa [23]. Likewise, compressive strength of these materials has ranged from 13 MPa [24] to 150 MPa [23]. Tensile moduli for elastomer composites have ranged from 586 MPa [25] to 3800 MPa [26]. A polymer fiber-reinforced composite (FRC) material developed for load-bearing orthopedic implants had a shear modulus of 378 ± 80 MPa and shear strength of 13.7 ± 5.0 MPa [27]. Bending moduli of elastomer composites go from 1000 MPa for extrusion-molded materials [28] to 12 GPa for FRCs [29]. However, testing of new composites for bone tissue engineering usually does not encompass all mechanical properties.
In this work, we hypothesized that modifying a 4-arm PLA network with urethane segments, provided by hexamethylene diisocyanate (HDI), and a crosslinking moiety, provided by maleic acid (MA), would create a new elastomer, which can be used to fabricate ceramic composites with improved mechanical properties as load-bearing materials for bone tissue engineering. We first synthesized and characterized the resulting 4-arm poly(lactic acid urethane)-maleate, henceforth named 4PLAUMA. According to our design, the PLA polymer network was initiated by erythritol, a four-arm polyol approved by the FDA as a food additive. Since erythritol provides four possible initiation points, it will allow the formation of branched macromolecules that can act as building materials for networked polymer matrices, which will reinforce the mechanical properties of the composite on the nanoscale [30, 31]. The chains would be alcohol-terminated, so both isocyanates and carboxylic groups from HDI and MA, respectively, can react with these groups and potentially link the 4-arm PLA into larger, mechanically-resilient networks, with urethane links and double bonds interspersed in the structure. By adjusting the composition of 4PLAUMA, these biocompatible and biodegradable polymer networks could provide for a high capacity of embedded particles in order to significantly enhance mechanical strength. To test the ability of 4PLAUMA to form composites of high strength, a nanosized HA ceramic was mixed into the polymer and mechanical performance was comprehensively tested for different compositions. To assess its compatibility, 4PLAUMA formulations and its composites were tested for cell attachment, morphology, and growth. With this work, we expected to provide, with this new elastomer and its composite, a promising elastomer for bone tissue engineering.
2. Materials and methods
2.1. Materials
D,L-lactide monomer was purchased from Ortec (Piedmont, SC). Erythritol (ET) was obtained from Alfa Aesar (Ward Hill, MA). Maleic (MA) and acrylic acid, ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), and hexamethylene diisocyanate (HDI), toluene, anhydrous ethanol, deuterated chloroform (CDCL3, 99.8%at. deuterated), 4′,6-diamidino-2-phenylindole (DAPI) and dimethylsulfoxide (DMSO) were obtained from Fisher (Pittsburgh, PA). Phalloidin-rhodamine was obtained from Cytoskeleton (Denver, CO). The tetrazole 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Sigma. Tin(II)-2-ethylhexanoate (TOC) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Nano-hydroxyapatite (nHA, Ca10(PO4)6(OH)2) was obtained from Nanocerox (Ann Arbor, MI). An MSCGM™ BulletKit™, EBM™ (endothelial basal medium), and an EGM™ (endothelial growth media) SingleQuots™ Kit were purchased from Lonza. Dulbecco’s Modified Eagle media (DMEM), fetal bovine serum (FBS), L-glutamine, antibiotic-antimycotic solution (penicillin-streptomycin-glutamine, PSG), phosphate buffer saline (PBS), and Hank’s balanced salt solution (HBSS) were obtained from Invitrogen (Grand Island, NY).
2.2. 4-arm poly(lactide urethane maleic acid) (4PLAUMA) synthesis and characterization
The 4PLAUMA macromer was synthesized using the protocol described by Wang et al. [32] with the modifications depicted in Figure 1. For the synthesis of the 4-arm PLA macromer, 30g of D,L-lactide were added to a 500 mL flame-dried Erlenmeyer flask. A small magnetic stirring bar was used throughout the experiment to stir the contents. Sixty mL of toluene were then added to completely dissolve the lactide under stirring. While dissolving, 1.22 g ET and 0.012g TOC were added to the beaker. The reaction vessel was carefully heated to 100°C under nitrogen atmosphere until all the ET dissolved. The temperature was then lowered and kept at 70°C under nitrogen for 24 hours. After the reaction was completed, the PLA-polyol product was precipitated in cold ethanol and dried under vacuum for 24 hours. The theoretical calculated yield was 99.3%; the final yield for reactions was 96.1%.
Figure 1.

Schematic for 4PLAUMA synthesis. Numbers in bold refer to the locations for the methyl (1) and methine (2) hydrogens of the lactide chains, the methylene hydrogens of erythritol (3), the vinyl groups of maleic acid (4), and the outer (5) and inner (6) hydrogens of HDI.
To obtain 4PLAUMA, 30g of 4-arm PLA was dissolved in 50 mL of toluene in a flask and heated to 70°C. Afterwards, 1 mL HDI, 1g MA, and 0.012g TOC were added to the reaction flask and allowed to react, under nitrogen, for 6 hours. The reaction solution was then quickly filtered to remove microgels and clusters, which were expected to form according to the Flory theory of gelation [33], which states that gelation might occur when rp2 > 1/(f-1), where r, p, and f are the molar ratio of the reactants, the extent of reaction of the multifunctional monomer, and functionality, respectively. The final polymer yield per reaction averaged 81%. The filtered polymer solution was precipitated in cold ethanol, filtered again, and dried under vacuum for 24 hours. For further purification, the products were then dialyzed against distilled deionized water using a Spectra/Por® dialysis membrane (3500 MWCO, Spectrum Labs, Rancho Dominguez, CA) and lyophilized. The original formulations were also modified with 2 times and 5 times MA or HDI to test the effect of these compounds on the material properties of the composites.
1H-NMR was performed on the 4PLAUMA polymer using a Varian Inova 300 instrument at ambient conditions. The polymer was dissolved in CDCL3 at a concentration of 10 mg/mL. Pure CDCL3 was used as an internal standard. ATR-FTIR was performed on the polymer using a Bruker Vertex 70 spectrometer with a deuterated triglycine sulfate (DTGS) detector at room temperature. About 30μL of polymer solution was added to the ATR well and pressed against the diamond window for sample analysis at a resolution of 2 cm−1.
2.3. Composite sample preparation
Prepolymer composite mixtures were made by adding 50% w/v polymer 4PLAUMA to a 25% v/v DMSO in water solution. These mixtures were combined with nHA to generate polymer/nHA weight ratios of 1:1, 1:2, 2:5, and 1:3. To start crosslinking, 6% v/v acrylic acid, 1% v/v, APS and 0.1% v/v TEMED were added into the mix. The mixture was then immediately cast into Teflon molds and allowed to crosslink for at least 6 hours (all samples showed fair consistency after this time). Afterwards, samples were cured in an oven for 48 hours at 80°C to finish the curing process. Afterwards, samples were cured in an oven for 48 hours at 80°C. Controls were made with crosslinked and cured prepolymer mixtures without nHA addition.
2.4. SEM imaging, EDS scanning, and AES analysis
Top and cross-sectional samples of 4PLAUMA and the 4PLAUMA 1:3 composite were cut, fixed on Al pin mounts, and sputter-coated with gold for observation under SEM. The surface features of the composites were observed using a FEI XL30 Sirion SEM at an accelerating voltage of 15 keV. The chamber was kept in vacuum to avoid surface charging during the observation. The chemical composition was analyzed using an energy dispersive spectroscopy detector (EDS) attached to the SEM. Auger electron spectroscopy (AES) was conducted on the 1:3 4PLAUMA composite using a PHI 700 Scanning Auger Nanoprobe (Physical Electronics) equipped with a low-energy ion gun. The composite was cut into small slabs for AES analysis. The top 2 mm of the slabs was sanded down and the resulting surface was polished and cleaned. Since the composites are non-conductive, the slabs were placed on the AES stage at an angle of approximately 90° for superficial scanning (10 nm scanning depth). The surface was sputter cleaned with the ion gun. The sample was then scanned in vacuum at a voltage of 5 keV.
