

Abstract
Early brain injury (EBI) during the first 72 h after subarachnoid hemorrhage (SAH) is an important determinant of clinical outcome. A hallmark of EBI, global cerebral ischemia, occurs within seconds of SAH and is thought to be related to increased intracranial pressure (ICP). We tested the hypothesis that ICP elevation and cortical hypoperfusion are the result of physical blockade of cerebrospinal fluid (CSF) flow pathways by cisternalmicrothrombi. In mice subjected to SAH, we measured cortical blood volume (CBV) using optical imaging, ICP using pressure transducers, and patency of CSF flow pathways using intracisternally injected tracer dye. We then assessed the effects of intracisternal injection of recombinant tissue plasminogen activator (tPA). ICP rose immediately after SAH and remained elevated for 24 h. This was accompanied by a decrease in CBV and impaired dye movement. Intracisternal administration of tPA immediately after SAH lowered ICP, increased CBV, and partially restored CSF flow at 24 h after SAH. Lowering ICP without tPA, by draining CSF, improved CBVat 1 h, but not 24 h after SAH. These findings suggest that blockade of CSF flow by microthrombi contributes to the early decline in cortical perfusion in an ICP-dependent and ICP-independent manner and that intracisternal tPA may reduce EBI and improve outcome after SAH.
Keywords: Early brain injury, Global ischemia, Intracranial pressure, Subarachnoid hemorrhage, Tissue plasminogen activator, Cerebral blood flow
Introduction
Cerebrospinal fluid (CSF) resides within the subarachnoid space and plays critical roles in both the regulation of intracranial pressure (ICP) and maintenance of the brain’s extracellular environment [1]. Blood leakage into that space, as occurs during subarachnoid hemorrhage (SAH), causes a multitude of complications that contribute to a 50 % mortality and even higher morbidity among survivors [2]. These complications are biphasic in nature, with some occurring immediately after ictus and others presenting several days later. The complications occurring within the first 72 h after SAH are collectively referred to as early brain injury (EBI) [3]. Because early complications are powerful predictors of delayed complications [4] and overall outcome [5], EBI is becoming a major focus of research aimed at improving the outcomes of SAH patients.
EBI describes a number of pathological processes, which are likely interrelated and thus are difficult to study in isolation. Broad categories of EBI include blood brain barrier disruption [6], altered metabolic homeostasis [7], cortical spreading depression [8], and neuronal cell death [9, 10]. Global cerebral hypoperfusion, which occurs within the first minutes of SAH, is thought to be one of the primary contributors to EBI and is a strong predictor of outcome [5]. Closely associated with hypoperfusion is the rapid elevation of ICP that occurs at the time of hemorrhage [11] and, depending on severity, can remain elevated from hours to days, or even indefinitely [12] following SAH. While elevation of ICP is known to impinge on cerebral perfusion, evidence suggests that hypoperfusion after SAH is multifactorial with both ICPdependent and ICP-independent components [13].A common pathology linking these components has not been established.
CSF moves out of the subarachnoid space and into the brain parenchyma alongside penetrating arterioles within the paravascular space [14]. These continuous extensions of the Virchow-Robin space permit movement of CSF through the neuropil and are thought to be integral to extracellular homeostasis. When blood flows out of the vasculature and into these spaces during SAH, the coagulation cascade is initiated, leading to clot formation [15]. This prevents additional bleeding from occurring but likely also blocks these CSF flow pathways. Blockade of CSF flow could contribute to the increase in ICP and prevention of normal CSF function. The exact nature of impaired CSF flow within these spaces after SAH and its role in early cortical hypoperfusion is unknown.
The current study was designed to test the hypothesis that early cerebral hypoperfusion is due in part to impaired CSF flow by microthrombi in the subarachnoid and paravascular space and that intracisternal administration of tPA restores CSF flow and cortical perfusion in the early phase of SAH. We also sought to determine the relative contribution of ICP to cerebral hypoperfusion.
As a surrogate for tissue perfusion, we used optical microangiography (OMAG), an optical coherence tomography (OCT)-based technique, to quantify cortical blood volume (CBV) in perfused cortical vessels noninvasively through an intact skull [16]. OMAG also allows us to obtain serial measurements of CBV in the same animal over time. The three-dimensional volumetric data produced by OMAG contains blood volume information fromactively perfused vessels both on the cortical surface and several hundred microns below the surface and thus is referred to as cortical perfusion. We have previously validated OMAG measurements of perfusion in mouse brain using [14C]-iodoantipyrine autoradiography [17] and have used this approach previously in studies of cerebral ischemia [17] and traumatic brain injury [18].
Materials and Methods
Animals
Experimental animal procedures performed in this study conform to the guidelines of the US National Institutes of Health, and the animal protocol was approved by the Institutional Animal Care and Use Committee of Oregon Health and Science University, Portland, OR, USA. Mice were housed in a room with a light:dark every 12 h and given free access to standard rodent chow and water. Studies were performed on 8- to 12-week-old wild-type (WT) C57BL/6J male mice obtained from Jackson Laboratories.
Induction of SAH
SAH was induced in mice using the endovascular perforation technique adapted from Sozen et al. [19]. Briefly, mice were anesthetized with isoflurane (1.5–2 % in O2-enriched air by face mask) and maintained at 37±0.5 °C rectal temperature using warm water pads. Two small laser Doppler probes (Moor Instruments) were affixed bilaterally over the parietal bones to monitor cerebrocortical perfusion and confirm vascular rupture. To induce hemorrhage, a nylon suture (5–0) was introduced into the internal carotid artery via the external carotid artery and advanced ~10 mm beyond the carotid bifurcation and into the Circle of Willis. The suture was then advanced slightly further to induce a hemorrhage and then removed. The common carotid artery wasmaintained patent at all times to maximize flow to the ruptured artery immediately following arterial perforation. Mortality throughout the study was <10 % and most often occurred immediately following SAH induction. Only mice who survived the initial surgery were included in this study. In sham-operated animals, the suture was advanced into the internal carotid artery and then removed without arterial perforation.
Optical Microangiography (OMAG)
To monitor changes in CBV in vivo, we used OCT-based OMAG [16]. Briefly, at baseline and after SAH or sham surgery, mice were anesthetized with isoflurane (1.5–2 % in O2-enriched air by face mask). The skin over the skull was reflected, the cortex was illuminated through the skull with a 1,310-nm light source, and the resulting backscattered and reference light was detected to produce spectral interferograms. Volumetric imaging data were collected by scanning the probe beam through a 1,000×500×512 voxel cube, representing 2.5×2.5×2 mm3 (x-y-z) of tissue. After scanning, the skin over the skull was closed, and the animal was allowed to recover. CBV images within the scanned tissue volume were rendered in 3-D (AMIRA, Visual Imaging GmbH), then thresholded and analyzed for mean pixel intensity changes over time using Image-J software. The 72-h time course study presented in Fig. 1 is a combination of six scans covering the entire surface of the mouse cortex. Subsequent time-sensitive studies focused on a single scan over the MCA territory ipsilateral to the hemorrhage site.
Fig. 1.
