

Toll-like receptor (TLR) signaling stimulated by diverse microbial components plays a pivotal role in innate immunity by eliciting a powerful proinflammatory response that is essential for pathogen elimination. However, uncontrolled inflammation can result in tissue damage. Thus, TLR signaling has to be tightly controlled to maintain immune balance in the organism. The paradigm for modulating TLR signaling seems to center on the canonical nuclear factor-κB (NF-κB) pathway, most likely because of the central role of NF-κB in the production of various proinflammatory cytokines.1 In a recent issue of Nature Immunology, Yuk et al. have reported the identification of a novel endogenous NF-κB inhibitor in TLR signaling—small heterodimer partner (SHP)2 Interestingly, SHP functions in a self-regulating system: TLR signaling induces the expression of SHP in macrophages through Ca2+-dependent activation of AMP-activated protein kinase (AMPK), which has anti-inflammatory effects3 SHP in turn decreases the expression of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) by physically binding to two key components of the canonical NF-κB pathway, namely, RelA/p65 and TNF receptor-associated factor 6 (TRAF6) This work demonstrates an essential role for SHP in the negative control of TLR signaling and provides a novel underlying molecular mechanism.
SHP is an atypical orphan member of the nuclear receptor superfamily Lacking a DNA-binding domain, SHP is a transcriptional corepressor that is involved in endocrine regulation.4 By examining the susceptibility of SHP-deficient mice to endotoxin shock, Yuk et al. have made the notable observation that SHP protects mice against the lethality induced by experimental endotoxemia. The decreased survival of SHP-deficient mice in response to endotoxin is attributed to augmented expression of proinflammatory cytokines from bone marrow-derived cells. With bone marrow-derived macrophages (BMDMs) as the major cell model, the authors showed that SHP participates in the regulation of inflammatory responses induced by various TLR agonists. Because several nuclear receptors exert anti-inflammatory effects by suppressing the transcription of genes encoding proinflammatory cytokines,5 it was reasonable for the authors to analyze p65, the major transactivating subunit of NF-κB.6 Indeed, they found that SHP hinders p65 nuclear translocation and DNA binding, which is associated with reduced Tnf promoter activity. Their results from coimmunoprecipitation and confocal analysis suggest an interaction between p65 and SHP in BMDMs. Even though these data help explain the anti-inflammatory effects of SHP, they indicate no novel molecular mechanism. Yuk et al. further showed that SHP interferes with the molecular events upstream of p65. The degradation of NF-κB inhibitor IκBα and the activation of the IκB kinase upon TLR stimulation correlate inversely with the protein level of SHP. By examining the initial steps of TLR signaling, the authors next showed that the lys63 (K63)-linked polyubiquitination of TRAF6 upon TLR stimulation is enhanced with SHP deficiency, but is impaired with SHP overexpression. Their results from coimmunoprecipitation and confocal analysis suggest that cytoplasmic SHP interacts with p65, but not TRAF6, in unstimulated resting BMDMs, and that TLR signaling leads to the binding of SHP to TRAF6, which is associated with reduced SHP–p65 interaction. The authors propose that the binding of SHP to TRAF6 hinders TRAF6 polyubiquitination because the interaction of TRAF6 with SHP depends on an amino-terminal RING domain, while this domain is required for the binding of TRAF6 to an E2 ubiquitin-conjugating enzyme and for TRAF6 to act as an E3 ubiquitin ligase.7 Moreover, the authors provide data indicating that TLR signaling induces endogenous SHP expression in an AMPK-dependent manner, consistent with previous findings that show that AMPK contributes to the induction of shp expression by various extracellular stimuli.8, 9 TLR-dependent activation of AMPK requires intracellular Ca2+ influx, whereas other extracellular stimuli might trigger AMPK activation in a Ca2+-independent manner. AMPK activation mediates SHP expression via transcription factor upstream stimulatory factor-1. Collectively, the complicated and challenging series of experiments performed identify previously unrecognized dual regulatory functions of SHP in TLR signaling and provide new insights into the important post-translational regulatory roles of nuclear receptors.