2.5. Mechanical testing
Samples were made and tested adapting guidelines indicated in American Society for Testing and Materials (ASTM) methods D638, D695, D790, and D6272. Samples were created for compression, tension, torsion, and 3-point bending tests. An ElectroPuls E10000 test system (Instron) was used for testing of samples. All samples were tested on dry condition at room temperature. For compression and bending, testing was performed at a strain rate of 1%/s until sample failure, or a maximum of 15% strain, whichever happened first. For compression and tension, strain was described as the displaced length over the initial length of the material. For bending, strain was measured as the ratio between deformation (actuator displacement) and radius of curvature to the span between supports. Torsional strain was calculated as rotated displacement over the length of the material. Samples were run until failure for torsion and tension. Elastic moduli, maximum strength, and maximum strain were determined for the samples.
2.6. Degradation study
Composite samples with a dry volume of 0.5 cm3 were crosslinked and cured as described above, using different nHA, MA, and HDI ratios. The 4PLAUMA polymer without nHA was used as a positive control. The samples were carefully weighed and placed in 2 mL of HBSS at 37°C for 8 weeks under sterile conditions, with time points every week. At each time point, wet and dry weights were recorded. Degradation was then determined by calculating the percentage of remaining mass at each time point. Swelling ratios (Q) of the samples were calculated after 4 hours in HBSS (time zero) and after 3 weeks by using Q=(Ws−Wd)/Wd, where Ws and Wd represent the swollen and dry weights of the samples. Sample HBSS supernatants were collected at 8 weeks for cell viability testing.
2.7. Cell culture
Human bone marrow-derived mesenchymal cells (hBMSC) were purchased from Lonza Inc. (Allendale, NJ). Human umbilical vein endothelial cells (HUVEC) constitutively expressing green fluorescent protein (GFP) were generously gifted from the late Dr. J. Folkman, Children’s Hospital, Boston. hBMSCs were cultured in basal media consisting of DMEM supplemented with 10% FBS, 1% L-glutamine (200 mM), and 1% PSG, and HUVECs were cultured in endothelial basal EBM-2 with EGM-2 supplements under standard conditions (5% CO2, 95% humidity, and 37 °C).
2.8. Evaluation of cell adhesion
Thin polymer films of 4PLAUMA and 4PLAUMA 1:3 were created in 48-well plates by coating under a cell-culture hood with 100 μL of the prepolymer solution each, with crosslinking provided by chemical initiators as described above. After crosslinking, the polymer layers were washed with 500 μL PBS three times, then cleaned with 200 μL 70% ethanol in water for 2 hours, and washed again three times with PBS. To test acidity, media was added to control well and the pH monitored for three hours to verify that the pH was stable at a value 7. If the value was not 7, then washing was repeated until the value stabilized.
HUVECs at a seeding density of 1×104 cells per well were added and cultured for 24 hours, with time points at 1, 2, 4, 6, 8, 12, 18, and 24 hours. At each time point, the media was removed and the attached cells were then trypsinized and resuspended in 200 μL media. The cells were then counted using a Z Series Coulter Counter (Beckman Coulter).
To visualize the cells, the cells were seeded on thin polymer films in 48-well plates and observed by light microscopy (Zeiss Axio Observer) after 24 hours. Cells seeded directly on the well plates were used as a control. Wells were washed three times with PBS and cells were fixed with 1 mL per well of 4% paraformaldehyde in PBS at 37°C for 20 min. After fixation, the paraformaldehyde solution was removed and cells were permeabilized using PBS containing 0.1% Triton X-100. For visualization of the cell cytoskeleton, the wells were rinsed with PBS and 200 μL of 100nM rhodamine phalloidin solution (excitation and emission wavelength of 540 and 565 nm) was added to each well for 30 min at ambient conditions to stain the actin filaments. Then, wells were washed with PBS and the cell nuclei were stained with 200 μL of DAPI solution (excitation and emission wavelength of 358 and 461 nm) for 30 seconds. The wells were washed once and kept in PBS. An inverted fluorescent microscope (Zeiss Axioplan 2) was used to take images of the cultured cells.
2.9. Cell viability studies
Thin 4PLAUMA polymer layers were fabricated in 48-well plates as described above. Next, HUVEC or hBMSCs were trypsinized and resuspended in their respective media at a concentration of 5×105 cells/mL. Then, 20 μL of cell suspension and 180 μL of media were directly added to each well to attain a final concentration of 1×104 cells per well. Cells were then allowed to grow for 7 days on the polymer films, with time points at 1, 3, 5, and 7 days. Wells without polymer layers were used as controls. At each time point, cell proliferation was determined by using an MTT assay. A 5 mg/mL MTT stock solution in PBS was directly added to the media in the wells until diluted to a concentration of 0.5 mg/mL. The cells were then incubated for 3 hours at 37°C to allow reduction of MTT to purple formazan crystals. The media was then carefully removed, and crystals were dissolved in 200 μL DMSO for 30 min at 37°C. Each well was thoroughly flushed to aid in formazan dissolution. Sample absorbance was then measured using an UV plate reader (BioTek) at 490 nm. All values were expressed as optical density, which indicates a measure of relative cell viability. To test the proliferation of the cells exposed to degradation products, cells at a concentration of 1×104 cells per well were cultured in suspensions consisting of 50% HBSS with degradation products of 8 weeks and 50% of EBM media for 3 days, with time points every 24 hours. Cells cultured in 50% pure HBSS and 50% EBM were used as a control group. Viability of cells was then determined by the MTT assay described above.
2.10. Statistical analysis
All data is presented as average values ± standard deviation. Experiments were performed in triplicate (n=3). Statistical difference was determined using one way ANOVA analysis with Systat 12 (Systat Software Inc, Chicago, IL) followed by Tukey’s post-hoc test (α=0.05).
3. Results
3.1. NMR and FTIR of 4PLAUMA
Figure 2 shows the NMR of 4PLAUMA with corresponding peaks. Peaks at 1.6 and 5.3 ppm were attributed to were attributed to the methyl hydrogens (−CH3) of the lactide repeat unit and the methine hydrogen of the lactide, respectively [34]. PLA-ET shared another quartet with PLAUMA at around 4.4 ppm and was attributed to the methylene hydrogens of the ET. The average degree of polymerization (DP) and the molecular weight of the PLA-ET precursor polymer were calculated from end-group analysis of the NMR spectrum [35] by using the integral intensity ratio of the ET to the lactide. The DP was determined to be 25 and the molecular weight was determined to be 3.7 kDa. The small, sharp singlets at 6.5 and 6.9 ppm were attributed to the vinyl protons of the MA, observed as a faint trace in the pure MA spectrum due to poor dissolution of MA in CDCL3 [36–38]. The doublet at 3.2 and singlet at 1.4 were attributed to the outermost and innermost methylene hydrogens of the HDI units [39, 40] prominently observed in the pure HDI NMR. FTIR-ATR spectra for the 4PLAUMA polymer and components are also shown in Figure 3. The absence of an isocyanate signal peak at 2200 cm−1 and the presence of small secondary amide signals at 1650, 1300, and 1100 cm−1 indicated the incorporation of HDI into the polymer and a high degree of reaction. Ester links were observed at 1750 cm−1, bonded alcohol C-O at 1150 cm−1, bonded carboxylic groups at around 2950 cm−1, and a broad N-H stretch band from 300–3500 cm−1. The peak around 725 cm−1 indicates the presence of the cis C=C double bond of the maleate. The increase in the free carboxylic group peak at 1420 cm−1 suggests an increase of acid groups by MA and presence of unreacted terminal acid groups.
Figure 2.