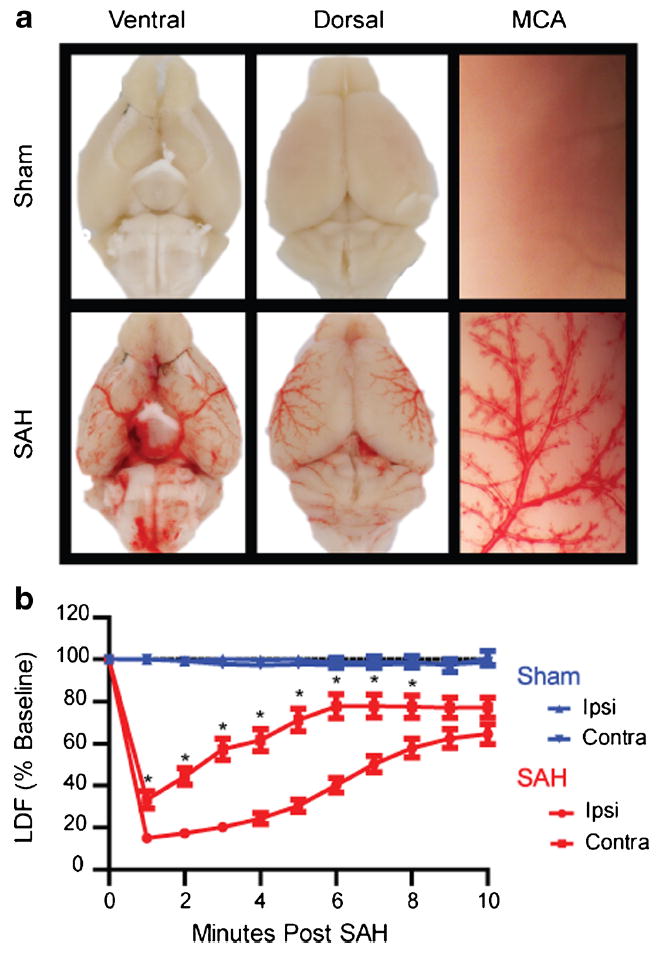
Validation of endovascular puncture model. a Mouse brains were perfused with heparinized saline to clear any intravascular blood and imaged 30 min after endovascular puncture. SAH mice show a wide distribution of extravasated blood throughout ventral (lower left) and dorsal (lower middle) brain surfaces. The magnified image of a perfused MCA (lower right) shows movement of extravasated blood along the paravascular pathway. b CBF measured by laser Doppler in both the ipsilateral (ipsi) and contralateral (contra) MCA territories of sham (blue) and SAH (red) mice. SAH mice (n=31) experience a rapid drop in CBF following puncture. Contralateral CBF recovers more rapidly than ipsilateral CBF, but both reach 65–75 % of baseline by 10 min (*p<0.05 contralateral different from ipsilateral)
Experimental Manipulation of ICP
Following SAH, mice were mounted on a stereotaxic frame, and the skull was exposed. For simultaneous measurement and manipulation of ICP, burr holes were placed bilaterally 0.5 mm rostral to bregma and 1 mm lateral to midline over the lateral ventricles. The dura mater was removed, and custommade 21-gauge steel guide tubes with dummy cannulas were placed 1.5 mm below the skull and sealed using dental cement and cyanoacrylate glue. One tube served as the experimental cannula for drainage, and the opposite tube served as a port for measurement of ICP using a closed system pressure transducer connected to a pressure monitor (Hewlett Packard). The dummy was removed (0-min time) from the experimental tube in the “open” group, and CSF was allowed to drain freely. In the “closed” group, the dummy cannula remained in place. After ICP measurements, the PE catheter was removed from the measurement tube, and the dummy was replaced to prevent further drainage of CSF. The skin was replaced around the cannulas, and the animal was allowed to recover. Twentyfour hours following SAH, the animals were again anesthetized, and ICP was measured again. Cannula flow in the open group was assessed at 24 h, and only animals with freely flowing cannulas at 24 h were included in the study. For studies looking at manipulation of ICP and its effect on cortical perfusion, a single tube was placed in the left lateral ventricle as the experimental cannula, and the right hemisphere was left untouched for OMAG image acquisition.
CSF Flow and Hemorrhage Assessment
At 30 min (Fig. 4) or 24 h (Fig. 5) after SAH or sham surgery, animals were anesthetized using isoflurane (1.5–2 %) and placed on a stereotaxic apparatus. Mice received 10 μl of dye (0.4 % Evan’s Blue (Fisher) and 0.1 % bovine serum albumin (Fisher) in saline) injected into the cisternamagna at a rate of 2 μl/min over 5 min. The dye was allowed to circulate for 30 min after injection. The animals were then deeply anesthetized and perfused in cold heparinized saline before the brains were removed and taken for imaging. Images of the ventral surface were obtained using a Nikon digital camera and capture software (ACT-1 Nikon). The images were normalized to background, and the red channel or blue channel was isolated and thresholded for analysis of blood or dye present in the basal cisterns, respectively. Dye movement was used as a surrogate for CSF flow, which was assessed by a blinded investigator in two ways. The first was a categorical six-point distribution score that described the distribution of tracer over the ventral surface of the brain. To generate the score, the ventral surface image was divided into six regions and given a score of 0 for no dye present or 1 for dye present in each region. This was then summed (Fig. 5b). The secondmethodwas a quantification of total pixel area that reached beyond the brainstem (the site of injection and dye pooling) and into the basal cisterns. Blood in the basal cisterns was also quantified by a blinded investigator as total pixel area beyond the brainstem. For immunolabeling of fibrin strand deposition, mice were deeply anesthetized with isoflurane (5 %) and transcardialy perfused with cold heparinized saline, followed by 4 % paraformaldehyde and postfixed overnight. The whole brain was removed and blocked in 4%normal goat serum for 3 h at room temperature, then incubated in primary antibody overnight at 4 °C (chicken anti-mouse fibrinogen, 1:200; Immunology Consultants Laboratory). Secondary detection was with Alexa Fluor® 594 conjugated goat antichicken secondary antibody (1:800, Invitrogen) incubated for 3 h at roomtemperature. After washing, whole brains were imaged using a fluorescent dissecting microscope (Leica Microsystems) equipped with camera and imaging software (LAS EZ, Leica).
Fig. 4.
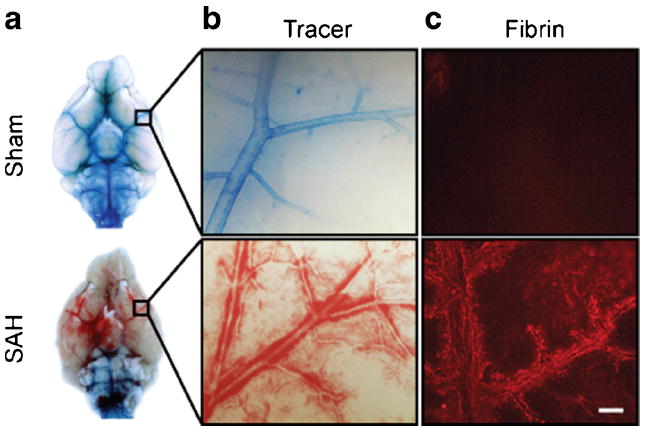
CSF flow is blocked immediately following SAH. a Images were taken of the ventral surface of the perfused mouse brain 1 h after SAH or sham surgery and 30 min after intracisternal tracer dye injection. In sham mice, tracer dye distributes throughout the basal cistern and paravascular routes. In SAH mice, tracer dye pools around the brainstem and does not enter paravascular routes. b High-magnification images of MCA branches showing tracer dyemovement into paravascular spaces. In sham animals, tracer dye moves along MCA branches and out to penetrating vessels. In SAH animals, this space is occupied by blood, and no tracer dye is present. c Fibrinogen staining showing fibrin strand formation in paravascular spaces following SAH but not sham surgery. Scale bar=100 μm
Fig. 5.