Some questions arise from this study. First, what is the molecular mechanism by which SHP dissociates from p65 and interacts with TRAF6 upon TLR stimulation? A possible explanation is that TLR signaling leads to post-translational modifications and conformational changes of SHP which result in the shuttling. Second, how does SHP modulate TRAF6 polyubiquitination? SHP does not contain a deubiquitination enzyme domain. Moreover, SHP deficiency does not affect the expression of A20 and CYLD, two known deubiquitinases that target the canonical NF-κB pathway.10 Even though SHP and TRAF6 coimmunoprecipitated and colocalized upon TLR stimulation, no evidence of a direct interaction was provided by the authors. It is possible that SHP and TRAF6 enter the same complex in TLR signaling and SHP dampens TRAF6 ubiquitination indirectly through some mediator(s). If SHP indeed directly interacts with TRAF6 upon TLR stimulation, it may mask the binding site of the E2 ubiquitin-conjugating enzyme and consequently prevent TRAF6 from acting as an E3 ubiquitin ligase. Third, what is the physiological role of SHP–p65 interaction? The optimal SHP–p65 interaction seems to occur in unstimulated resting BMDMs, while TLR signaling leads to reduced SHP–p65 interaction. These facts suggest that the binding of SHP to p65 does not play a major role in the modulation of TLR signaling by SHP. By contrast, SHP–p65 interaction might be involved in the control of the basal level of NF-κB activity. Fourth, does SHP inhibit the activation of intracellular signaling pathway(s) other than the canonical NF-κB pathway? Adaptor TRAF6 transduces signals from activated TLRs to different intracellular signaling pathways. Theoretically, the modulation of other signaling pathways such as the mitogen-activated protein kinase pathways might also contribute to the anti-inflammatory effects of SHP. Future studies are required to address these issues.
Nevertheless, this is the first study to show that the orphan nuclear receptor SHP inhibits TLR signaling through a transcription-dependent mechanism and post-translational regulation. These findings illustrate the complexities of innate immunity and indicate possible future strategies for controlling systemic inflammation and endotoxic shock.
Figure 1.
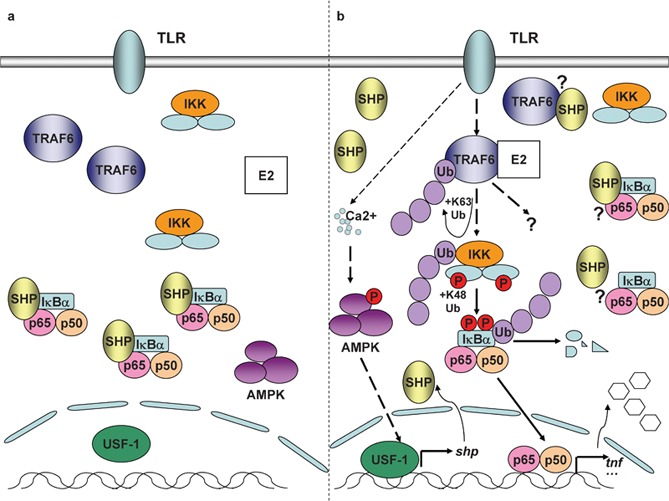
SHP in control of TLR signaling. (a) In unstimulated resting BMDMs, SHP interacts with p65, the major transactivating subunit of NF-κB. (b) TLR signaling induces dissociation of SHP from p65. TLR signaling simultaneously induces the expression of SHP in macrophages through Ca2+-dependent activation of AMPK. SHP in turn decreases TLR-triggered expression of proinflammatory cytokines such as TNF-α by physically binding to TRAF6 and dampening TRAF6 ubiquitination. AMPK, AMP-activated protein kinase; BMDM, bone marrow-derived macrophage; IKK, IκB kinase; NF-κB, nuclear factor-κB; SHP, small heterodimer partner; TLR, Toll-like receptor; TNF-α, tumor-necrosis factor-α TRAF6, TNF receptor-associated factor 6; USF-1, upstream stimulatory factor-1.
References
- Liew FY, Xu D, Brint EK, O'Neill LAJ. Negative regulation of Toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol. 2011;12:742–751. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- Sag D, Carling D, Stout RD, Shuttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavner A, Sabyal S, Gustafsson JA, Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab. 2005;16:478–488. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissink S, van Heerde EC, Schmitz ML, Kalkhoven E, vander Burg B, et al. Distinct domains of the RelA NF-κB subunit are required for negative cross-talk and direct interaction with the glucocorticoid receptor. J Biol Chem. 1997;272:22278–22284. doi: 10.1074/jbc.272.35.22278. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Chanda D, Li T, Song KH, Kim TH, Sim J, Lee CH, et al. Hepatocyte growth factor family negatively regulates hepatic gluconeogenesis via induction of orphan nuclear receptor small heterodimer partner in primary hepatocytes. J Biol Chem. 2009;284:28610–28521. doi: 10.1074/jbc.M109.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–314. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8:501–511. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]