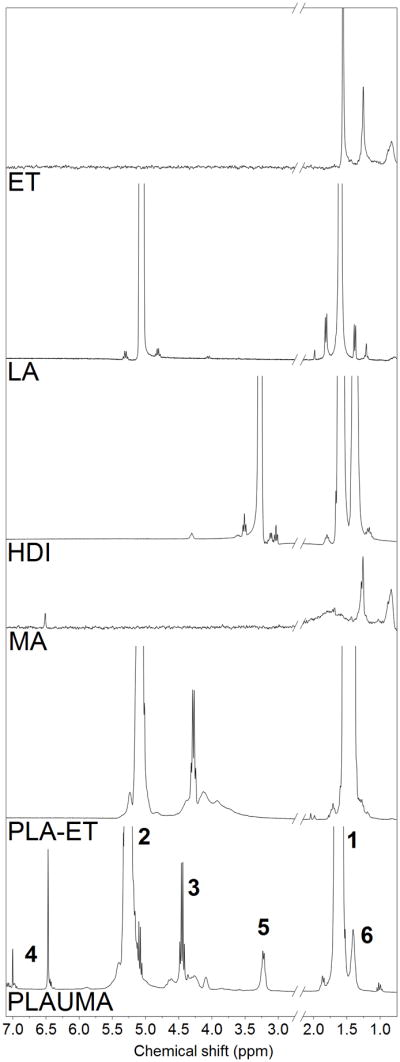
NMR spectra for the 4PLAUMA polymer and its components. Numbers in bold correspond to the hydrogens in the 4PLAUMA structure shown in Figure 1.
Figure 3.
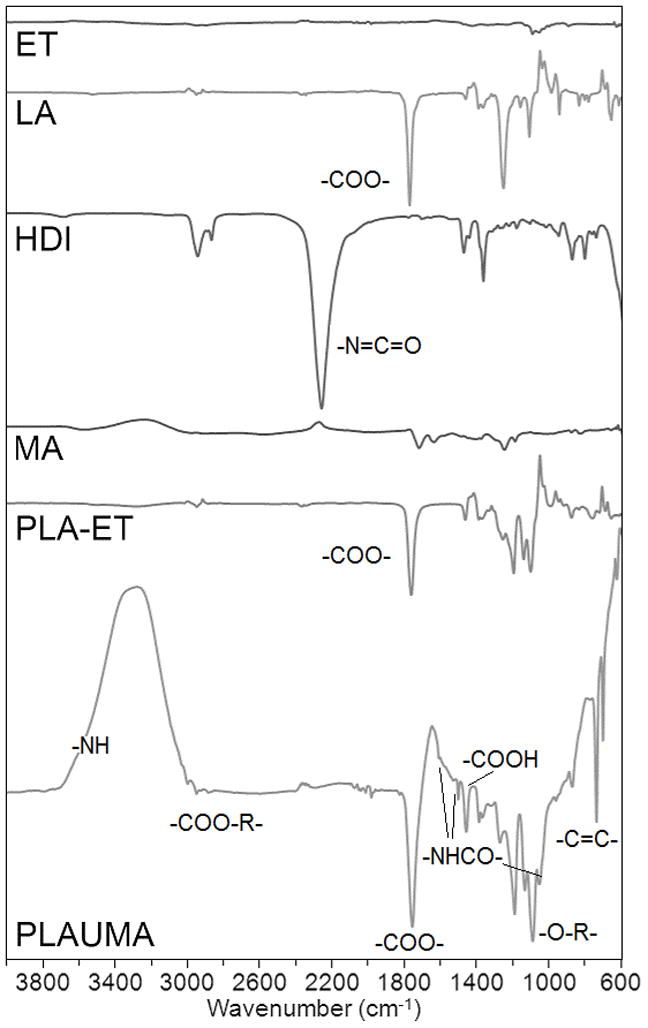
FTIR spectra for the 4PLAUMA polymer and its components.
We also tested the effect of increasing the concentration of MA or HDI on the polymer structure. When the MA concentration was raised, the signal at 6.5 ppm increased (Figure 4). Similarly, the signal intensity of the peaks at 1.4 and 3.2 increased with HDI loading amount. The integrated area ratios of MA/HDI (1.8) and MA/lactide (0.3) do not significantly change with an increase of MA, indicating a limit to the extent of reactivity of MA to hydroxyl groups. However, the MA/HDI ratio decreases two- and tenfold with an increase to 2 and 5 times the HDI loading amount, respectively. This suggested a stronger dependence to concentration on HDI reactivity towards the hydroxyl groups at the PLA-ET ends, compared to MA. However, since the IR signal intensities for amide and vinyl groups are subtle, the FTIR spectra of formulations showed no significant differences (Figure 5).
Figure 4.

NMR spectra of 4PLAUMA and its formulations. Numbers in bold correspond to the hydrogens in the 4PLAUMA structure.
Figure 5.
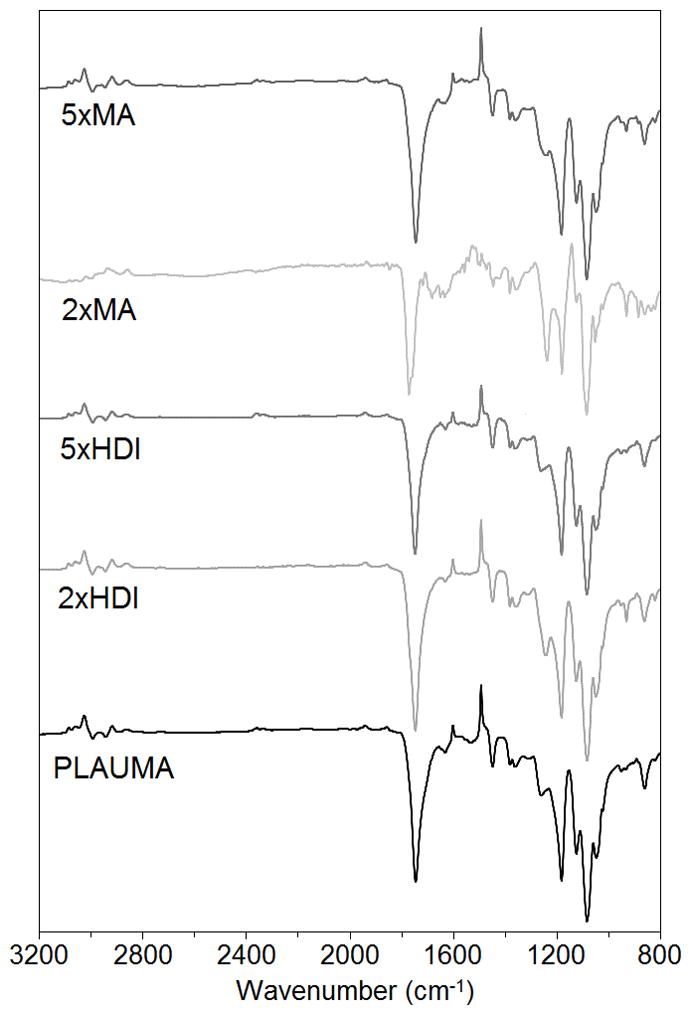
FTIR spectra of 4PLAUMA and its formulations.
3.2. EM imaging and spectroscopy analyses
SEM imaging of the 4PLAUMA and its composite is shown in Figure 6. The 1:3 composite was slightly rough at its surface (Figure 6A). A more pronounced roughness was observed in its cross-sectional area, with spherical-like structures (Figure 6B) indicative of particulate dispersion within the matrix. EDS analysis demonstrated that HA was present on both the surface and cross-section of this material, as evidenced by Ca/P ratios close to that of HA. The 4PLAUMA polymer, by itself, showed a smoother surface (Figure 6C), suggesting that HA increased the surface roughness of the material. Figure 7 shows the results of AES scanning. On the surface (Figure 7A), Ca and P were detected (Figure 7B).
Figure 6.

Representative SEM and EDS analysis of 4PLAUMA and 1:3 4PLAUMA composites. (A) 4PLAUMA 1:3-top surface (B) 4PLAUMA 1:3-cross-sectional area (C) 4PLAUMA. Ca/P atomic ratio in the 1:3 composite was similar to that of natural HA (1.67).
Figure 7.