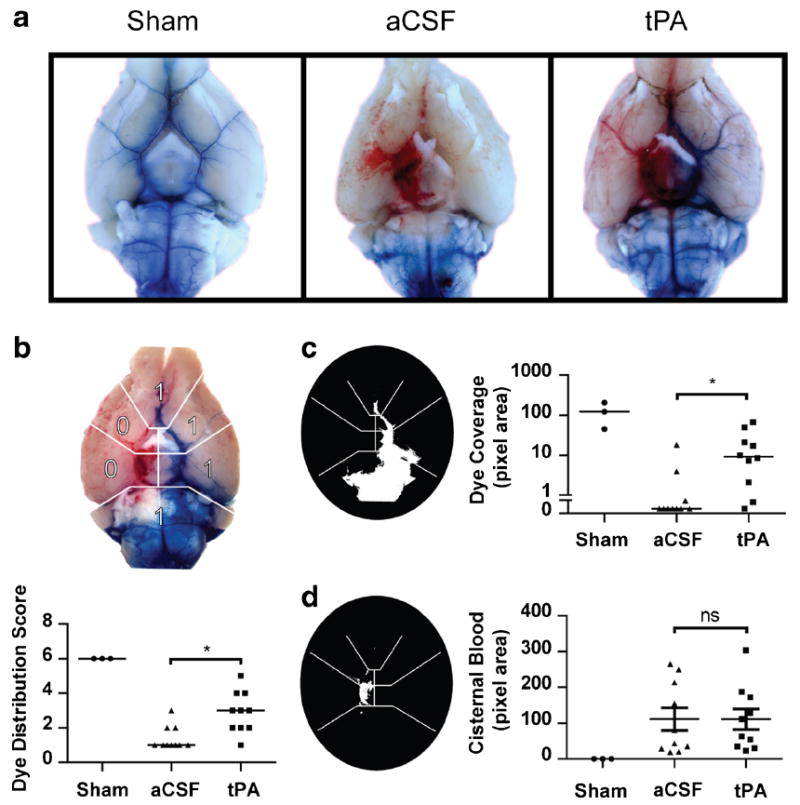
Intracisternal tPA can restore CSF flow within 24 h. a Images were taken of the ventral surface of the perfused mouse brain 24 h after SAH or sham surgery and 30 min after intracisternal dye injection. Dye moves freely in sham animals but not aCSF-treated SAH animals. Intracisternally injected tPA 1 h after SAH partially restores CSF flow to the basal cistern and paravascular routes by 24 h. b To assess effect of tPA dye movement within the basal cisterns, the blue channel was isolated and thresholded for categorical quantification of dye distribution over the ventral surface of the brain and c quantification of pixel area coverage of dye beyond the brainstem. Data are expressed as medians. d To assess the effect of tPA on hemorrhage size, the red channel was isolated and thresholded for quantification of pixel area of blood beyond the brainstem. Sham (n=3), SAH aCSF (n=10), SAH tPA (n=10), *P<0.05
Intracisternal Injections of tPA and ICP Measurement
Following SAH or sham surgery, animals were mounted on a stereotaxic frame under isoflurane anesthesia (1.5–2 %). An incision was made above the occipital bone, and the posterior neck musculature was partially reflected to reveal the atlantooccipital membrane. A custom-made 30-gauge cannula connected to PE-10 tubing (B-D) was then inserted into the cisterna magna and affixed to the skull using dental cement (Lang Dental) and cyanoacrylate glue. ICP was assessed through the cannula using a pressure transducer connected to a pressure monitor (Hewlett Packard). After the monitor was connected, ICP was recorded after 15 min of equilibration. Following ICP measurements, 10 μl of CSF was drained out of the cisterna magna to accommodate subsequent infusion of 10-μl solution containing either 15-μg recombinant tissue plasminogen activator (tPA) in sterile water (Cathflo, Alteplase, Genentech), artificial CSF (aCSF) (containing NaCl 73.05 mg/ml, KCl 1.86 mg/ml, MgCl2-6H2O 2.03 mg/ml, CaCl2-2H2O 2.94 mg/ml, NaH2PO4-H2O 1.73 mg/ml, NaHCO3 21 mg/ml, and glucose 4.5 mg/ml) or vehicle (l-arginine 52.5 mg/ml, H3PO4 15 mg/ml, and polysorbate 80 0.17 mg/ml in sterile water). Intracisternal injection was performed using an infusion pump (Harvard Apparatus) at a rate of 2 μl/min for 5 min. Sham-operated animals also received tPA injections. After injection, the tubing was heat-sealed to prevent backflow, the muscle and skin were sutured, and the animal was allowed to recover for 24 h. After 24 h, this same cannula was used for the final ICP measurement and dye injection.
Statistics
Comparison of CBVand ICP changes between treatment groups and over time was made using a two-way within-between ANOVA. Comparison of CBVand ICP changes over time were made using a one-way repeated measure ANOVA. Comparison of cisternal blood between groups was made using one-way ANOVA. All post hoc comparisons were made using Bonferroni’s correction. Comparison of dye region data between groups was made using a Kruskal-Wallis test with Dunn’s post hoc comparison. All data analysis was performed using Prism 5.0 (GraphPad). Data are expressed as mean±SEM unless otherwise noted. Statistical significance was set at P<0.05.
Results
Blood Distribution in the Endovascular Perforation Model
By perfusing the mice with saline immediately after the hemorrhage, we were able to observe where extravasated blood from the endovascular puncture model distributes immediately following induction of the hemorrhage. Within 30 min of the endovascular puncture, blood has moved into the basal cistern and along paravascular spaces on the outside of the middle cerebral artery (MCA) (Fig. 1a).We confirmed induction of the hemorrhage using CBF measured by laser Doppler probes placed against the skull over the MCA territory during the surgery. We found that at the time of the hemorrhage, an abrupt drop in CBF occurs bilaterally and slowly returns to ~65–75 % of baseline by 10 min. The contralateral side experiences a less severe drop than the ipsilateral side to the hemorrhage (P<0.05 at 1–8 min, n=31), but CBF values ultimately converge by the end of the 10-min period (Fig. 1b).