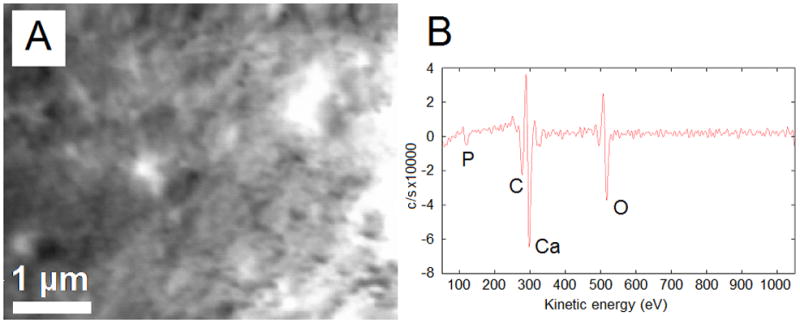
AES analysis of the 1:3 4PLAUMA composite. (A) SEM of the region of interest, (B) AES spectrum of the composite.
3.3. Mechanical testing
Results for mechanical testing of samples are shown in Table 1. The samples were run until failure, or until samples were seen to not show signs of failure with increasing strain. For compression and bending, only the samples of polymer:nHA ratios of 1:3 and 2:5 failed at 15% strain; the lower concentrations (1:2 and 1:1) showed compliance with increasing strain. All samples were tested until failure for tension and torsion. The measured maxima for moduli and strength of the 4PLAUMA/nHA composites were, respectively, 1973.31 ± 298.53 MPa and 78.10 ± 3.82 MPa for compression, 3630.46 ± 528.32 MPa and 6.23 ± 1.44 MPa for tension, 1810.42 ± 86.10 MPa and 13.00 ± 0.72 for bending, and 282.46 ± 24.91 MPa and 5.20 ± 0.85 MPa for torsion. The maximum tensile strains of the polymer and composites ranged from 5% to 93%, and their maximum torsional strains varied from 0.26 to 0.90. The polymer by itself showed both the highest ultimate tensile strain (93%) and the maximum torsion strain (0.90 ± 0.07, corresponding to a strain angle of torsion at ~250º). The data in Table 1 show that all moduli and maximum strengths increased with increasing concentration of nHA. However, as expected, torsion and tensile strains decreased with increasing nHA content. There were no significant effects when increasing the MA or HDI content in the maximum properties of 1:3 composites.
Table 1.
Elastic moduli and ultimate strength of 4PLAUMA composites. Maximum tensile and shear strains are shown for all samples. Ratios are expressed as mass polymer: mass nHA. Values are given as average ± standard deviation.
| Polymer/nHA ratio
|
||||||
|---|---|---|---|---|---|---|
| 1:3 | 2:5 | 1:2 | 1:1 | 1:0 | ||
| Compression | Modulus (MPa) | 1973.31 ± 298.53 | 1675.23 ± 145.83 | 508.63 ± 107.34 | 198.64 ± 41.58 | - |
| Strength (MPa) at 15% | 78.10 ± 3.82 | 48.76 ± 10.34 | 36.37 ± 12.13* | 22.25 ± 4.45* | - | |
|
| ||||||
| Tension | Modulus (MPa) | 3630.46 ± 528.32 | 1485.31 ± 442.17 | 822.47± 72.81 | 149.82 ± 53.62 | 3.70 ± 0.34 |
| Strength (MPa) | 6.23 ± 1.44 | 4.25 ± 0.22 | 3.89 ± 0.08 | 2.11 ± 0.35 | 1.11 ± 0.16 | |
| Maximum strain (%) | 5.33 ± 1.53 | 14.34 ± 0.58 | 22.04 ± 2.39 | 40.11 ± 2.07 | 93.33 ± 4.04 | |
|
| ||||||
| Bending | Modulus (MPa) | 1810.42 ± 86.10 | 1479.19 ± 224.91 | 310.97 ± 106.14 | 125.56 ± 39.46 | - |
| Strength (MPa) at 15% | 13.00 ± 0.72 | 6.76 ± 1.68 | 4.40 ± 0.78* | 2.03 ± 0.54* | - | |
|
| ||||||
| Torsion | Modulus (MPa) | 282.46 ± 24.91 | 223.37 ± 15.36 | 159.40 ± 38.33 | 46.28 ± 32.53 | 0.12 ± 0.01 |
| Strength (MPa) | 5.20 ± 0.85 | 5.03 ± 0.33 | 4.33 ± 0.06 | 2.67 ± 1.10 | 0.05 ± 0.01 | |
| Maximum strain (−) | 0.26 ± 0.03 | 0.34 ± 0.06 | 0.56 ± 0.03 | 0.71 ± 0.05 | 0.90 ± 0.07 | |
One star (*) indicates samples that did not fail at 15% strain. 1:0 indicates pure 4PLAUMA.
3.4. Degradation studies
Degradation profiles for the 4PLAUMA polymer and the 4PLAUMA/nHA composites are shown in Figure 8. The rates of degradation of the 4PLAUMA/nHA composites were significantly slower than that of the 4PLAUMA, with remainders on 8 weeks at approximately 43% for 4PLAUMA, 61% mass remaining for the 1:1 composite, 72% for the 1:2 composite, 70% for the 2:5 composite, and 75% for the 1:3 composite (Figure 8A). Significant difference was observed for the 1:1 group and other composite groups after 5 weeks (p < 0.042), but there were no significant differences between the remaining composite groups through the duration of the experiment. The degradation profiles for polymer composites made with 2 and 5 times as much HDI and MA were also examined shown in Figure 8B. However, there was no statistical difference between groups during the duration of the experiment. After 8 weeks, the 2x and 5x HDI had approximately 68% and 69% of their initial mass remaining, respectively, while for the 2x and 5x HDI the initial mass had decreased to 72% and 74%. The pH of all solutions with composite degradation products was generally higher for all time points than those with only polymer degradation products (data not shown). The swelling ratio of the polymers after 4 hours and 3 weeks is shown in Table 2. The polymer and composites showed very little swelling at both time points.
Figure 8.
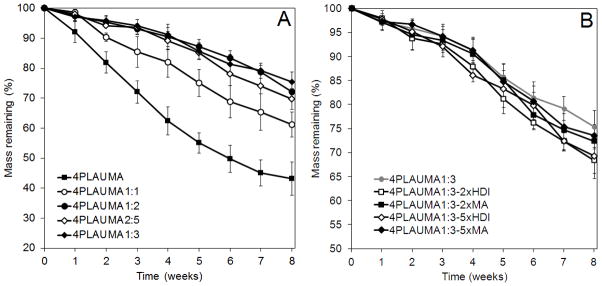
Degradation patterns of the 4PLAUMA polymers. (A) Comparison between 4PLAUMA with 4PLAUMA with different nHA ratios. Ratios expressed as Mass polymer: Mass nHA. (B) Comparison between the 4PLAUMA 1:3 polymers with different MA and HDI amounts. Amounts expressed as times increase of component. Samples were done in triplicate.
Table 2.
Swelling ratio of the elastomer composites with time. Values are given with ± one standard deviation. Initial value refers to swelling 4 hours after start of experiment.
| Polymer/composite | Initial | 3 weeks |
|---|---|---|
| 4PLAUMA | 1.24 ± 0.18 | 2.56 ± 0.43 |
| 4PLAUMA1:1 | 0.26 ± 0.11 | 0.35 ± 0.14 |
| 4PLAUMA1:2 | 0.15 ± 0.08 | 0.26 ± 0.05 |
| 4PLAUMA2:5 | 0.09 ± 0.05 | 0.16 ± 0.04 |
| 4PLAUMA1:3 | 0.05 ± 0.02 | 0.06 ± 0.01 |
| 4PLAUMA1:3-2xHDI | 0.03 ± 0.01 | 0.03 ± 0.03 |
| 4PLAUMA1:3-2xMA | 0.07 ± 0.04 | 0.07 ± 0.01 |
| 4PLAUMA1:3-5xHDI | 0.04 ± 0.03 | 0.04 ± 0.02 |
| 4PLAUMA1:3-5xMA | 0.04 ± 0.01 | 0.05 ± 0.02 |
3.5. Cell behavior
The adherence of HUVECs on the polymer films was assessed by determining the number of cells present on the polymer films several hours after cell seeding. The results are shown in Figure 9. The profile shows a rapid attachment of cells and cell adhesion reaches a plateau only after 4–6 hours. No significant differences were observed at any time point compared to cells seeded directly on well plates. Figure 10 shows imaging of the cells attached to thin polymer layers. GFP-tagged HUVEC attached to the pure polymer after 2 hours. Cell morphology suggests that cells can adhere and spread well on the polymer.
Figure 9.
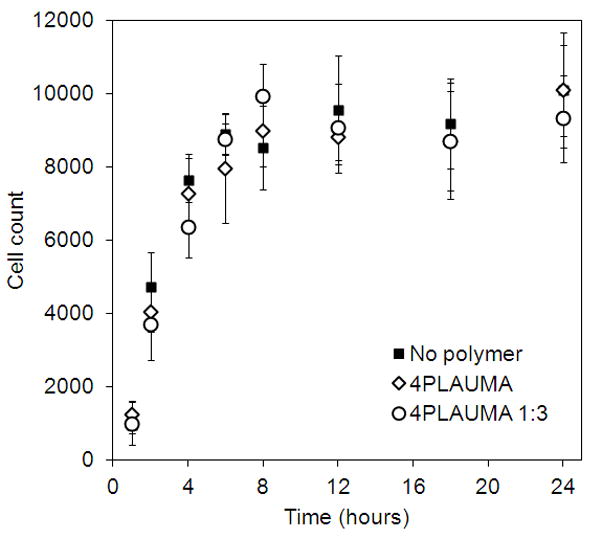
Cell attachment trends for thin 4PLAUMA layers. Cells were trypsinized and resuspended in media. Numbers were then calculated directly on a cell counter. No statistical differences were observed among groups. Samples were done in triplicate.
Figure 10.

Images of HUVEC attached to polymer films. (A) Control (no polymer film) (B) 4PLAUMA film. Scale bar size for these images 100 microns. (C) Fluorescence imaging of GFP-tagged HUVEC attached to the polymer. Red=rhodamine-phalloidin, cytoskeleton; Blue=DAPI, nuclei; green=GFP. The green hue was due to the slight autofluorescence of polymer in the green channel. Scale bar is 50 microns.
Figure 11 shows viability of cells directly seeded on polymer films as determined by an MTT assay. The 4PLAUMA thin films showed excellent compatibility for both GFP-tagged HUVECs and hMSCs (Figure 11A). Cells were viable, with steady growth over the course of 7 days incubation. During the experiment, no significant differences were observed between cells seeded on the films and cells directly seeded on well plates. HUVECs were seeded on top of 2x HDI and 2x MA 4PLAUMA films to test if the viability of the cells would change with an increase of these components (Figure 11B). The results show cells could grow on top of the polymer films. Again, no significant differences were observed between groups at all time points.
Figure 11.
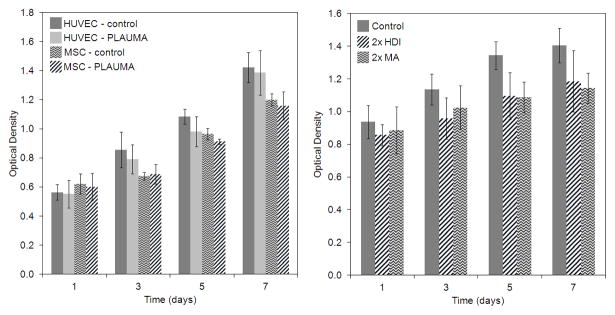
Quantification of cell growth on 4PLAUMA films as determined by an MTT assay. (A) Proliferation of HUVECs and hBMSCs seeded on thin 4PLAUMA layers. (B) Proliferation of HUVECs seeded on 2x MA and 2x HDI films. Control groups refer to cells seeded on the well plates without a polymer layer. No statistical differences were observed among groups. Samples were done in triplicate.
HUVECs were cultured with 8-week degradation products of the polymer and composites to test their cytotoxicity to test the most extreme conditions for the new materials. The results of MTT activity is shown in Figure 12. The 3-day incubation experiment shows that cells grew in all the testing groups, but cell metabolic activities in presence of degradation products of polymer or composites were lower than those in absence of degradation products.
Figure 12.
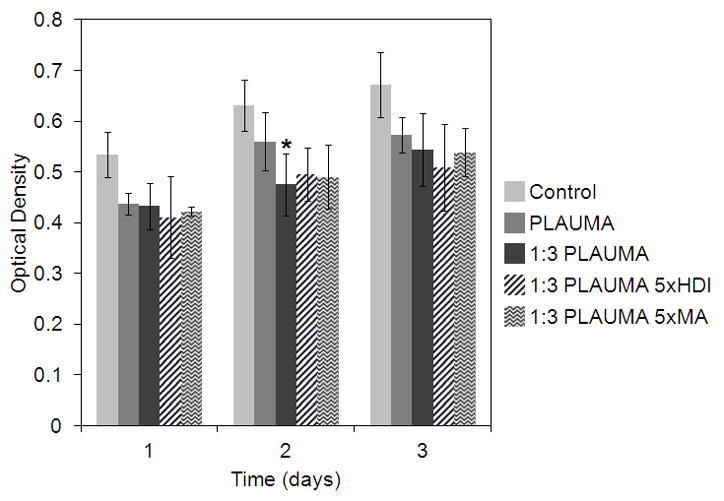
Proliferation of HUVECs seeded with 50% EBM and 50% HBSS with degradation products at 8 weeks. Control refers to 50% pure HBSS and 50% EBM. One star (*) indicates statistical difference with control group (p<0.05). Samples were done in triplicate.
4. Discussion
The goal of this study was to develop a novel elastomer as a matrix in a polymer/nHA composite for use as a bone void filler with sufficient mechanical properties to withstand loads comparable to those seen during post-operative mobilization and rehabilitation. We thoroughly characterized both the chemical and mechanical properties of the materials. In vitro cytotoxicity of the materials and their degradation products were also examined. An obvious advantage of this design is versatility in matrix solidification for a single-component polyester urethane, including crosslinking of double bonds and heat-curing of the urethane phase. Moreover, the mechanical properties of the elastomer are tunable by simple and cost-effective variation in composition or processing. Unlike the conventional method for synthesizing polymers with intended high mechanical strength, the current fabrication process does not require high pressure or high temperature. This feature potentially allows for incorporation of biological agents.
NMR and FTIR measurements demonstrated that the polymer obtained is the 4PLAUMA polymer that we designed, consisting of a PLA backbone with maleate and urethane units incorporated to the polyester chains. According to our methods, the polymer composition did not change significantly with increasing HDI or MA loading. This could be due to several reasons, such as saturation of reactive sites, interinhibition of MA and HDI during the reaction time, and slow reactivity of low-solubility initial reactants. It was expected, then, that all polymer formulations would exhibit similar behavior, and this was the trend observed.
EDS and AES results showed Ca and P were evenly dispersed throughout the composite, verifying the presence and homogeneous distribution of nHA in the composite. The interaction of polymers with ceramic particles has four potential mechanisms: embedding, chemical bonding, interpenetration, and copolymerization [41]. HA, in particular, has been shown to interact with polyesters and urethanes by calcium ion interaction with carboxyl [42, 43], hydroxyl [44], and isocyanate [45, 46] groups, all of which are present in this polymer. The homogeneous dispersion of the ceramic in the 3D polymeric network is essential to keep the strength of the material. Agglomerates can disrupt the strength of the material due to phase discontinuities and addition of stress concentration points [47, 48]. However, no large agglomerates were observed in the bulk of the material under observation.
In our study, we evaluated a full scope of the mechanical properties of our material. Compression, tension, torsion, and bending moduli of the polymer/nHA composite are as high as 1.97, 3.63, 0.28 and 1.81 GPa, respectively, with greatest strengths at 78.10, 6.23, 5.2, and 13 MPa. Torsion and tensile properties, together with its observed ability to recover its original shape after deformation, evidence 4PLAUMA is elastomeric. The compressive moduli and strength of 4PLAUMA composites are similar to injectable two-component cements [49] and pressure-molded fillers [50] previously designed, or exceeding the properties of preceding materials [24, 51]. Our 4PLAUMA composites have a tension moduli comparable to the high-end of reported values for pressure-molded elastomeric composites [25, 26], show no significant difference with FRC torsional moduli [27]. and are within the range for reported values for bending moduli of other proposed elastomer composites [28, 29].