Cortical Perfusion is Impaired Following Subarachnoid Hemorrhage
To study changes in cortical perfusion over days after SAH, we scanned a large area of the cortical surface using OMAG (larger box in Fig. 2a) at baseline before the animals were subjected to SAH or sham surgery. Following a 6-h recovery period, animals were rescanned at 6, 12, 24, and 72 h following SAH. Cortical perfusion was decreased compared with sham animals at 6 h (Fig. 2b, 73.0±3.3%vs 99.3±2.1 %, n=5 for sham and 10 for SAH), 12 h (79.1±3.1 % vs 103.4± 2.2 %), 24 h (80.0±2.5 % vs 102.2±1.4 %), and 72 h (87.8 ±3.3 % vs 102.1±3.0 %) after SAH. Cortical perfusion was reduced bilaterally across middle cerebral and anterior cerebral flow territories indicating that the effect was global (Fig. 2c). Further studies using OMAG were limited to the scan area indicated by the smaller box in Fig. 2a.
Fig. 2.
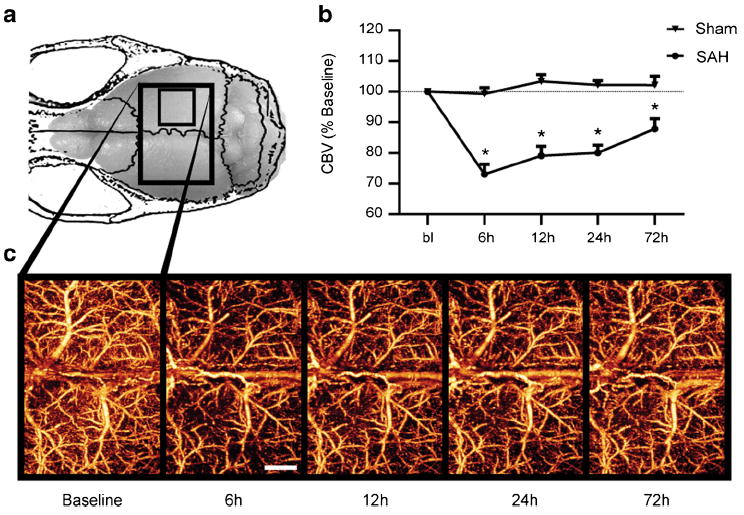
Cortical perfusion is impaired globally early after SAH. a Changes in perfused CBV were tracked in mice for several days following SAH or sham surgery using OMAG. The scan area of the dorsal cortex was 5× 7.5 mm (large box). The smaller box indicates the scan area for all subsequent studies. b Cortical perfusion was reduced in SAH animals as early as 6 h after SAH and persisting for at least 72 h. c Representative OMAG images of SAH mouse showing changes in perfused CBV over time. Decreased intensity corresponds to a reduction in perfused CBV. Values are mean±SEM. SAH (n=10), sham (n=5), *P<0.01. Scale bar= 1 mm
Cortical Perfusion Deficits are Due in Part to Elevated ICP
There is evidence to suggest that early hypoperfusion following SAH is independent of ICP [20]. To test whether ICP contributes to the hypoperfusion seen in our model, we developed single- and double-cannula methods for measuring CBVand ICP, respectively, before and after ventricular drainage. At the 1-h time point following SAH induction, we measured ICP in one lateral ventricle and drained CSF out of the other. ICP immediately decreased when the cannula was opened and CSF allowed to drain (from 34.8±1.36 to 25.4± 1.8 mm Hg, n=5, Fig. 3b). In another group of animals, CSF was drained from one cannula while OMAG scans were performed over the MCA territory on the opposing side without a cannula. Figure 3b shows that CBV was also increased from 45.7±4.9 to 66.0±7.1 % of baseline (n=5) when the cannula was opened and CSF was allowed to drain. This finding suggests that the decrease in CBV present 1 h after SAH is at least, in part, due to increased ICP.
Fig. 3.
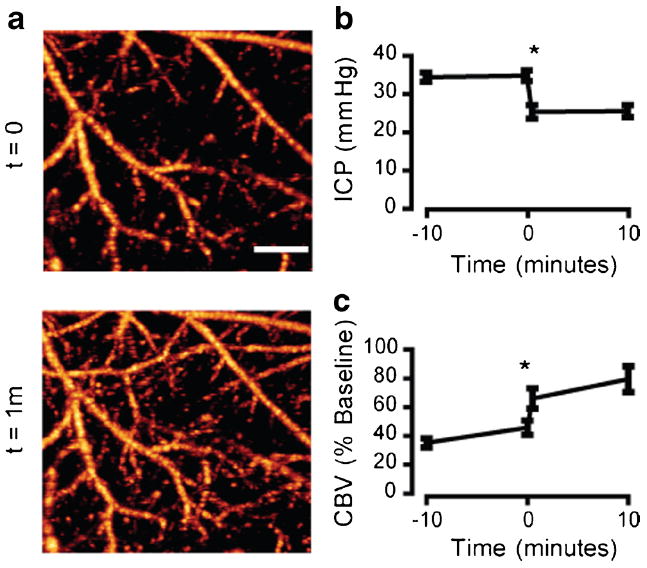
Elevated ICP contributes to the early decrease in cortical perfusion after SAH. a Representative OMAG scans of the cortex within the MCA territory 1 h after SAH. A ventricular cannula was placed on the contralateral side and opened to drain CSF, leading to an immediate improvement of cortical perfusion. b ICP measurements 1 h post-SAH. Cannula was opened to allow CSF drainage at time=0. n=5, *P<0.05. c Quantification of perfused CBV 1 h post-SAH. Cannula was opened at time=0. n=5, *P<0.05. Scale bar=500 μm
CSF Flow is Impaired Immediately Following SAH
Impaired CSF flow is a likely cause of increased ICP, so we sought to determine whether CSF flow is impaired immediately following SAH. Thirty minutes after SAH induction, we assessed CSF flow by tracking tracer dye injected into the cisterna magna. In sham-operated mice, the tracer dye moves into the basal cistern and out along paravascular routes alongside the MCA (Fig. 4a, b). In the SAH mice, tracer dye pools around the brainstem and does not enter into the basal cistern or paravascular spaces which are occupied by blood (Fig. 4a, b). The third most abundant protein in the plasma is fibrinogen, which is rapidly converted into insoluble fibrin strands upon extravasation. To determine if paravascular fibrin deposition takes place after SAH, we fixed the whole brains 30 min after SAH and labeled the surface using a fibrinogen antibody. Labeling shows fibrin strand deposition in the paravascular spaces of the MCA (Fig. 4c). This finding suggests that CSF flow is blocked immediately following SAH and that fibrin deposition is occurring in the paravascular space.
tPA Partially Restores CSF Flow Pathways Blocked by Subarachnoid Thrombi
To determine if CSF movement was impeded by microthrombi obstructing CSF flow pathways, we examined the effect of intracisternally injected tPA on tracer dye movement 24 h after SAH or sham surgery. One hour after SAH, mice received an injection of 10 μl of either a CSF or 1.5 mg/ml of tPA into the cisterna magna. Sham-operated mice also received tPA to rule out the effects of tPA unrelated to SAH. The mice were recovered until 24 h later, when all mice received injections of tracer dye into the cisterna magna (Fig. 5a). Movement of dye was assessed semiquantitatively by subdividing the ventral surface into six regions and assigning a value of 1 or 0 for the presence or absence of dye in any given region (Fig. 5b). In sham-operated mice, the dye moved freely from the cisterna magna into the basal cisterns and out along major paravascular routes (Fig. 5c, n= 3 with all three animals scoring 6, suggesting free dye movement and presence of the dye in all regions). In aCSF-injected SAHmice, the tracer dye pooled around the brainstem and did not enter into the basal cisterns or along paravascular routes, resulting in dye absence from most areas and a median score of 1, suggesting a restricted dye movement (Fig. 5c, n=10). Finally, SAH mice injected with tPA showed partial restoration of dye movement into the basal cisterns and out along paravascular routes, resulting in a median score of 3 (Fig. 5c, n=10). This observation suggests that CSF flow impairment after SAH is, in part, due to clot formation, which can be partially reversed with tPA.