It is worth noting that compressive, tension, torsion, and bending moduli of bone are reported to be as high as 19, 20, 3 and 16 GPa, respectively, with strengths going up to 215, 107, 53, and 181 MPa [52]. It is therefore clear that the ultimate mechanical properties of bone are much higher than those of the new composites we synthesized. In addition, the biodegradable bone void filler requires interconnected pores for facilitating host tissue ingrowth and integration. This porous structure will further reduce the ultimate strength of the final products made of the new composites. Fortunately, considering the limited ambulatory activity and the use of fixation plates during recovery period, the loading on the bone void fillers will be much lower than its ultimate mechanical properties of bones [53, 54]. Moreover, studies have shown that either sporadic high levels of strain [55] or frequent low levels of strain [54] would be required to elicit cellular responses for bone formation. Therefore, the objective of the new elastomer/ceramic composite will be the retention of the structural integrity and stabilization of the interface during the mechanical loading for integration with host bone tissue and facilitating bone regeneration, aided by the porous structure of the bone void filler.
The material degradation properties are tunable and exhibit a broad range of degradation rates, which will affect the material’s mechanical performance over time. A slow degradation rate of synthetic bone grafts in load-bearing application can avoid early failure in mechanical support and promote integration between bone grafts and the site of injury due to interface stabilization and maintenance of structural integrity [53]. There was no significant effect of the polymer composition in the material degradability when the MA/PLA and HDI/PLA ratios changed, suggesting a weak correlation of crosslinking density and urethane composition with degradation. Since the ratios of MA or HDI to PLA are relatively low, degradation will be driven mainly by hydrolytic ester degradation of the major component, PLA. Degradation of the strong urethane bonds may not be significant [56], so it not expected to have considerable impact in determining the overall degradation of the matrix, especially at low ratios. On the other hand, the nHA ceramic content seemed to strongly regulate the degradability of the material. The degradation rate of HA can be substantially lower than other ceramic compounds, such as bi-phase calcium phosphate (BCP) and TCP [57]. It is possible HA nanocrystals stabilized the polymer network with electrostatic and weak interactions, which decreases the impact of water intake and hydrolysis [58]. Moreover, the increase in pH for all composite degradation solutions might have decreased the impact of acid erosion. Since the ability of the material to withstand loads is decreased with fluid content in the material, as the liquid phase expands the polymer network and increases volume and flexibility, it is not desired to have a material that can swell considerably in water. Our results show a very low swellability of the polymer after 3 weeks
The ceramic composites support excellent cell attachment and significant proliferation on its surface and in the presence of degradation products. Polyester HDI polymer materials have been successfully used in vivo with little or no immune and inflammatory response, due to their compatibility and hydrophobic nature [59–61]. The results suggested that the material allows cell adhesion, possibly due to the biostability of the polymer network. Crosslinking moieties in the polymer structure can also provide sites for the addition of functional factors and molecules that could be used to enhance attachment, differentiation, or other cell signaling [62–64].
In the future, we will continue to optimize the synthesis of composites and fabricate a functionally-graded bone void filler using the new composites by spatially manipulating its chemistry, pore size and porosity to promote cell ingrowth and tissue integration as well as providing load-bearing mechanical support. We will characterize our composites under degradation to directly assess how HA affects the degradation rates, and optimize dispersion of material for improved mechanical properties. We will also evaluate the biocompatibility and functionality of the elastomer/ceramic composite in vivo.
5. Conclusions
A new crosslinkable and heat-curable poly(D,L-lactide urethane maleic acid) (4PLAUMA) and its nHA composites were synthesized and characterized to evaluate their potential use as load-bearing bone fillers. The material showed tensile and torsion properties consistent with elastomeric materials, but these decreased with increasing ceramic content. The addition of nHA strengthened the material and reduced its degradation over time due to interactions with surrounding polymer chains. The material also showed excellent compatibility and attachment properties for both stem cells and endothelial cells. Within the scope of this work, the novel polymer and its ceramic composites could be tailored for prospective development of weight-bearing bone void fillers.
Highlights.
A polyester urethane for load-bearing bone tissue engineering was synthesized
Chemical characterization evidenced successful synthesis of polymer
Hydroxyapatite composites were fabricated by mixing, crosslinking and curing
Comprehensive mechanical testing was conducted on the composites
In vitro data showed biocompatibility of our material
Acknowledgments
This work was supported by grants from the following agencies: NIH R01AR057837 (NIAMS), NIH R01DE021468 (NIDCR), DOD W81XWH-10-1-0966 (PRORP), W81XWH-10-200-10 (Airlift Research Foundation), W81XWH-11-2-0168-P4 (Alliance of NanoHealth) and Wallace H. Coulter Foundation. We would also like to thank Anthony Behn for his assistance in mechanical testing of samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marra KG, Szem JW, Kumta PN, DiMilla PA, Weiss LE. In vitro analysis of biodegradable polymer blend/hydroxyapatite composites for bone tissue engineering. J Biomed Mater Res. 1999;47(3):324–335. doi: 10.1002/(sici)1097-4636(19991205)47:3<324::aid-jbm6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 2.Kavlock KD, Pechar TW, Hollinger JO, Guelcher SA, Goldstein AS. Synthesis and characterization of segmented poly(esterurethane urea) elastomers for bone tissue engineering. Acta Biomater. 2007;3(4):475–484. doi: 10.1016/j.actbio.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J, Webb AR, Ameer GA. Novel Citric Acid-Based Biodegradable Elastomers for Tissue Engineering. Adv Mater. 2004;16(6):511–516. [Google Scholar]
- 4.Ryszkowska JL, Auguścik M, Sheikh A, Boccaccini AR. Biodegradable polyurethane composite scaffolds containing Bioglass® for bone tissue engineering. Compos Sci Technol. 2010;70(13):1894–1908. [Google Scholar]
- 5.Qiu H, Yang J, Kodali P, Koh J, Ameer GA. A citric acid-based hydroxyapatite composite for orthopedic implants. Biomaterials. 2006;27(34):5845–5854. doi: 10.1016/j.biomaterials.2006.07.042. [DOI] [PubMed] [Google Scholar]
- 6.Bennett S, Connolly K, Lee DR, Jiang Y, Buck D, Hollinger JO, Gruskin EA. Initial biocompatibility studies of a novel degradable polymeric bone substitute that hardens in situ. Bone. 1996;19(1, Supplement 1):S101–S107. doi: 10.1016/s8756-3282(96)00130-5. [DOI] [PubMed] [Google Scholar]