tPA Reduces ICP and Improves Cortical Perfusion After SAH
Because impaired CSF flow may increase ICP, which decreases cortical perfusion, we determined if intracisternal tPA, which improves CSF flow dynamics, can also decrease ICP and increase cortical perfusion after SAH. We used OMAG to measure perfused CBVover the MCA territory at baseline and at 1 h after SAH (before tPA injection). Mice were then injected with tPA and survived for 24 h, when CBV was measured again before sacrifice (Fig. 6a). ICP was measured using a cannula inserted prior to injection at 1 h after SAH, and the measurement was repeated at 24 h after SAH. Prior to treatment (1 h after SAH), both tPA and aCSF groups had similarly elevated ICP (Fig. 6b, 28.7±3.2 and 28.5± 4.8 mm Hg in aCSF (n=5) and tPA (n=6) groups, respectively). Both groups also had equally decreased CBV (Fig. 6c, 35.0±5 % in aCSF (n=5) and 32.2±5 % in tPA group (n=6)) at 1 h after SAH prior to treatment. Twenty-four hours after SAH, tPA-treated animals had significantly higher CBV than aCSF-treated mice (Fig. 6c, 80.0±4.7 % (n=6) vs 51.7±9 %, n=5, respectively). Mice treated with tPA also had lower ICP than aCSF-treated mice (Fig. 6b, 17.5±3.0 (n=6) vs 34.8± 3.7 mm Hg n=5, respectively). Because l-arginine is required for stability of tPA in solution and may serve as a precursor for nitric oxide synthesis and subsequent vasodilation, we included an additional vehicle group containing the equivalent dosage of l-arginine. We found no differences in CBV between this vehicle group and the aCSF group 24 h after SAH (Fig. 6c (n=4)).We additionally measured ICP in three vehicle-treated animals and found no difference from aCSF-treated animals 24 h after SAH. There was no effect of tPA injections alone on ICP or CBV in sham-operated mice (Fig. 6b, c).
Fig. 6.
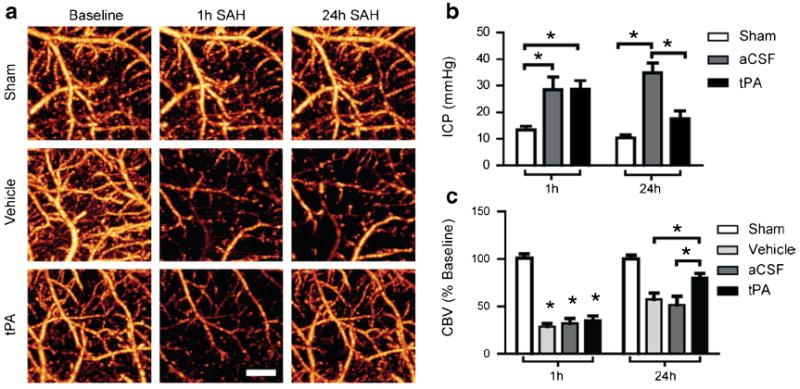
tPA reduces ICP and improves cortical perfusion 24 h after SAH. a Representative OMAG images of the cortex within the MCA territory at baseline, 1 h post-SAH/sham (pretreatment) and 24 h post-SAH/Sham (post-treatment). b ICP measured at the cisterna magna 1 h post SAH/ sham (pre-treatment) and 24 h post SAH/sham (post-treatment). Intracisternal tPA reduced ICP 24 h after SAH compared with aCSF-treated mice. c Quantification of perfused CBV 1 h post-SAH/Sham (pretreatment) and 24 h post-SAH (posttreatment). Intracisternal tPA increased perfused CBV compared with aCSF or vehicle-treated mice. Sham (n=3), SAH aCSF (n=5), SAH vehicle (n=4), SAH tPA (n=6), *P<0.05. Scale bar=500 μm
Management of ICPAlone Does Not Improve Cortical Perfusion at 24 h
Since there are other functions of CSF besides ICP regulation [1], and there is evidence of ICP independent contributors to impaired cortical hypoperfusion, we asked the question of whether the ICP reduction by tPA was sufficient in itself to explain the improvement in cortical perfusion at 24 h. To answer this question, we extended the time of CSF drainage through the ventricular cannula to the 24-h time point.We first measured ICP before CSF drainage, then immediately after, and again at 24 h after opening the cannula. As a control, we included a group with an inserted cannula that was kept closed. Prior to cannula manipulation, both “open cannula” and “closed cannula” groups had similarly elevated ICP (Fig. 7b, 37.3±3.0 vs 37.0±2.3 mm Hg in open vs closed groups, respectively, n=4 per group). Both groups also had similar reductions in perfused CBVafter SAH (Fig. 7c, 34.2± 4.2 % in open-cannula group (n=8) vs 28.3±3.3 % in closedcannula (n=6) group). Consistent with Fig. 4, immediately after cannula manipulation, ICP was decreased (from 37.3± 2.4 to 26.2±1.9 mm Hg, n=4, Fig. 7b) and CBVover the MCA territory was increased (from 31.5±4 % (n=8) to 49.6± 5.5 % (n=8) of baseline, Fig. 7c) in the open-cannula group compared with the closed-cannula group. Interestingly, at 24 h after SAH, while ICP remained lower in the open compared with the closed group (13.5±4.3 vs 26±1 mm Hg, n=4, Fig. 7b), there was no significant difference in CBV between the open- and closed-cannula groups (96.1±7.1 % (n=8) vs 79±2.1 % (n=6), Fig. 7c). This observation suggests that the improvement in cortical perfusion by tPA treatment is only partly due to reduced ICP and that improved CSF flow contributes to cortical perfusion improvement by an additional mechanism unrelated to ICP.
Fig. 7.
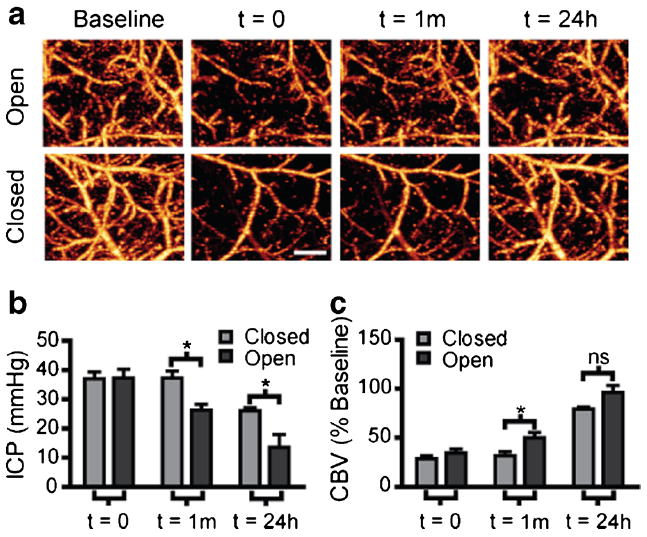
CSF drainage without tPA does not improve cortical perfusion at 24 h. a Representative OMAG images showing changes in perfused CBV. A ventricular cannula was placed on the contralateral side and opened 1 h after SAH then allowed to drain for 24 h. Scans were taken at baseline, 1 h post-SAH before cannula manipulation (time=0), immediately after cannula manipulation (time=1 m), and 24 h later (time=24 h). b Quantification of ICP 1 h post-SAH before cannula manipulation (time=0), immediately after cannula manipulation (time=1 m), and 24 h later (time= 24 h). Open cannula reduced ICP at both the 1 h and 24 h time points after SAH. Closed (n=4), open (n=4), *P<0.05. c Quantification of perfused CBV changes expressed as percent of baseline. Open cannula increased perfused CBV over closed group at 1 h but not 24 h after SAH. Closed (n=6), open (n=8), *=P<0.05, ns no significance. Scale bar=500 μm
Discussion
Review of Findings
In the current study, we demonstrated that SAH in the mouse induces rapid and sustained decrease in cortical perfusion, associated with increased ICP and impaired CSF dynamics. Furthermore, intracisternal injection of tPA reduced ICP and restored CSF flow and cortical perfusion. However, when we drained CSF through a ventricular cannula to decrease ICP without tPA treatment, we were able to improve perfusion at 1 h, but not 24 h after SAH. These findings suggest that the early decrease in cortical perfusion after SAH is, in part, due to impaired CSF flow by blood clots and that intracisternal administration of tPA improves CSF flow and cortical perfusion which cannot be fully explained by decreased ICP.
Relationship Between ICP and CBV
In our model, elevated ICP is an important contributor to early hypoperfusion within the first hours of SAH, but our results suggest that this role diminishes over 24 h. Despite that finding, restoration of CSF flow with tPA can improve cortical perfusion 24 h after SAH in a manner that must be separate or additive to its effect on ICP. The mechanism by which restoration of CSF flow can improve cortical perfusion remains to be determined, especially given that the consequences of impaired CSF function, aside from elevated ICP, are unknown.
Impaired CSF Flow and Early Brain Injury
The brain parenchyma lacks a traditional lymphatic system for the clearance of interstitial solutes. It has been long suggested that the CSF system serves that role by clearing interstitial fluid by bulk flow along preferential pathways, especially paravascular spaces and axon tracts [21]. More recently, a specific paravascular pathway has been described in detail [14]. Once CSF is produced in the choroid plexus, it moves from within the ventricles through the cisterns and out over the surface of the brain along periarterial pathways [22]. CSF then penetrates the brain parenchymal tissue via a continuous extension of the Virchow-Robin space along the penetrating arterioles known as the paravascular space [14]. Once it passes through the parenchyma and equilibrateswith the extracellular space, CSF then drains fromthe cranium through three known pathways [1]: (1) the cervical lymphatic pathway, (2) the arachnoid granulations, and finally, (3) across postcapillary venules in the paravascular space. If we make the assumption that these flow pathways must be patent for the proper maintenance of extracellular homeostasis by CSF, we can infer from our findings in this study that this function is also impaired following SAH. Clinical and animal studies that have shown a buildup of metabolic waste products, decreased available glucose, and altered ionic concentrations [7, 13, 23]—all hallmarks of EBI—support this idea.We also cannot ignore that a functional and patent CSF systemis likely critical to the clearance of toxic blood products from the initial hemorrhage itself. Any or all of these perturbations of normal physiology could contribute to the early ICP-independent vascular dysfunction that is documented in both animal [11] and clinical studies [20, 23]. Further, as we gain a better understanding of the importance of CSF function, coupled with our finding that blockade of CSF flow occurs within minutes of an SAH, we are obliged to examine how disruption of normal CSF function could contribute to the wide range of pathologies collectively referred to as EBI [3].
Previous Work Studying Thrombolytic Therapy After SAH
Both thrombolytic and antithrombolytic drugs are still actively being studied for the treatment of SAH. While these would seem to be contradictory, there is rationale and evidence for both. Specifically, intrathecal thrombolytic therapy has been shown to reduce delayed vasospasm in dogs [24], nonhuman primates [25-27], and rabbits [28, 29]. However, these studies did not examine the effect of tPA on early injury after SAH and did not link the beneficial effect of tPA to CSF dynamics. Other studies showed improved CSF flow after SAH in cats by tPA but did not link these changes to early changes in cortical perfusion [30, 31]. In clinical studies, intrathecal thrombolysis has been shown to improve outcome and reduce the need for shunt placement [32-37]. In contrast to tPA, antithrombolytic therapy has been shown to increase the incidence of cerebral ischemia and worsen outcome after SAH [38]. More recent work suggests that antithrombolytic in the ultraearly stage of SAH may be beneficial in preventing rebleeding [39], yet detrimental in the later stages when clot clearance and restoration of CSF flow become more important [40]. Together, these studies are consistent with our findings in support of a beneficial role for disrupting subarachnoid thrombi and improving CSF flowin the early stages following SAH.
Risk of Aneurysmal Rerupture After Thrombolytic Therapy
The intended scope of this study is limited to improving our understanding of mechanisms behind early cortical hypoperfusion and impaired CSF flow in the early stages of SAH. Nevertheless, we will consider the implications of an early intervention involving a thrombolytic agent. The most dangerous perceived complication of intrathecal thrombolytic therapy after SAH is secondary bleeding, which, even without thrombolytic therapy, is a major cause of death after SAH [2]. However, our understanding of the physical forces that affect the action of thrombolytic agents do not support the idea that intrathecal tPA would cause substantial clot lysis at the aneurysm site, in contrast to intravenous administration which is strongly contraindicated in SAH. This is due to the effect of pressure-driven permeation, where the pressure gradient across the clot is the major determinant of the rate at which clot lysis occurs [41]. Intravenously, tPA is most effective in the arterial circulation where the pressure gradient across a given clot is high and much less effective for venous thrombi where low pressure gradients exist [42]. At the site of an aneurysm clot, the pressure gradient is in the completely opposite direction to support pressure-driven permeation of tPA from the subarachnoid space and would substantially impair the rate of clot lysis. This idea is supported by clinical evidence, which to date have found no increase in the incidence of aneurysm rerupture following thrombolytic therapy [32, 34, 36]. Experiments designed to address the question of whether substantial clot lysis can occur at the aneurysm site from intrathecal tPA administration are warranted.
Conclusion
Blood clots within the subarachnoid space impair CSF flow, which contributes to the increased ICP and impaired cortical perfusion during the early phase (24 h) after SAH. Intracisternal tPA administration immediately after SAH partially restores CSF flow, reduces ICP, and improves cortical perfusion. Improvement of CSF flow dynamics may serve as an important therapeutic target for SAH.
Acknowledgments
Funding Sources Dominic A. Siler—NHLBI F30 HL108624, Oregon Brain Institute.