- 7.Dumas JE, Davis T, Holt GE, Yoshii T, Perrien DS, Nyman JS, Boyce T, Guelcher SA. Synthesis, characterization, and remodeling of weight-bearing allograft bone/polyurethane composites in the rabbit. Acta Biomater. 2010;6(7):2394–2406. doi: 10.1016/j.actbio.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 8.Adhikari R, Gunatillake PA, Griffiths I, Tatai L, Wickramaratna M, Houshyar S, Moore T, Mayadunne RTM, Field J, McGee M, Carbone T. Biodegradable injectable polyurethanes: Synthesis and evaluation for orthopaedic applications. Biomaterials. 2008;29(28):3762–3770. doi: 10.1016/j.biomaterials.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Guelcher SA, Srinivasan A, Dumas JE, Didier JE, McBride S, Hollinger JO. Synthesis, mechanical properties, biocompatibility, and biodegradation of polyurethane networks from lysine polyisocyanates. Biomaterials. 2008;29(12):1762–1775. doi: 10.1016/j.biomaterials.2007.12.046. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J-Y, Doll BA, Beckman EJ, Hollinger JO. Three-Dimensional Biocompatible Ascorbic Acid-Containing Scaffold for Bone Tissue Engineering. Tissue Eng. 2003;9(6):1143–1157. doi: 10.1089/10763270360728053. [DOI] [PubMed] [Google Scholar]
- 11.Chen Q-Z, Quinn JMW, Thouas GA, Zhou X, Komesaroff PA. Bone-Like Elastomer-Toughened Scaffolds with Degradability Kinetics Matching Healing Rates of Injured Bone. Adv Eng Mater. 2010;12(11):B642–B648. [Google Scholar]
- 12.Song J, Xu J, Filion T, Saiz E, Tomsia AP, Lian JB, Stein GS, Ayers DC, Bertozzi CR. Elastomeric high-mineral content hydrogel-hydroxyapatite composites for orthopedic applications. J Biomed Mater Res A. 2009;89A(4):1098–1107. doi: 10.1002/jbm.a.32110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han DK, Hubbell JA. Lactide-Based Poly(ethylene glycol) Polymer Networks for Scaffolds in Tissue Engineering. Macromolecules. 1996;29(15):5233–5235. [Google Scholar]
- 14.Zhao F, Grayson WL, Ma T, Bunnell B, Lu WW. Effects of hydroxyapatite in 3-D chitosan-gelatin polymer network on human mesenchymal stem cell construct development. Biomaterials. 2006;27(9):1859–1867. doi: 10.1016/j.biomaterials.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Jagur-Grodzinski J. Biomedical application of functional polymers. React Funct Polym. 1999;39:99–138. [Google Scholar]
- 16.Rezwan K, Chen QZ, Blaker JJ, Boccaccini AR. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials. 2006;27(18):3413–3431. doi: 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 17.Pitt GG, Gratzl MM, Kimmel GL, Surles J, Sohindler A. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (ε-caprolactone), and their copolymers in vivo. Biomaterials. 1981;2(4):215–220. doi: 10.1016/0142-9612(81)90060-0. [DOI] [PubMed] [Google Scholar]
- 18.Hu Y, Winn SR, Krajbich I, Hollinger JO. Porous polymer scaffolds surface-modified with arginine-glycine-aspartic acid enhance bone cell attachment and differentiation in vitro. J Biomed Mater Res A. 2003;64A(3):583–590. doi: 10.1002/jbm.a.10438. [DOI] [PubMed] [Google Scholar]
- 19.Santerre JP, Woodhouse K, Laroche G, Labow RS. Understanding the biodegradation of polyurethanes: From classical implants to tissue engineering materials. Biomaterials. 2005;26(35):7457–7470. doi: 10.1016/j.biomaterials.2005.05.079. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Doll BA, Beckman EJ, Hollinger JO. A biodegradable polyurethane-ascorbic acid scaffold for bone tissue engineering. J Biomed Mater Res A. 2003;67A(2):389–400. doi: 10.1002/jbm.a.10015. [DOI] [PubMed] [Google Scholar]
- 21.Grad S, Kupcsik L, Gorna K, Gogolewski S, Alini M. The use of biodegradable polyurethane scaffolds for cartilage tissue engineering: potential and limitations. Biomaterials. 2003;24(28):5163–5171. doi: 10.1016/s0142-9612(03)00462-9. [DOI] [PubMed] [Google Scholar]
- 22.Peroglio M, Gremillard L, Eglin D, Lezuo P, Alini M, Chevalier J. Evaluation of a new press-fit in situ setting composite porous scaffold for cancellous bone repair: Towards a “surgeon-friendly” bone filler? Acta Biomater. 2010;6(9):3808–3812. doi: 10.1016/j.actbio.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Guelcher SA, Dumas JE, Boyce TM. Office UP, editor. Weight-Bearing Polyurethane Composites and Methods Theoreof. United States: Osteotech Inc., Vanderbilt University; 2010. [Google Scholar]
- 24.Dumas JE, Zienkiewicz K, Tanner SA, Prieto EM, Bhattacharyya S, Guelcher SA. Synthesis and Characterization of an Injectable Allograft Bone/Polymer Composite Bone Void Filler with Tunable Mechanical Properties. Tissue Eng Part A. 2010;16(8):2505–2518. doi: 10.1089/ten.TEA.2009.0672. [DOI] [PubMed] [Google Scholar]
- 25.Rakmae S, Ruksakulpiwat Y, Sutapun W, Suppakarn N. Physical properties and cytotoxicity of surface-modified bovine bone-based hydroxyapatite/poly(lactic acid) composites. J Compos Mater. 2010;45(12):1259–1269. [Google Scholar]
- 26.Hong H, Wei J, Yuan Y, Chen F-P, Wang J, Qu X, Liu C-S. A novel composite coupled hardness with flexibleness—polylactic acid toughen with thermoplastic polyurethane. J Appl Polym Sci. 2011;121(2):855–861. [Google Scholar]
- 27.Zhao DS, Moritz N, Laurila P, Mattila R, Lassila LVJ, Strandberg N, Mäntylä T, Vallittu PK, Aro HT. Development of a multi-component fiber-reinforced composite implant for load-sharing conditions. Med Eng Phys. 2009;31(4):461–469. doi: 10.1016/j.medengphy.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Jaggi HS, Kumar Y, Satapathy BK, Ray AR, Patnaik A. Analytical interpretations of structural and mechanical response of high density polyethylene/hydroxyapatite bio-composites. Mater Des. 2012;36:757–766. [Google Scholar]
- 29.Ahmed I, Jones I, Parsons A, Bernard J, Farmer J, Scotchford C, Walker G, Rudd C. Composites for bone repair: phosphate glass fibre reinforced PLA with varying fibre architecture. J Mater Sci Mater Med. 2011;22(8):1825–1834. doi: 10.1007/s10856-011-4361-0. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Zheng S. Polyurethane Networks Nanoreinforced by Polyhedral Oligomeric Silsesquioxane. Macromol Rapid Commun. 2005;26(3):196–200. [Google Scholar]
- 31.Zlatanić A, Petrović ZS, Dušek K. Structure and Properties of Triolein-Based Polyurethane Networks. Biomacromolecules. 2002;3(5):1048–1056. doi: 10.1021/bm020046f. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Ping P, Chen X, Jing X. Shape memory effect of poly(L-lactide)- based polyurethanes with different hard segments. Polym Int. 2007;56(7):840–846. [Google Scholar]
- 33.Flory PJ. Principles of Polymer Chemistry. Ithaca, NY: Cornell University Press; 1953. [Google Scholar]
- 34.He X, Jabbari E. Material Properties and Cytocompatibility of Injectable MMP Degradable Poly(lactide ethylene oxide fumarate) Hydrogel as a Carrier for Marrow Stromal Cells. Biomacromolecules. 2007;8(3):780–792. doi: 10.1021/bm060671a. [DOI] [PubMed] [Google Scholar]
- 35.Izunobi JU, Higginbotham CL. Polymer Molecular Weight Analysis by 1H NMR Spectroscopy. J Chem Educ. 2011;88(8):1098–1104. [Google Scholar]