Nabil J. Alkayed—NINDS R01 NS044313 and NS070837
Ruikang K. Wang—NHLBI R01 HL093140 and NIBIB R01 EB009682
Footnotes
Conflict of Interest Dominic A. Siler declares that he has no conflict of interest.
Jorge A. Gonzalez declares that he has no conflict of interest.
Ruikang K. Wang declares that he has no conflict of interest.
Justin S. Cetas declares that he has no conflict of interest.
Nabil J. Alkayed declares that he has no conflict of interest.
All institutional and national guidelines for the care and use of laboratory animals were followed.
Contributor Information
Dominic A. Siler, Department of Anesthesiology and Perioperative Medicine, Oregon Health and Science University, 3181 S.W. Sam Jackson Pk. Rd., UHN-2, Portland, OR 97239-3098, USA Department of Neurological Surgery, Oregon Health and Science, University, Portland, OR 97239-3098, USA.
Jorge A. Gonzalez, Department of Anesthesiology and Perioperative Medicine, Oregon Health and Science University, 3181 S.W. Sam Jackson Pk. Rd., UHN-2, Portland, OR 97239-3098, USA
Ruikang K. Wang, Department of Bioengineering, University of Washington, Seattle, WA 98195, USA
Justin S. Cetas, Department of Neurological Surgery, Oregon Health and Science, University, Portland, OR 97239-3098, USA Neurological Surgery, Portland VA Medical Center, Portland, OR 97239-3098, USA.
Nabil J. Alkayed, Department of Anesthesiology and Perioperative Medicine, Oregon Health and Science University, 3181 S.W. Sam Jackson Pk. Rd., UHN-2, Portland, OR 97239-3098, USA, alkayedn@ohsu.edu Department of Neurological Surgery, Oregon Health and Science, University, Portland, OR 97239-3098, USA; The Knight Cardiovascular Institute, Oregon Health and Science, University, Portland, OR 97239-3098, USA.
References
- 1.Johanson C, Duncan J, Klinge P, Brinker T, Stopa E, Silverberg G. Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 2008;5(1):10. doi: 10.1186/1743-8454-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Gijn J, Kerr RS, Rinkel GJE. Subarachnoid haemorrhage. Lancet. 2007;369(9558):306–18. doi: 10.1016/S0140-67360760153-6. [DOI] [PubMed] [Google Scholar]
- 3.Caner B, Hou J, Altay O, Fuj M, 2nd, Zhang JH. Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem. 2012;123(Suppl 2):12–21. doi: 10.1111/j.1471-4159.2012.07939.x. [DOI] [PubMed] [Google Scholar]
- 4.de Rooij NK, Greving JP, Rinkel GJE, Frijns CJM. Early prediction of delayed cerebral ischemia after subarachnoid hemorrhage: development and validation of a practical risk chart. Stroke. 2013;44(5):1288–94. doi: 10.1161/strokeaha.113.001125. [DOI] [PubMed] [Google Scholar]
- 5.Kreiter KT, Copeland D, Bernardini GL, Bates JE, Peery S, Claassen J, et al. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke. 2002;33(1):200–9. doi: 10.1161/hs0102.101080. [DOI] [PubMed] [Google Scholar]
- 6.Yan J, Li L, Khatibi NH, Yang L, Wang K, Zhang W, et al. Blood–brain barrier disruption following subarchnoid hemorrhage may be faciliated through PUMA induction of endothelial cell apoptosis from the endoplasmic reticulum. Exp Neurol. 2011;230(2):240–7. doi: 10.1016/j.expneurol.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 7.Sarrafzadeh A, Haux D, Sakowitz O, Benndorf G, Herzog H, Kuechler I, et al. Acute focal neurological deficits in aneurysmal subarachnoid hemorrhage: relation of clinical course, CT findings, and metabolite abnormalities monitored with bedside microdialysis. Stroke. 2003;34(6):1382–8. doi: 10.1161/01.str.0000074036.97859.02. [DOI] [PubMed] [Google Scholar]
- 8.Sakowitz OW, Santos E, Nagel A, Krajewski KL, Hertle DN, Vajkoczy P, et al. Clusters of spreading depolarizations are associated with disturbed cerebral metabolism in patients wwith aneurysmal subarachnoid hemorrhage. Stroke. 2013;44(1):220–3. doi: 10.1161/strokeaha.112.672352. [DOI] [PubMed] [Google Scholar]
- 9.Lee J-Y, He Y, Sagher O, Keep R, Hua Y, Xi G. Activated autophagy pathway in experimental subarachnoid hemorrhage. Brain Res. 2009;1287:126–35. doi: 10.1016/j.brainres.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Endo H, Nito C, Kamada H, Yu F, Chan PH. Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of Akt/glycogen synthase kinase-3beta survival signaling. J Cereb Blood FlowMetab. 2007;27(5):975–82. doi: 10.1038/sj.jcbfm.9600399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westermaier T, Jauss A, Eriskat J, Kunze E, Roosen K. Acute vasoconstriction: decrease and recovery of cerebral blood flow after various intensities of experimental subarachnoid hemorrhage in rats. J Neurosurg. 2009;110(5):996–1002. doi: 10.3171/2008.8.JNS08591. [DOI] [PubMed] [Google Scholar]
- 12.Woernle CM, Winkler KML, Burkhardt J-K, Haile SR, Bellut D, Neidert MC, et al. Hydrocephalus in 389 patients with aneurysmassociated subarachnoid hemorrhage. J Clin Neurosci. 2013;20(6):824–6. doi: 10.1016/j.jocn.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 13.Schubert G, Seiz M, Hegewald A, Manville J, Thomé C. Hypoperfusion in the acute phase of subarachnoid hemorrhage. In: Feng H, Mao Y, Zhang J, editors. Early brain injury or cerebral vasospasm Acta neurochirurgica supplements. Vienna: Springer; 2011. pp. 35–8. [DOI] [PubMed] [Google Scholar]
- 14.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4(147):147ra11. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackowski A, Crockard A, Burnstock G, Russell RR, Kristek F. The time course of intracranial pathophysiological changes following experimental subarachnoid haemorrhage in the rat. J Cereb Blood Flow Metab. 1990;10(6):835–49. doi: 10.1038/jcbfm.1990.140. [DOI] [PubMed] [Google Scholar]
- 16.Wang RK, Jacques SL, Ma Z, Hurst S, Hanson SR, Gruber A. Three dimensional optical angiography. Opt Express. 2007;15(7):4083–97. doi: 10.1364/oe.15.004083. [DOI] [PubMed] [Google Scholar]
- 17.Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ. Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab. 2009;29(8):1475–81. doi: 10.1038/jcbfm.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia Y, Alkayed N, Wang RK. Potential of optical microangiography to monitor cerebral blood perfusion and vascular plasticity following traumatic brain injury in mice in vivo. J Biomed Opt. 2009;14(4):040505. doi: 10.1117/1.3207121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40(7):2519–25. doi: 10.1161/STROKEAHA.109.549592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schubert GA, Seiz M, Hegewald AA, Manville J, Thome C. Acute hypoperfusion immediately after subarachnoid hemorrhage: a xenon contrast-enhanced CT study. J Neurotrauma. 2009;26(12):2225–31. doi: 10.1089/neu.2009.0924. [DOI] [PubMed] [Google Scholar]
- 21.Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45(4):545–52. doi: 10.1016/j.neuint.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Preston SD, Steart PV, Wilkinson A, Nicoll JAR, Weller RO. Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol Appl Neurobiol. 2003;29(2):106–17. doi: 10.1046/j.1365-2990.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 23.Schubert GA, Poli S, Mendelowitsch A, Schilling L, Thome C. Hypothermia reduces early hypoperfusion and metabolic alterations during the acute phase of massive subarachnoid hemorrhage: a laser-Doppler-flowmetry and microdialysis study in rats. J Neurotrauma. 2008;25(5):539–48. doi: 10.1089/neu.2007.0500. [DOI] [PubMed] [Google Scholar]
- 24.Chung WY, Lee LS. The effect of tissue plasminogen activator (t-PA) on cerebral vasospasm in canine subarachnoid hemorrhage. Zhonghua Yi Xue Za Zhi (Taipei) 1993;52(5):298–306. [PubMed] [Google Scholar]
- 25.Findlay JM, Weir BKA, Kanamaru K, Grace M, Baughman R. The effect of timing of intrathecal fibrinolytic therapy on cerebral vasospasm in a primate model of subarachnoid hemorrhage. Neurosurgery. 1990;26(2):201–6. doi: 10.1097/00006123-199002000-00003. [DOI] [PubMed] [Google Scholar]
- 26.Findlay JM, Weir BKA, Steinke D, Tanabe T, Gordon P, Grace M. Effect of intrathecal thrombolytic therapy on subarachnoid clot and chronic vasospasm in a primate model of SAH. J Neurosurg. 1988;69(5):723–35. doi: 10.3171/jns.1988.69.5.0723. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki H, Kanamaru K, Kuroki M, Sun H, Waga S, Miyazawa T. Effects of unilateral intrathecal administrations of low dose tissuetype plasminogen activator on clot lysis, vasospasm and brain phospholipid hydroperoxidation in a primate model of bilateral subarachnoid hemorrhage. Neurol Res. 1998;20(7):625–31. doi: 10.1080/01616412.1998.11740574. [DOI] [PubMed] [Google Scholar]
- 28.Asada M, Kong J, Nakamura M, Tamaki N. Thrombolytic therapy with tissue plasminogen activator for prevention of vasospasm in experimental subarachnoid hemorrhage: its efficacy and problems. Neurol Res. 1996;18(4):342–4. doi: 10.1080/01616412.1996.11740433. [DOI] [PubMed] [Google Scholar]
- 29.Kawada S, Kinugasa K, Meguro T, Hirotsune N, Tokunaga K, Kamata I, et al. Experimental study of intracisternal administration of tissue-type plasminogen activator followed by cerebrospinal fluid drainage in the ultra-early stage of subarachnoid haemorrhage. Acta Neurochir (Wien) 1999;141(12):1331–8. doi: 10.1007/s007010050438. [DOI] [PubMed] [Google Scholar]
- 30.Brinker T, Seifert V, Dietz H. Subacute hydrocephalus after experimental subarachnoid hemorrhage: its prevention by intrathecal fibrinolysis with recombinant tissue plasminogen activator. Neurosurgery. 1992;31(2):306–11. doi: 10.1227/00006123-199208000-00016. discussion 11–2. [DOI] [PubMed] [Google Scholar]
- 31.Brinker T, Seifert V, Stolke D. Effect of intrathecal fibrinolysis on cerebrospinal fluid absorption after experimental subarachnoid hemorrhage. J Neurosurg. 1991;74(5):789–93. doi: 10.3171/jns.1991.74.5.0789. [DOI] [PubMed] [Google Scholar]
- 32.Litrico S, Almairac F, Gaberel T, Ramakrishna R, Fontaine D, Sedat J, et al. Intraventricular fibrinolysis for severe aneurysmal intraventricular hemorrhage: a randomized controlled trial and meta-analysis. Neurosurg Rev. 2013;36(4):523–30. doi: 10.1007/s10143-013-0469-7. [DOI] [PubMed] [Google Scholar]
- 33.Kinouchi H, Ogasawara K, Shimizu H, Mizoi K, Yoshimoto T. Prevention of symptomatic vasospasm after aneurysmal subarachnoid hemorrhage by intraoperative cisternal fibrinolysis using tissuetype plasminogen activator combined with continuous cisternal drainage. Neurol Med Chir. 2004;44(11):569–77. doi: 10.2176/nmc.44.569. [DOI] [PubMed] [Google Scholar]
- 34.Ramakrishna R, Sekhar LN, Ramanathan D, Temkin N, Hallam D, Ghodke BV, et al. Intraventricular tissue plasminogen activator for the prevention of vasospasm and hydrocephalus after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2010;67(1):110–7. doi: 10.1227/01.NEU.0000370920.44359.91. discussion 7. [DOI] [PubMed] [Google Scholar]
- 35.Varelas PN, Rickert KL, Cusick J, Hacein-Bey L, Sinson G, Torbey M, et al. Intraventricular hemorrhage after aneurysmal subarachnoid hemorrhage: pilot study of treatment with intraventricular tissue plasminogen activator. Neurosurgery. 2005;56(2):205–13. doi: 10.1227/01.neu.0000147973.83688.d8. discussion –13. [DOI] [PubMed] [Google Scholar]
- 36.Findlay JM, Jacka MJ. Cohort study of intraventricular thrombolysis with recombinant tissue plasminogen activator for aneurysmal intraventricular hemorrhage. Neurosurgery. 2004;55(3):532–7. doi: 10.1227/01.neu.0000134473.98192.b1. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 37.Amin-Hanjani S, Ogilvy CS, Barker FG., 2nd Does intracisternal thrombolysis prevent vasospasm after aneurysmal subarachnoid hemorrhage? A meta-analysis. Neurosurgery. 2004;54(2):326–34. doi: 10.1227/01.neu.0000103488.94855.4f. discussion 34–5. [DOI] [PubMed] [Google Scholar]
- 38.Roos YB, Rinkel GJ, Vermeulen M, Algra A, van Gijn J. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev. 2003;2 doi: 10.1002/14651858.CD001245. CD001245. [DOI] [PubMed] [Google Scholar]
- 39.Hillman J, Fridriksson S, Nilsson O, Yu Z, Säveland H, Jakobsson K-E. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg. 2002;97(4):771–8. doi: 10.3171/jns.2002.97.4.0771. [DOI] [PubMed] [Google Scholar]
- 40.Iplikcioglu AC, Berkman MZ. The effect of short-term antifibrinolytic therapy on experimental vasospasm. Surg Neurol. 2003;59(1):10–6. doi: 10.1016/S0090-30190200867-4. [DOI] [PubMed] [Google Scholar]
- 41.Diamond SL, Anand S. Inner clot diffusion and permeation during fibrinolysis. Biophys J. 1993;65(6):2622–43. doi: 10.1016/S0006-34959381314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diamond SL. Engineering design of optimal strategies for blood clot dissolution. Annu Rev Biomed Eng. 1999;1(1):427–61. doi: 10.1146/annurev.bioeng.1.1.427. [DOI] [PubMed] [Google Scholar]