- 36.Gyawali D, Nair P, Zhang Y, Tran RT, Zhang C, Samchukov M, Makarov M, Kim HKW, Yang J. Citric acid-derived in situ crosslinkable biodegradable polymers for cell delivery. Biomaterials. 2010;31(34):9092–9105. doi: 10.1016/j.biomaterials.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maniara G, Rajamoorthi K, Rajan S, Stockton GW. Method Performance and Validation for Quantitative Analysis by 1H and 31P NMR Spectroscopy. Applications to Analytical Standards and Agricultural Chemicals. Anal Chem. 1998;70(23):4921–4928. doi: 10.1021/ac980573i. [DOI] [PubMed] [Google Scholar]
- 38.Grobelny J. N.M.R study of maleate (cis)-fumarate (trans) isomerism in unsaturated polyesters and related compounds. Polymer. 1995;36(22):4215–4222. [Google Scholar]
- 39.Woo SI, Kim BO, Jun HS, Chang HN. Polymerization of aqueous lactic acid to prepare high molecular weight poly(lactic acid) by chain-extending with hexamethylene diisocyanate. Polym Bull. 1995;35(4):415–421. [Google Scholar]
- 40.Tian H, Deng C, Lin H, Sun J, Deng M, Chen X, Jing X. Biodegradable cationic PEG-PEI-PBLG hyperbranched block copolymer: synthesis and micelle characterization. Biomaterials. 2005;26(20):4209–4217. doi: 10.1016/j.biomaterials.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Kickelbick G. Concepts for the incorporation of inorganic building blocks into organic polymers on a nanoscale. Prog Polym Sci. 2003;28(1):83–114. [Google Scholar]
- 42.Tampieri A, Celotti G, Landi E, Sandri M, Roveri N, Falini G. Biologically inspired synthesis of bone-like composite: Self-assembled collagen fibers/hydroxyapatite nanocrystals. J Biomed Mater Res A. 2003;67A(2):618–625. doi: 10.1002/jbm.a.10039. [DOI] [PubMed] [Google Scholar]
- 43.Ellis J, Jackson AM, Scott BP, Wilson AD. Adhesion of carboxylate cements to hydroxyapatite: III. Adsorption of poly(alkenoic acids) Biomaterials. 1990;11(6):379–384. doi: 10.1016/0142-9612(90)90090-d. [DOI] [PubMed] [Google Scholar]
- 44.Shen J-W, Wu T, Wang Q, Pan H-H. Molecular simulation of protein adsorption and desorption on hydroxyapatite surfaces. Biomaterials. 2008;29(5):513–532. doi: 10.1016/j.biomaterials.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 45.Liu Q, de Wijn JR, van Blitterswijk CA. A study on the grafting reaction of isocyanates with hydroxyapatite particles. J Biomed Mater Res. 1998;40(3):358–364. doi: 10.1002/(sici)1097-4636(19980605)40:3<358::aid-jbm3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 46.Dong GC, Sun JS, Yao CH, Jiang GJ, Huang CW, Lin FH. A study on grafting and characterization of HMDI-modified calcium hydrogenphosphate. Biomaterials. 2001;22(23):3179–3189. doi: 10.1016/s0142-9612(01)00070-9. [DOI] [PubMed] [Google Scholar]
- 47.Boesel LF, Reis RL. A review on the polymer properties of Hydrophilic, partially Degradable and Bioactive acrylic Cements (HDBC) Prog Polym Sci. 2008;33(2):180–190. [Google Scholar]
- 48.Bakar MSA, Cheang P, Khor KA. Tensile properties and microstructural analysis of spheroidized hydroxyapatite-poly(etheretherketone) biocomposites. Mater Sci Eng A. 2003;345(1–2):55–63. [Google Scholar]
- 49.Bonzani IC, Adhikari R, Houshyar S, Mayadunne R, Gunatillake P, Stevens MM. Synthesis of two-component injectable polyurethanes for bone tissue engineering. Biomaterials. 2007;28(3):423–433. doi: 10.1016/j.biomaterials.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 50.Yoshii T, Dumas JE, Okawa A, Spengler DM, Guelcher SA. Synthesis, characterization of calcium phosphates/polyurethane composites for weight-bearing implants. J Biomed Mater Res B Appl Biomater. 2012;100B(1):32–40. doi: 10.1002/jbm.b.31917. [DOI] [PubMed] [Google Scholar]
- 51.Boxberger JI, Adams DJ, Diaz-Doran V, Akkarapaka NB, Kolb ED. Radius Fracture Repair Using Volumetrically Expanding Polyurethane Bone Cement. J Hand Surg Am. 2011;36(8):1294–1302. doi: 10.1016/j.jhsa.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 52.An YH. In: Mechanical properties of bone. An YH, Draughn RA, editors. Boca Raton, FL: CRC Press; 2000. pp. 45–46. [Google Scholar]
- 53.Bodde EWH, Habraken WJEM, Mikos AG, Spauwen PHM, Jansen JA. Effect of Polymer Molecular Weight on the Bone Biological Activity of Biodegradable Polymer/Calcium Phosphate Cement Composites. Tissue Eng Part A. 2009;15(10):3183–3191. doi: 10.1089/ten.TEA.2008.0694. [DOI] [PubMed] [Google Scholar]
- 54.Qin Y-X, Rubin CT, McLeod KJ. Nonlinear dependence of loading intensity and cycle number in the maintenance of bone mass and morphology. J Orthop Res. 1998;16(4):482–489. doi: 10.1002/jor.1100160414. [DOI] [PubMed] [Google Scholar]
- 55.Rubin C, Lanyon L. Regulation of bone mass by mechanical strain magnitude. Calcif Tissue Int. 1985;37(4):411–417. doi: 10.1007/BF02553711. [DOI] [PubMed] [Google Scholar]
- 56.Guelcher SA, Srinivasan A, Hafeman A, Gallagher K, Doctor J, Khetan S, McBride S, Hollinger J. Synthesis, In Vitro Degradation, and Mechanical Properties of Two-Component Poly(Ester Urethane)Urea Scaffolds: Effects of Water and Polyol Composition. Tissue Eng. 2007;13(9):2321–2333. doi: 10.1089/ten.2006.0395. [DOI] [PubMed] [Google Scholar]
- 57.Rojbani H, Nyan M, Ohya K, Kasugai S. Evaluation of the osteoconductivity of α-tricalcium phosphate, β-tricalcium phosphate, and hydroxyapatite combined with or without simvastatin in rat calvarial defect. J Biomed Mater Res A. 2011;98A(4):488–498. doi: 10.1002/jbm.a.33117. [DOI] [PubMed] [Google Scholar]
- 58.Neffe AT, Loebus A, Zaupa A, Stoetzel C, Müller FA, Lendlein A. Gelatin functionalization with tyrosine derived moieties to increase the interaction with hydroxyapatite fillers. Acta Biomater. 2011;7(4):1693–1701. doi: 10.1016/j.actbio.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 59.Bashur CA, Shaffer RD, Dahlgren LA, Guelcher SA, Goldstein AS. Effect of Fiber Diameter and Alignment of Electrospun Polyurethane Meshes on Mesenchymal Progenitor Cells. Tissue Eng Part A. 2009;15(9):2435–2445. doi: 10.1089/ten.tea.2008.0295. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z, Cheng S, Li Z, Xu K, Chen G-Q. Synthesis, Characterization and Cell Compatibility of Novel Poly(ester urethane)s Based on Poly(3-hydroxybutyrate-co-4-hydroxybutyrate) and Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) Prepared by Melting Polymerization. J Biomater Sci Polym Ed. 2009;20(10):1451–1471. doi: 10.1163/092050609X12457419007621. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Tran RT, Gyawali D, Yang J. Development of Photocrosslinkable Urethane-Doped Polyester Elastomers for Soft Tissue Engineering. Int J Biomater Res Eng. 2011;1(1):18–31. doi: 10.4018/ijbre.2011010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garty S, Kimelman-Bleich N, Hayouka Z, Cohn D, Friedler A, Pelled G, Gazit D. Peptide-Modified “Smart” Hydrogels and Genetically Engineered Stem Cells for Skeletal Tissue Engineering. Biomacromolecules. 2010;11(6):1516–1526. doi: 10.1021/bm100157s. [DOI] [PubMed] [Google Scholar]
- 63.Lavanant L, Pullin B, Hubbell JA, Klok H-A. A Facile Strategy for the Modification of Polyethylene Substrates with Non-Fouling, Bioactive Poly(poly(ethylene glycol) methacrylate) Brushes. Macromol Biosci. 2009;10(1):101–108. doi: 10.1002/mabi.200900205. [DOI] [PubMed] [Google Scholar]
- 64.Padavan DT, Hamilton AM, Millon LE, Boughner DR, Wan W. Synthesis, characterization and in vitro cell compatibility study of a poly(amic acid) graft/cross-linked poly(vinyl alcohol) hydrogel. Acta Biomater. 2011;7(1):258–267. doi: 10.1016/j.actbio.2010.07.038. [DOI] [PubMed] [Google Scholar]